The Self-Assembled Behavior of DNA Bases on the Interface



Abstract
:1. Introduction
2. Nanostructure Assembly in Two Dimensions (2D)
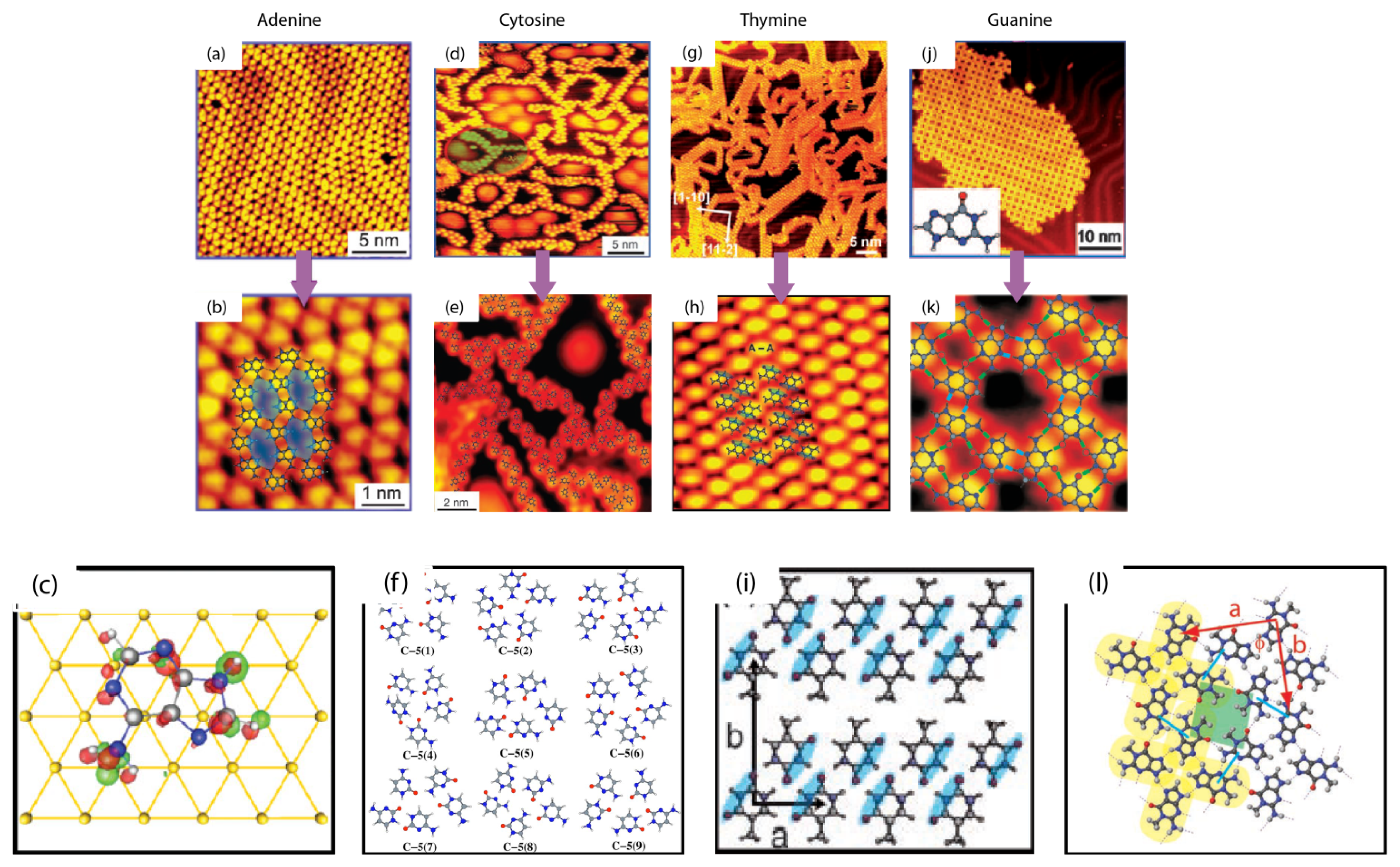
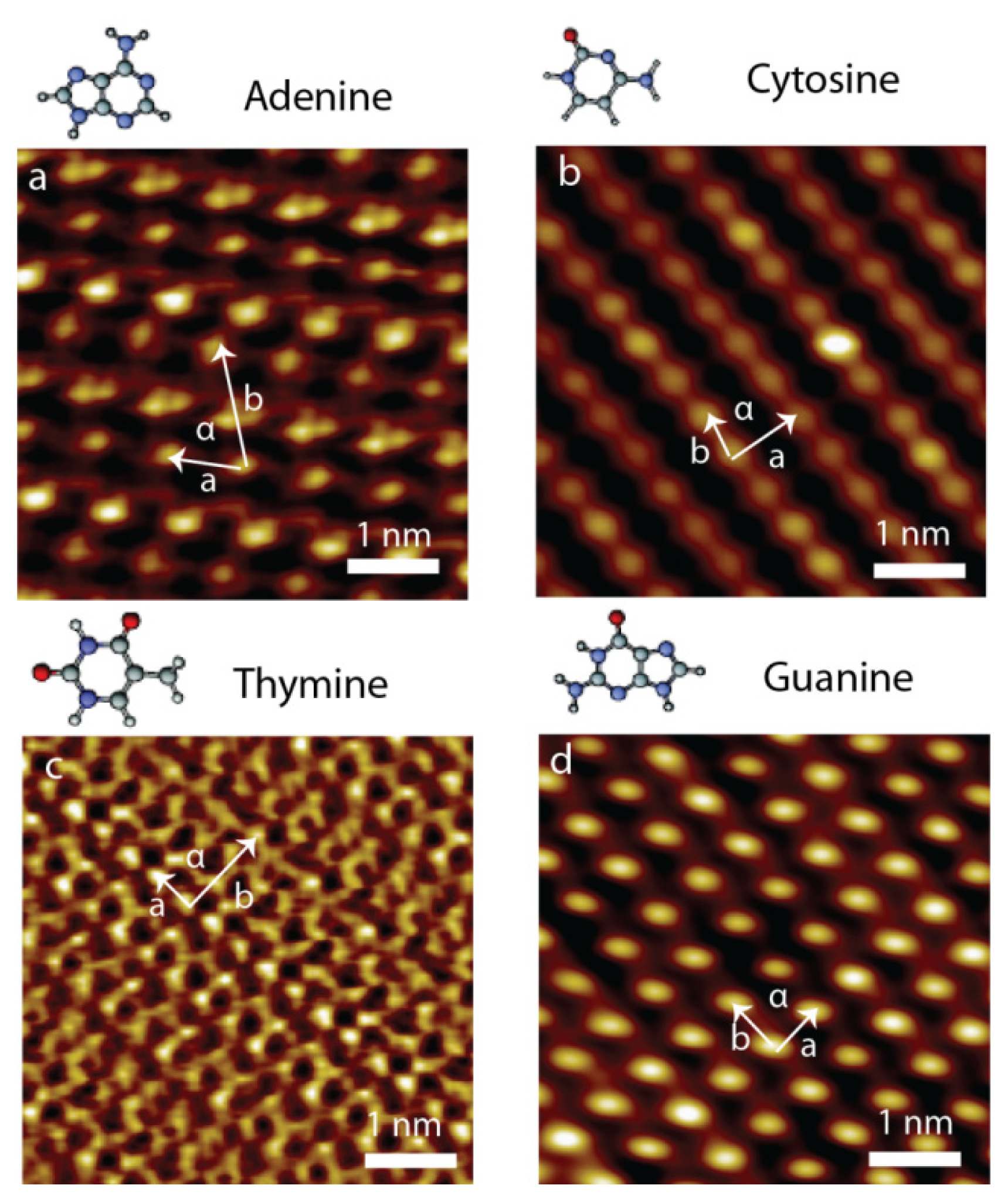
2.1. Self-Assembly of Individual Nucleic Acids Bases at Vacuum
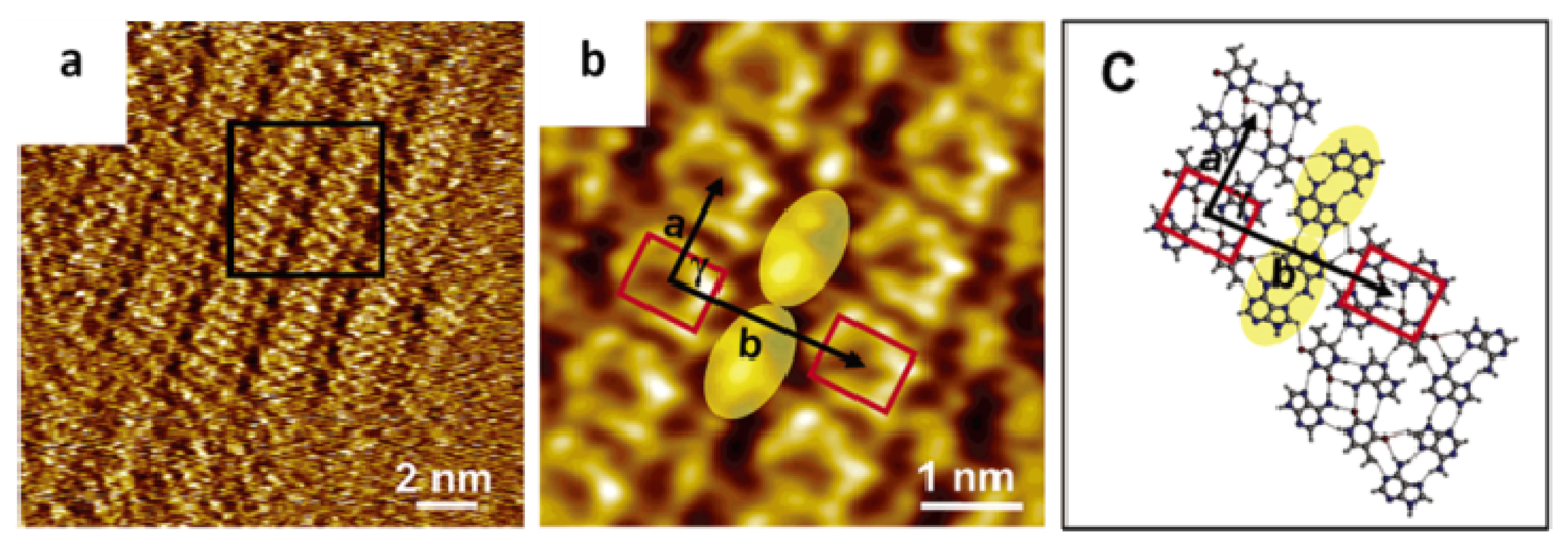
2.2. Self-Assembly of Complementary and Non-Complementary Nucleic Acids Bases at Vacuum
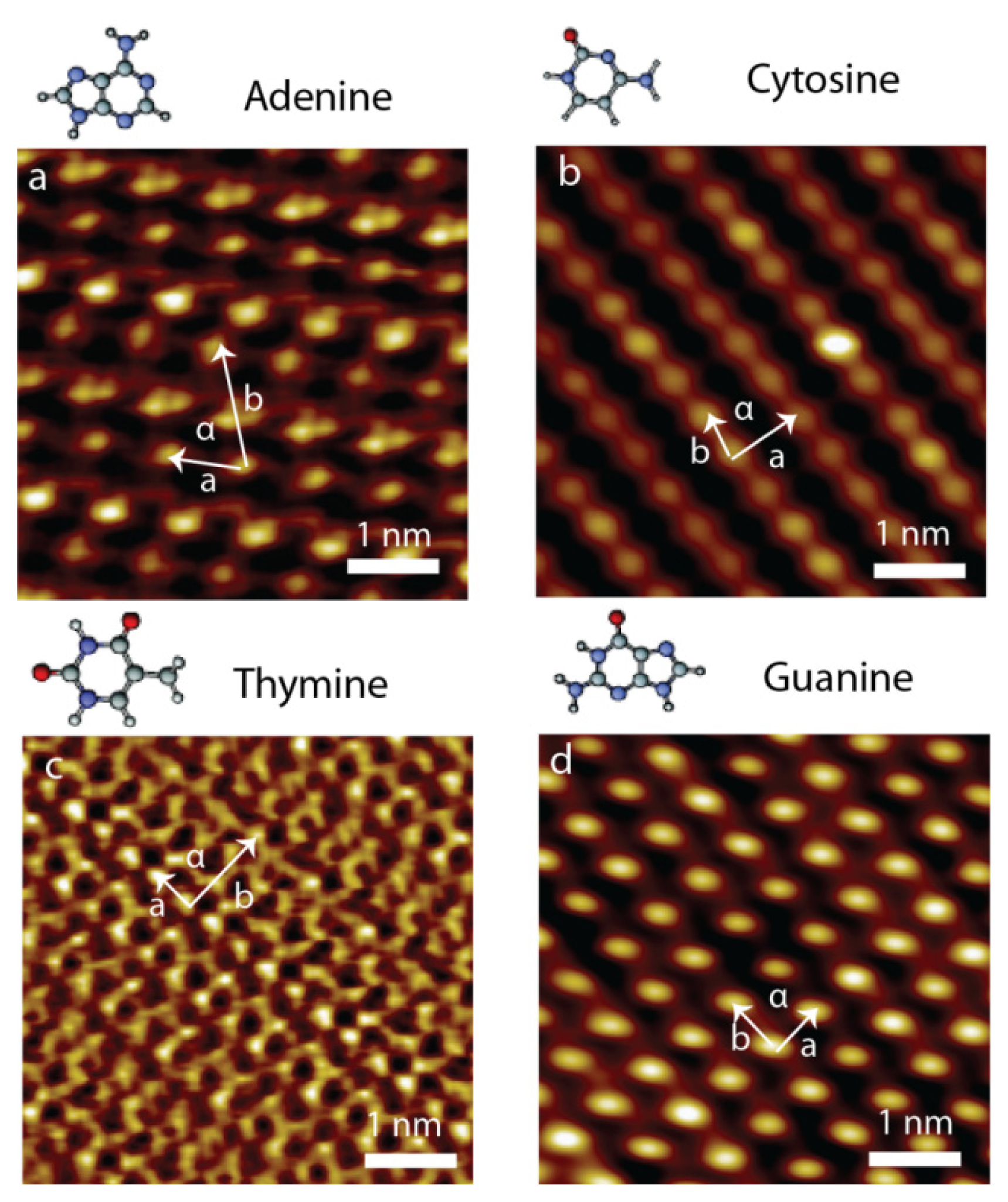
2.3. Self-Assembly of Individual Nucleic Acid Bases at Ambient Condition
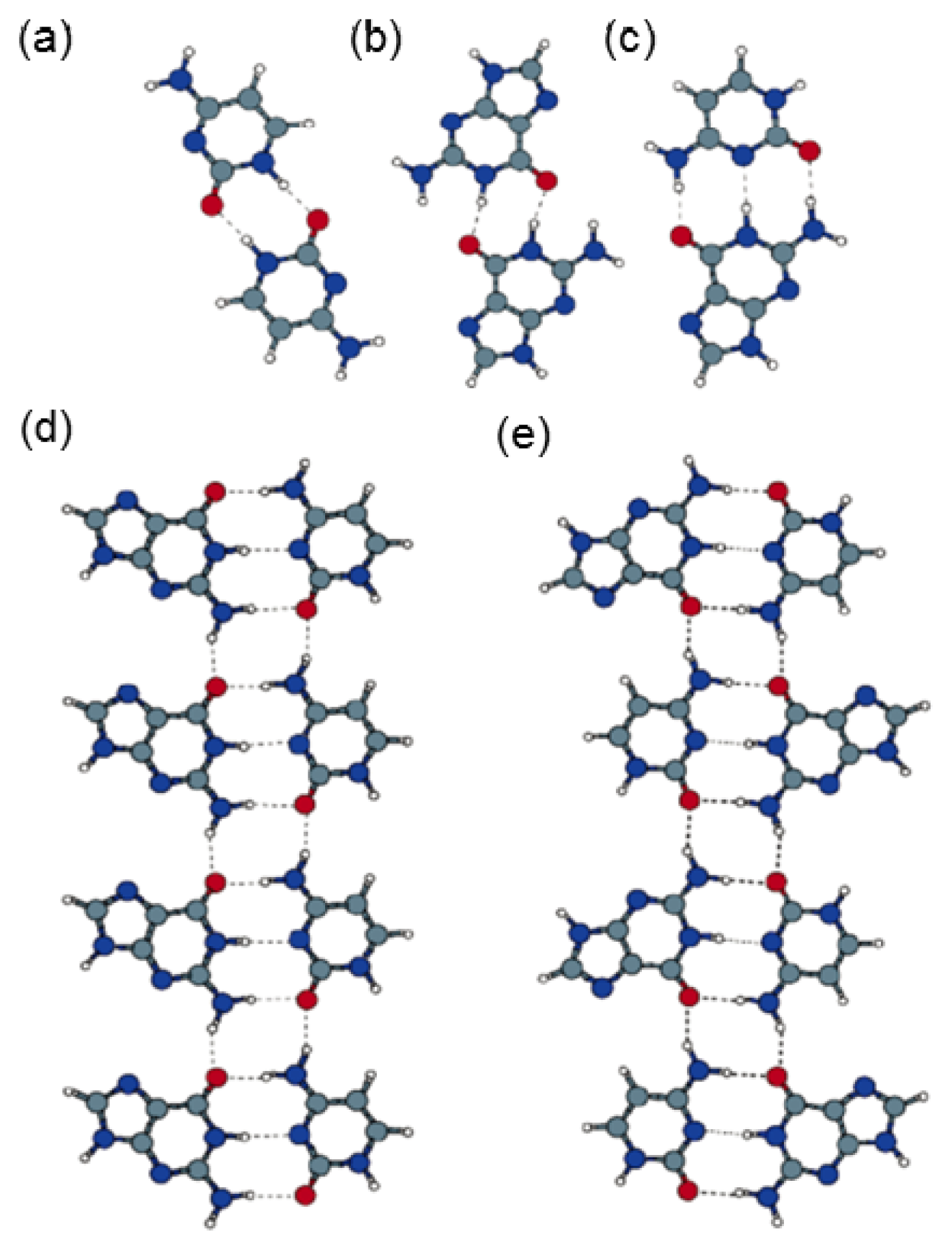
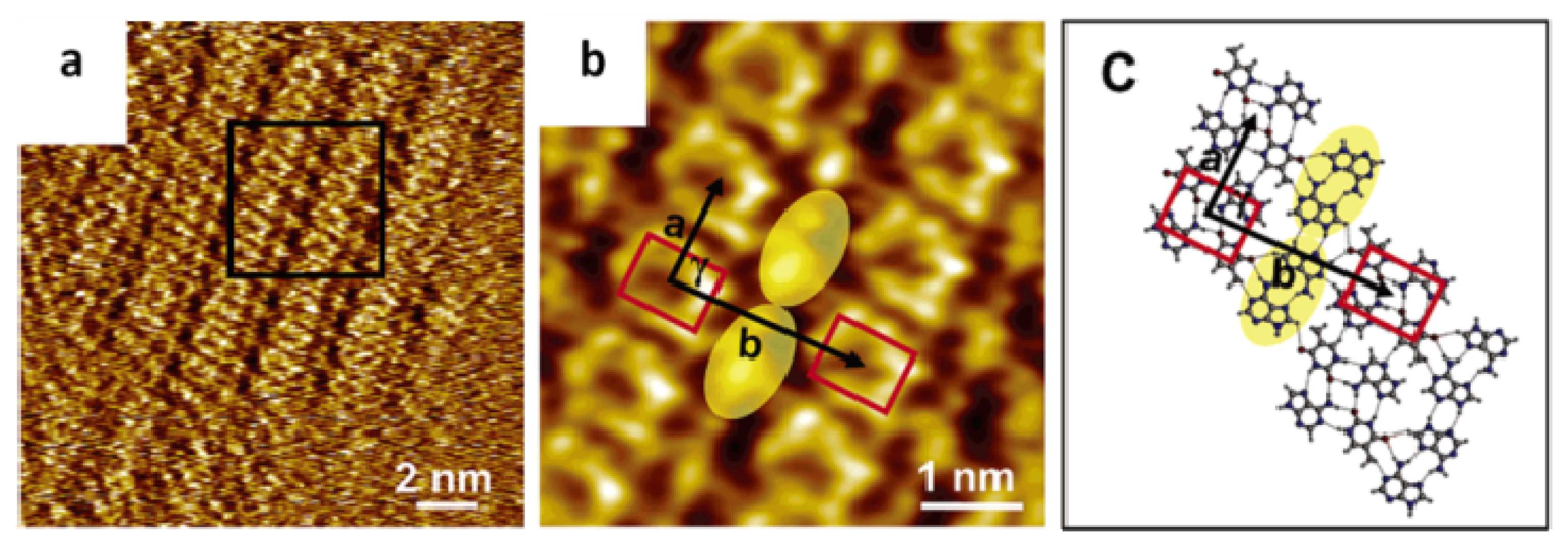
2.4. Self-Assembled Structure of Complementary Bases (G–C, A–T)
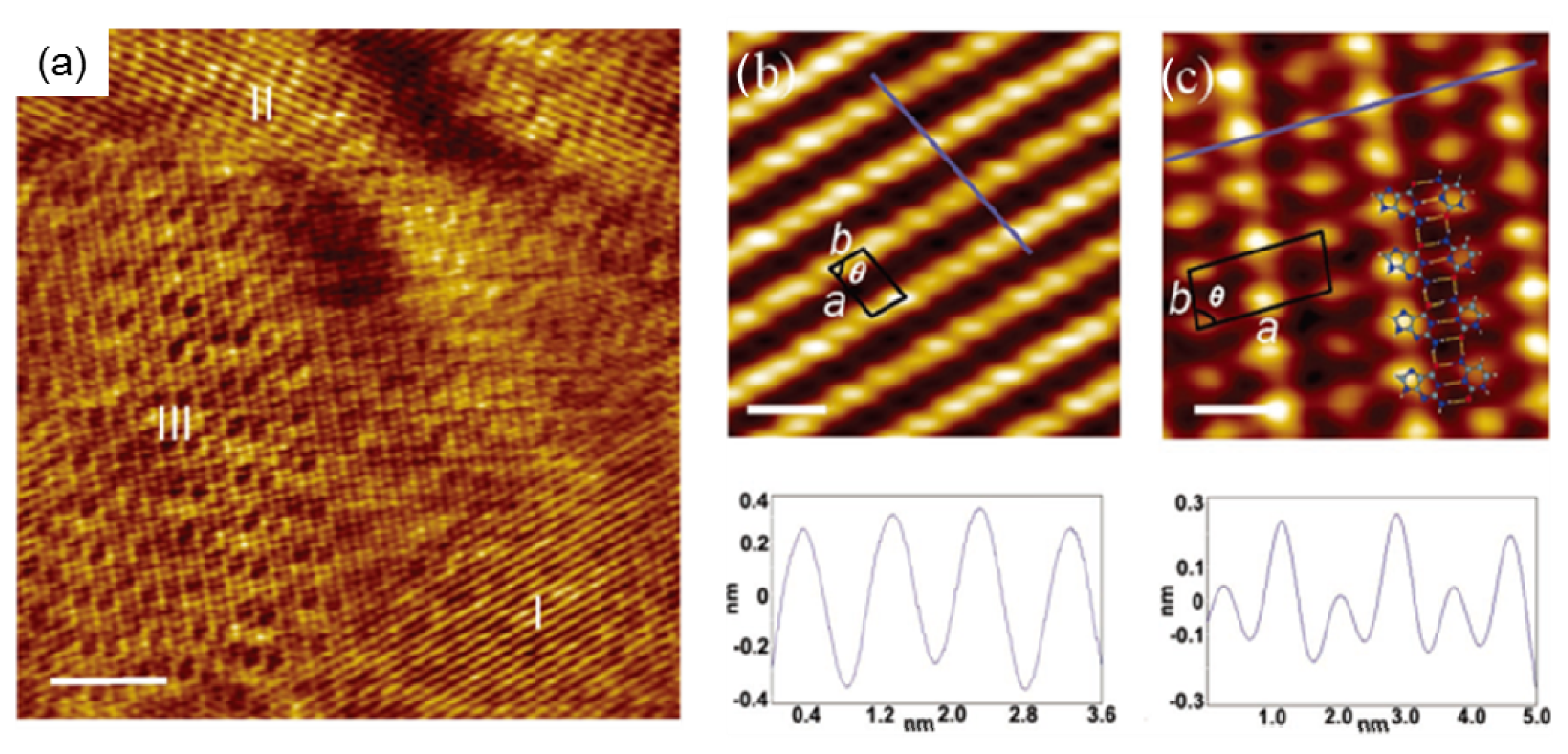
2.4.1. Self-Assembled Structure of Complementary Bases G–C
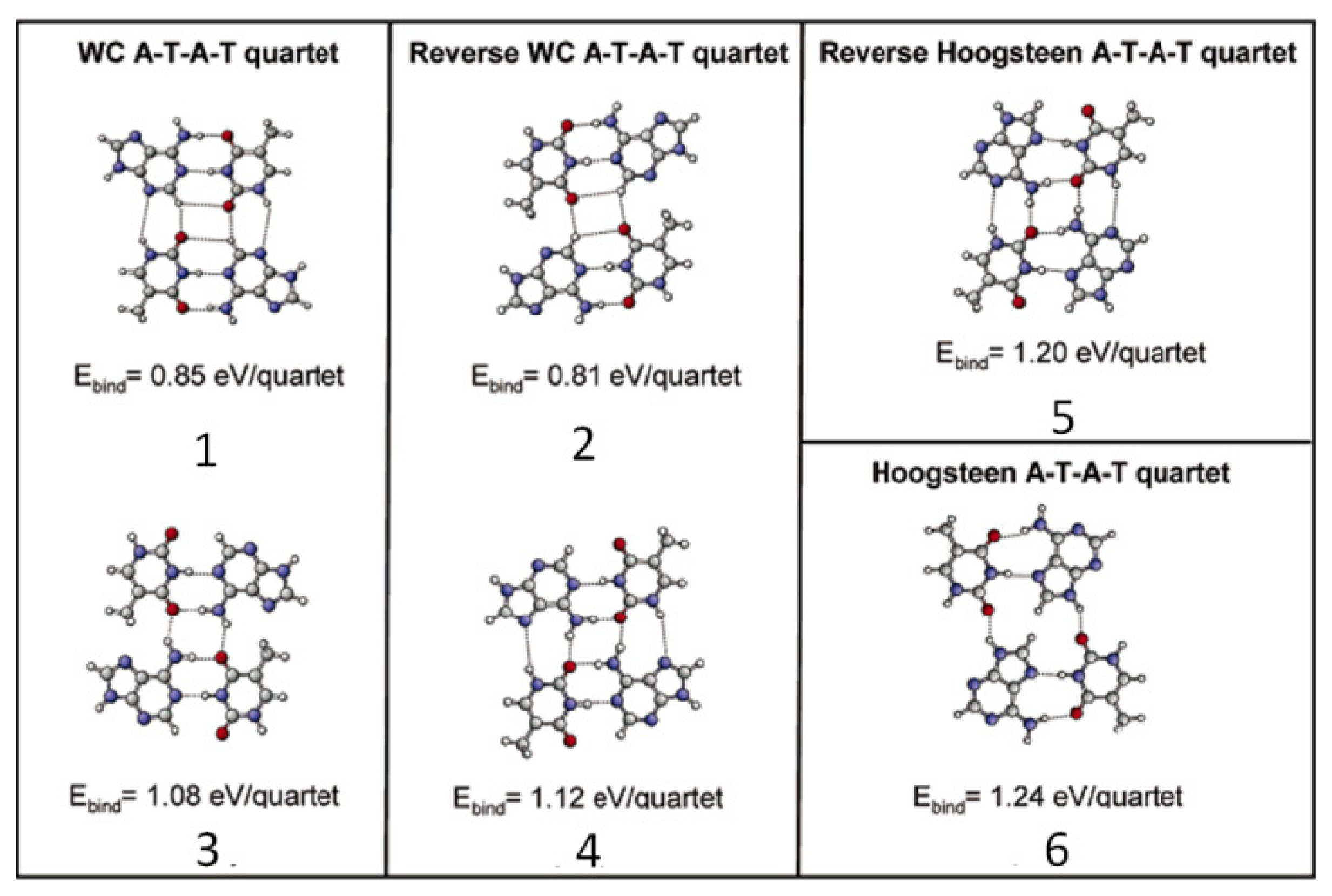
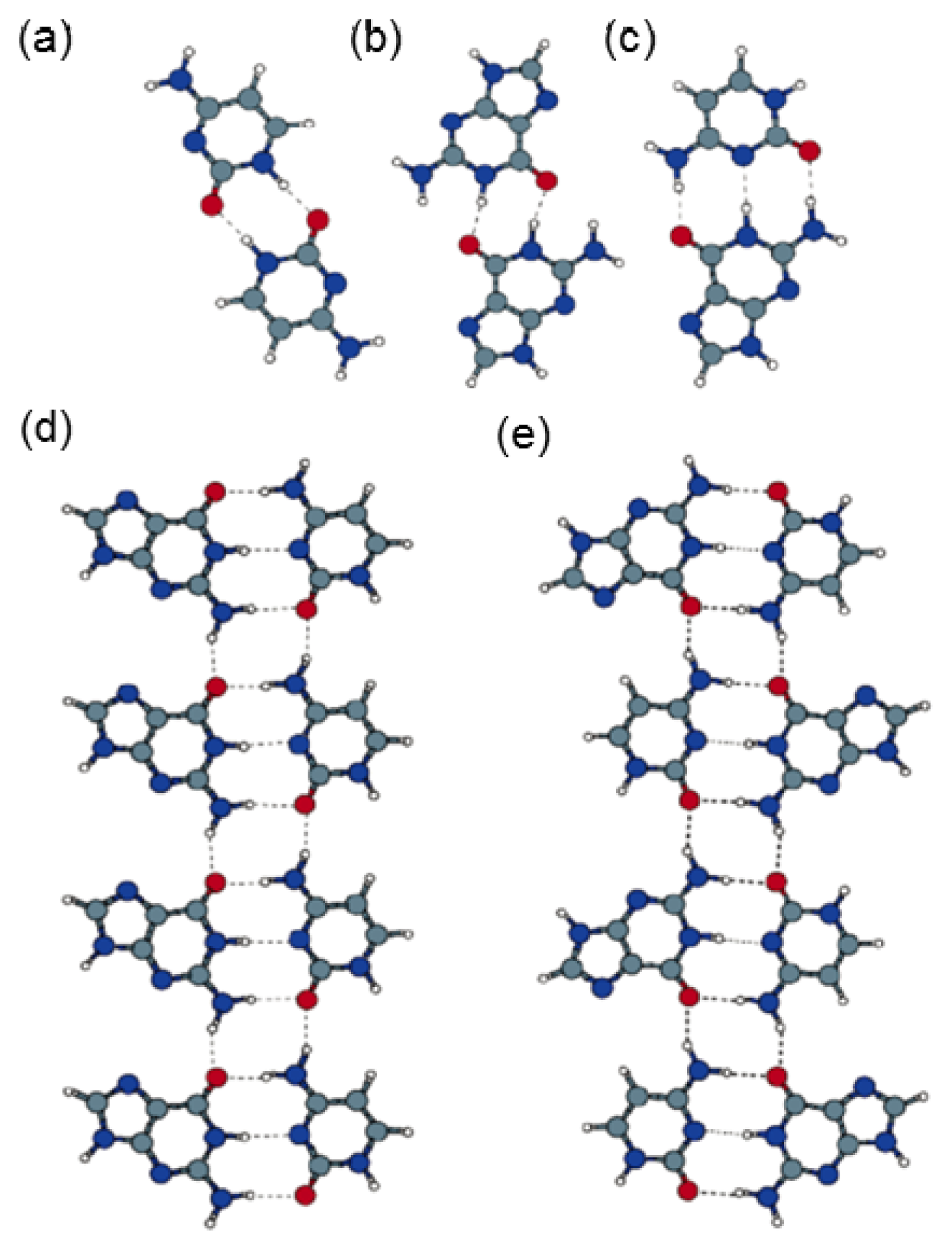
2.4.2. Self-Assembled Structure of Complementary Bases T–A
3. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
- Author ContributionsLei Liu and Mingdong Dong wrote the manuscript. Dan Xia and Lasse H. Klausen revised the manuscript and involved in discussion.
References
- Winfree, E.; Liu, F.R.; Wenzler, L.A.; Seeman, N.C. Design and self-assembly of two-dimensional DNA crystals. Nature 1998, 394, 539–544. [Google Scholar]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Hogberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2009, 459, 414–418. [Google Scholar]
- Saenger, W. Principles of Nucleic Acid Structures; Springer: New York, NY, USA, 1984. [Google Scholar]
- Watson, J.D.; Crick, F.H.C. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid (Reprinted from Nature, 1953, volume 171, page 737–738). Revist. Investig. Clin 2003, 55, 108–109. [Google Scholar]
- Arnott, S. Sprung from chains. Nature 1984, 312, 174. [Google Scholar]
- Perdigao, L.M.A.; Staniec, P.A.; Champness, N.R.; Kelly, R.E.A.; Kantorovich, L.N.; Beton, P.H. Experimental and theoretical identification of adenine monolayers on Ag-terminated Si (111). Phys. Rev 2006. [Google Scholar] [CrossRef]
- Otero, R.; Schock, M.; Molina, L.M.; Laegsgaard, E.; Stensgaard, I.; Hammer, B.; Besenbacher, F. Guanine quartet networks stabilized by cooperative hydrogen bonds. Angew. Chem. Int. Ed 2005, 44, 2270–2275. [Google Scholar]
- McNutt, A.; Haq, S.; Raval, R. RAIRS investigations on the orientation and intermolecular interactions of adenine on Cu (110). Surf. Sci 2003, 531, 131–144. [Google Scholar]
- Kelly, R.E.A.; Lee, Y.J.; Kantorovich, L.N. Homopairing possibilities of the DNA base adenine. J. Phys. Chem 2005, 109, 11933–11939. [Google Scholar]
- Kelly, R.E.A.; Lee, Y.J.; Kantorovich, L.N. Homopairing possibilities of the DNA bases cytosine and guanine: An ab initio DFT study. J. Phys. Chem 2005, 109, 22045–22052. [Google Scholar]
- Kelly, R.E.A.; Kantorovich, L.N. Homopairing possibilities of the DNA base thymine and the RNA base uracil: An ab initio density functional theory study. J. Phys. Chem 2006, 110, 2249–2255. [Google Scholar]
- Furukawa, M.; Tanaka, H.; Kawai, T. Formation mechanism of low-dimensional superstructure of adenine molecules and its control by chemical modification: A low-temperature scanning tunneling microscopy study. Surf. Sci 2000, 445, 1–10. [Google Scholar]
- Furukawa, M.; Tanaka, H.; Kawai, T. The role of dimer formation in the self-assemblies of DNA base molecules on Cu (111) surfaces: A scanning tunneling microscope study. J. Chem. Phys 2001, 115, 3419–3423. [Google Scholar]
- Kawai, T.; Tanaka, H.; Nakagawa, T. Low dimensional self-organization of DNA-base molecules on Cu (111) surfaces. Surf. Sci 1997, 386, 124–136. [Google Scholar]
- Nishimura, M.; Tanaka, H.; Kawai, T. Structure of linear double-stranded deoxyribonucleic acid adsorbed on Cu (111) surfaces: A low-temperature scanning tunneling microscopy study. Jpn. J. Appl. Phys 2002, 41, 7510–7511. [Google Scholar]
- Sowerby, S.J.; Heckl, W.M. The role of self-assembled monolayers of the purine and pyrimidine bases in the emergence of life. Orig. Life Evol. Biosph 1998, 28, 283–310. [Google Scholar]
- Wandlowski, T.; Lampner, D.; Lindsay, S.M. Structure and stability of cytosine adlayers on Au (111): An in situ STM study. J. Electroanal. Chem 1996, 404, 215–226. [Google Scholar]
- Kelly, R.E.A.; Xu, W.; Lukas, M.; Otero, R.; Mura, M.; Lee, Y.-J.; Lægsgaard, E.; Stensgaard, I.; Kantorovich, L.N.; Besenbacher, F. An investigation into the interactions between self-assembled adenine molecules and a Au (111) surface. Small 2008, 4, 1494–1500. [Google Scholar]
- Otero, R.; Lukas, M.; Kelly, R.E.A.; Xu, W.; Lægsgaard, E.; Stensgaard, I.; Kantorovich, L.N.; Besenbacher, F. Elementary structural motifs in a random network of cytosine adsorbed on a gold (111) surface. Science 2008, 319, 312–315. [Google Scholar]
- Kelly, R.E.A.; Lukas, M.; Kantorovich, L.N.; Otero, R.; Xu, W.; Mura, M.; Laegsgaard, E.; Stensgaard, I.; Besenbacher, F. Understanding the disorder of the DNA base cytosine on the Au (111) surface. J. Chem. Phys 2008. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Kelly, R.E.A.; Otero, R.; Schöck, M.; Lægsgaard, E.; Stensgaard, I.; Kantorovich, L.N.; Besenbacher, F. Probing the hierarchy of thymine–thymine interactions in self-assembled structures by manipulation with scanning tunneling microscopy. Small 2007, 3, 2011–2014. [Google Scholar]
- Otero, R.; Xu, W.; Lukas, M.; Kelly, R.E.A.; Lægsgaard, E.; Stensgaard, I.; Kjems, J.; Kantorovich, L.N.; Besenbacher, F. Specificity of Watson-Crick base pairing on a solid surface studied at the atomic scale. Angew. Chem 2008, 120, 9819–9822. [Google Scholar]
- Bald, I.; Wang, Y.-G.; Dong, M.; Rosen, C.B.; Ravnsbæk, J.B.; Zhuang, G.-L.; Gothelf, K.V.; Wang, J.-G.; Besenbacher, F. Control of self-assembled 2D nanostructures by methylation of guanine. Small 2011, 7, 939–949. [Google Scholar]
- Xu, S.L.; Dong, M.D.; Rauls, E.; Otero, R.; Linderoth, T.R.; Besenbacher, F. Coadsorption of Guanine and Cytosine on graphite: Ordered structure based on GC pairing. Nano Lett 2006, 6, 1434–1438. [Google Scholar]
- Leonard, G.A.; Zhang, S.; Peterson, M.R.; Harrop, S.J.; Helliwell, J.R.; Cruse, W.B.T.; Destaintot, B.L.; Kennard, O.; Brown, T.; Hunter, W.N. Self-association of a DNA loop creates a quadruplex: Crystal-structure of d(GCATGCT) at 1.8 Å resolution. Structure 1995, 3, 335–340. [Google Scholar]
- Mamdouh, W.; Dong, M.D.; Xu, S.L.; Rauls, E.; Besenbacher, F. Supramolecular nanopatterns self-assembled by adenine-hymine quartets at the liquid/solid interface. J. Am. Chem. Soc 2006, 128, 13305–13311. [Google Scholar]
- Zain, R.; Sun, J.S. Do natural DNA triple-helical structures occur and function in vivo? Cell. Mol. Life Sci 2003, 60, 862–870. [Google Scholar]
- Lamond, A.I. Molecular biology of the cell, 4th edition. Nature 2002, 417, 383–383. [Google Scholar]
- Sivakova, S.; Rowan, S.J. Nucleobases as supramolecular motifs. Chem. Soc. Rev 2005, 34, 9–21. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a (nm) | b (nm) | α (degree) | |
|---|---|---|---|
| Pure G | 0.64 ± 0.07 | 0.68 ± 0.07 | 90.1 ± 2.6 |
| Pure C | 0.82 ± 0.08 | 0.53 ± 0.06 | 77.3 ± 2.3 |
| Pure A | 0.80 ± 0.10 | 2.20 ± 0.20 | 76.0 ± 2.3 |
| Pure T | 0.80 ± 0.10 | 1.50 ± 0.20 | 87.0 ± 2.5 |
| a (nm) | b (nm) | θ (degree) | |
|---|---|---|---|
| Domain I and II | 0.87 ± 0.09 | 0.45 ± 0.05 | 76.7 ± 2.2 |
| Domain III | 1.71 ± 0.18 | 0.69 ± 0.07 | 84.1 ± 2.4 |
| Pure cytosine | 0.82 ± 0.08 | 0.53 ± 0.06 | 77.3 ± 2.3 |
| Pure guanine | 0.64 ± 0.07 | 0.68 ± 0.07 | 90.1 ± 2.6 |
| a (nm) | b (nm) | γ (degree) | |
|---|---|---|---|
| Pure adenine (A) | 0.8 ± 0.1 | 2.2 ± 0.2 | 76.0 ± 2.3 |
| Pure thymine (T) | 0.8 ± 0.1 | 1.5 ± 0.2 | 87.0 ± 2.5 |
| Mixture A + T | 1.0 ± 0.2 | 2.3 ± 0.2 | 88.0 ± 2.0 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, L.; Xia, D.; Klausen, L.H.; Dong, M. The Self-Assembled Behavior of DNA Bases on the Interface. Int. J. Mol. Sci. 2014, 15, 1901-1914. https://doi.org/10.3390/ijms15021901
Liu L, Xia D, Klausen LH, Dong M. The Self-Assembled Behavior of DNA Bases on the Interface. International Journal of Molecular Sciences. 2014; 15(2):1901-1914. https://doi.org/10.3390/ijms15021901
Chicago/Turabian StyleLiu, Lei, Dan Xia, Lasse H. Klausen, and Mingdong Dong. 2014. "The Self-Assembled Behavior of DNA Bases on the Interface" International Journal of Molecular Sciences 15, no. 2: 1901-1914. https://doi.org/10.3390/ijms15021901