Regulation of an Autoimmune Model for Multiple Sclerosis in Th2-Biased GATA3 Transgenic Mice



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Results
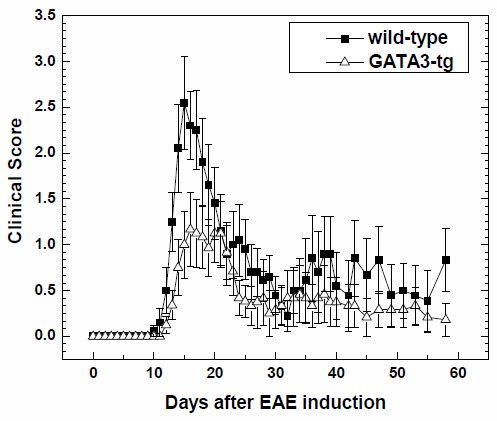
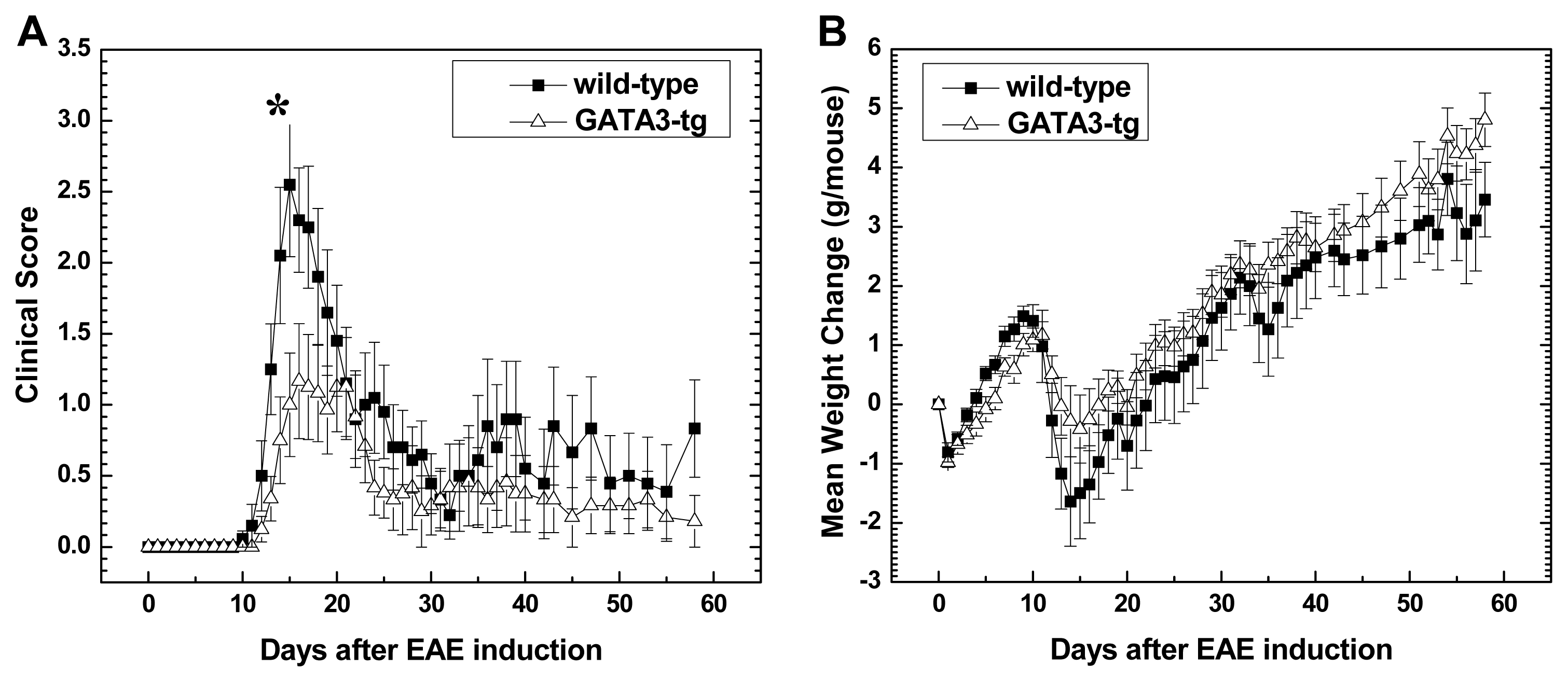
2.1.1. Suppression of Clinical Disease in GATA3-tg Mice with EAE
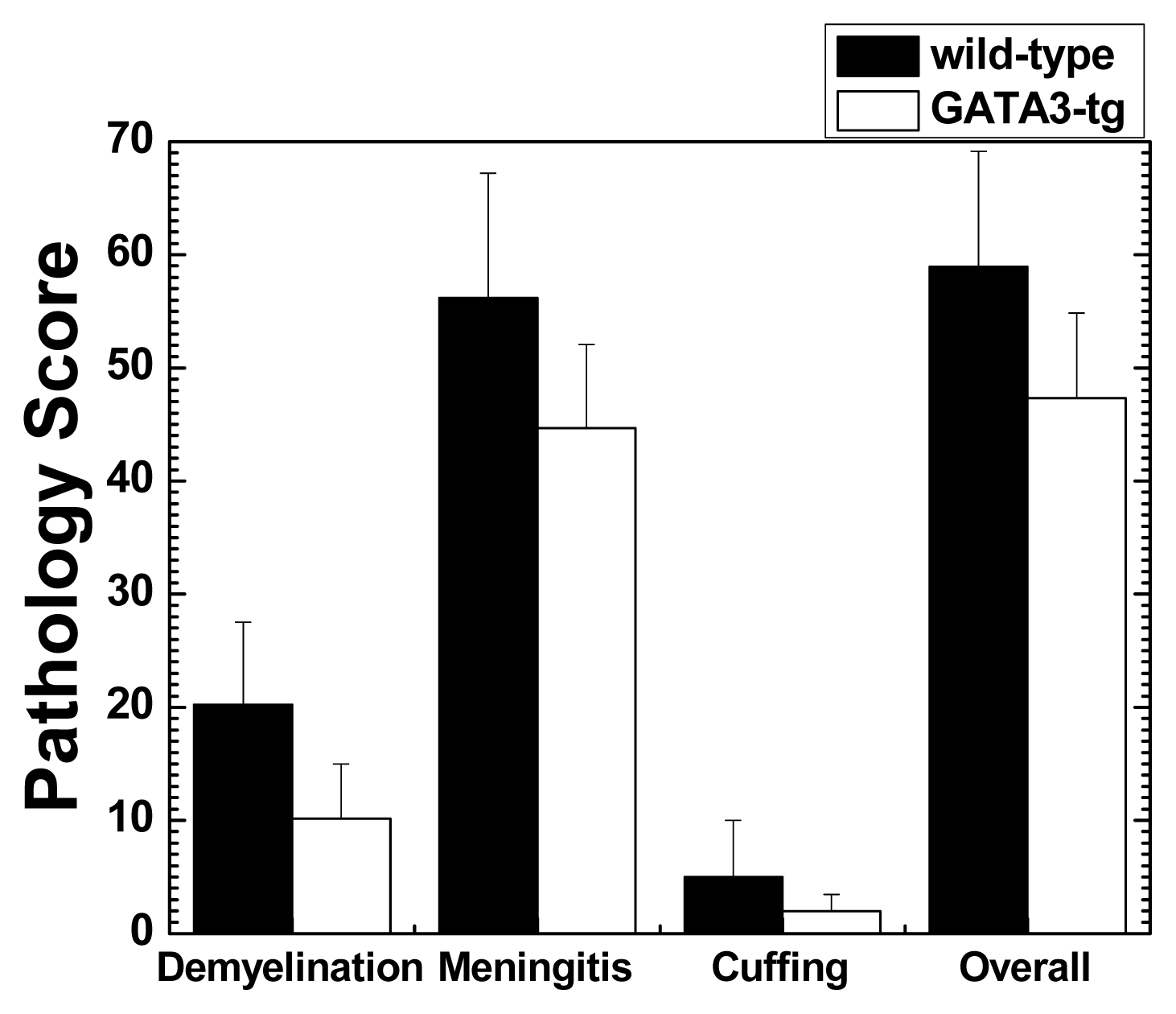
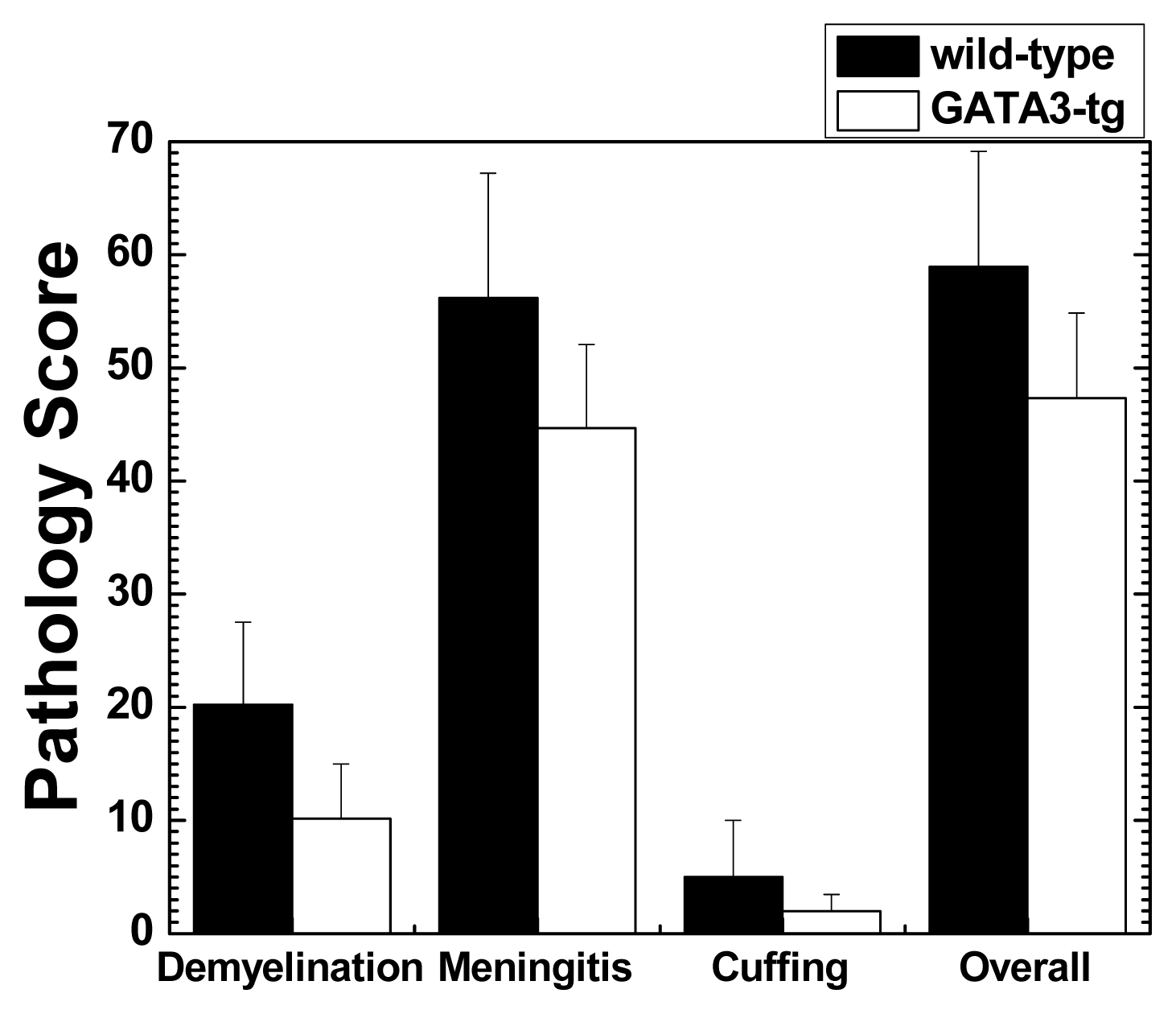
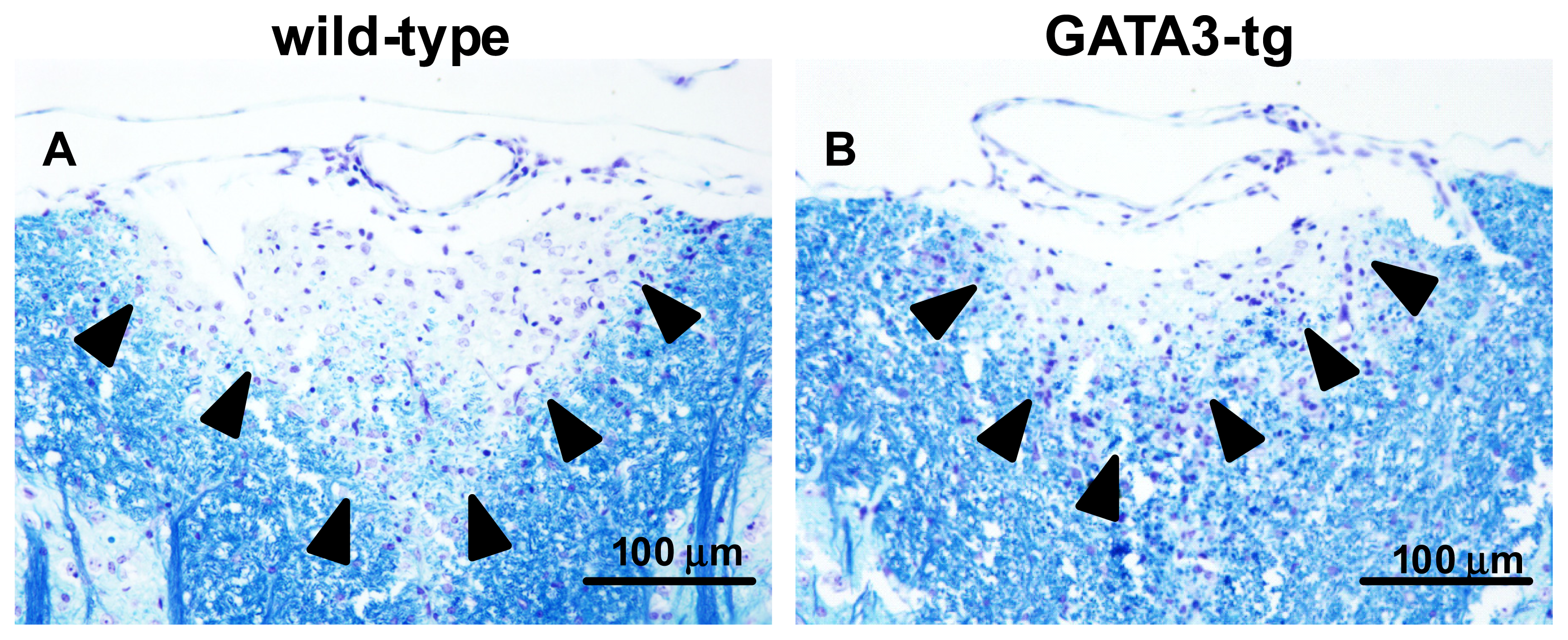
2.1.2. Suppression of Inflammatory Demyelination in GATA3-tg Mice
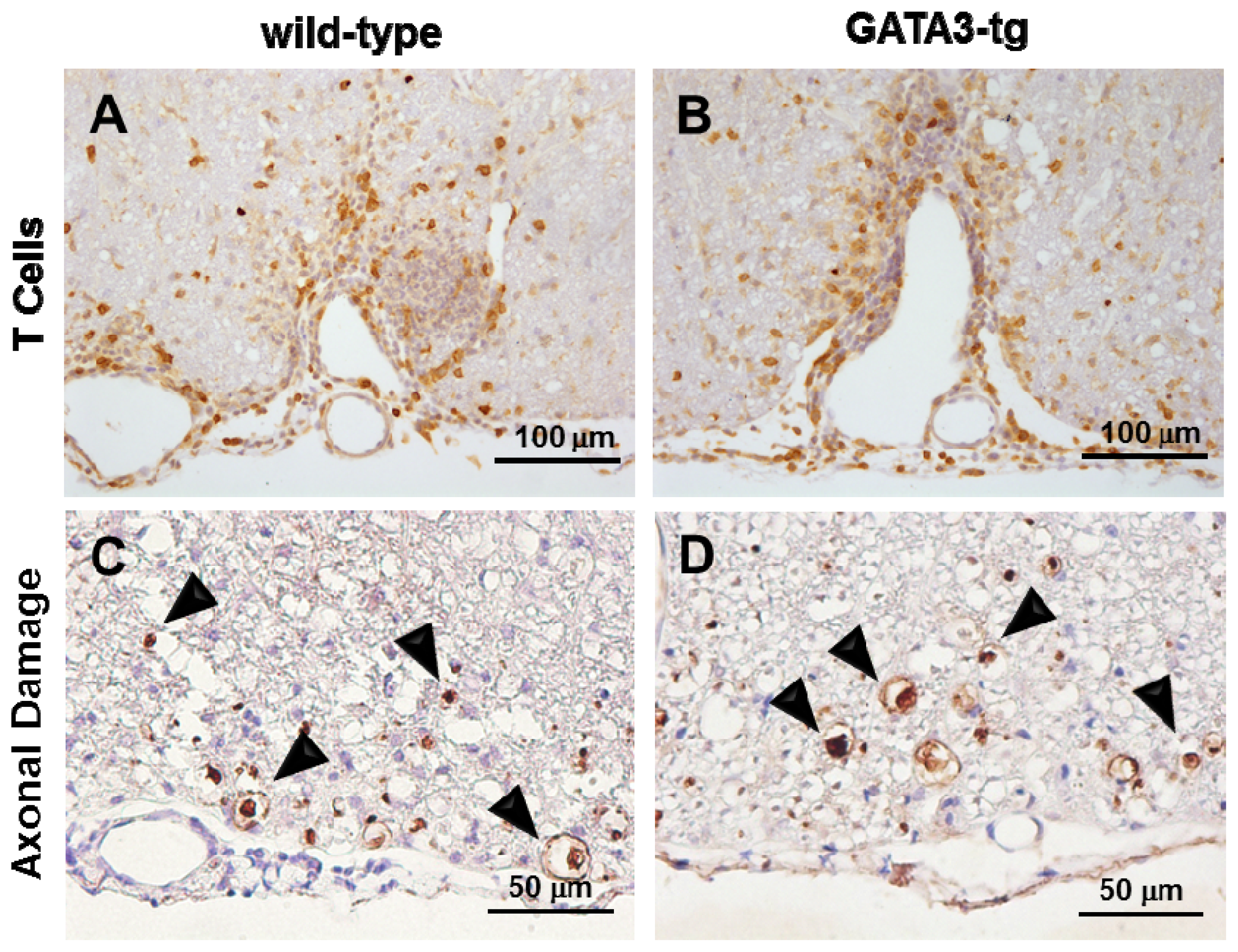
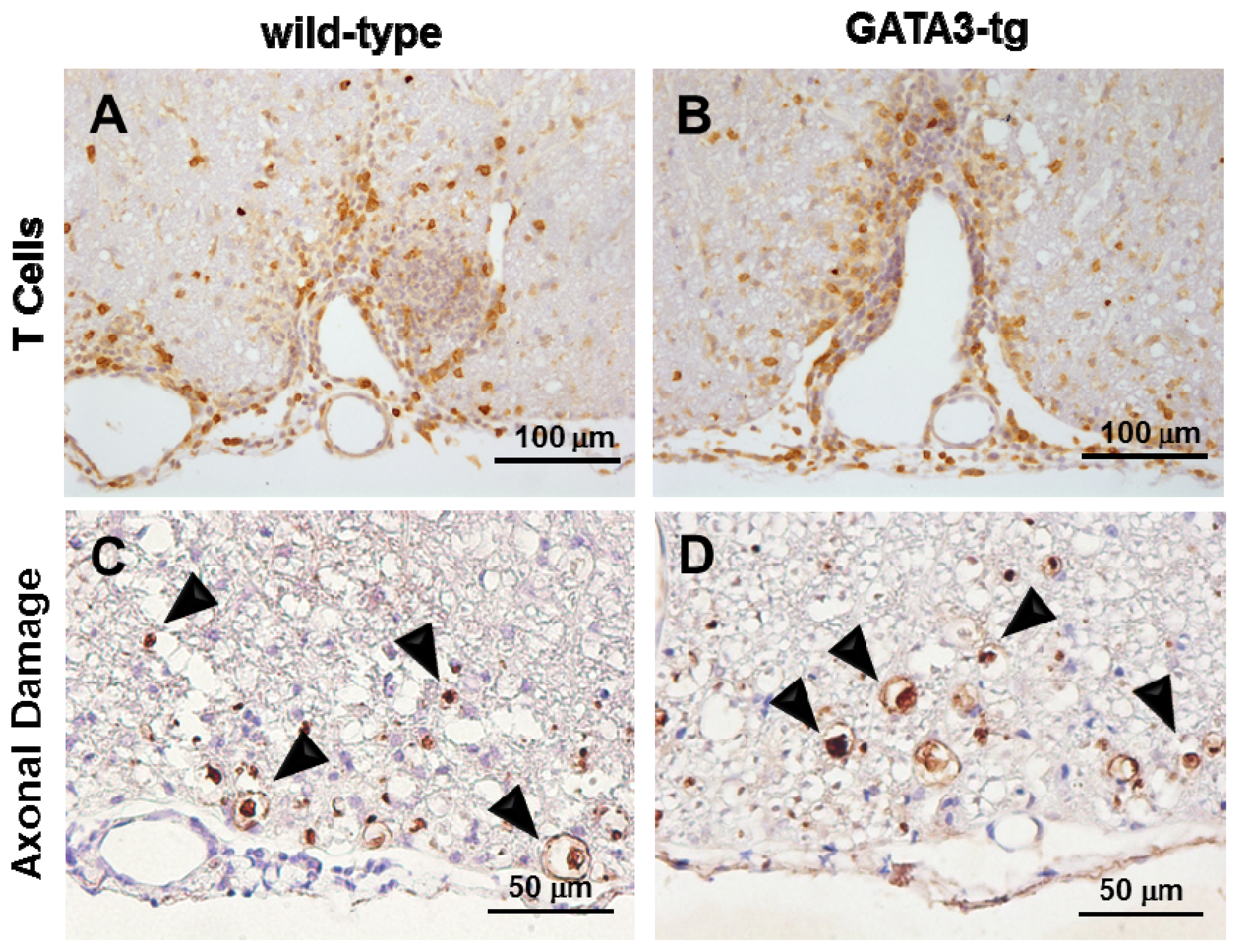
2.1.3. No Change in T Cell Ratio among Infiltrates or Axonal Degeneration in GATA3-tg Mice
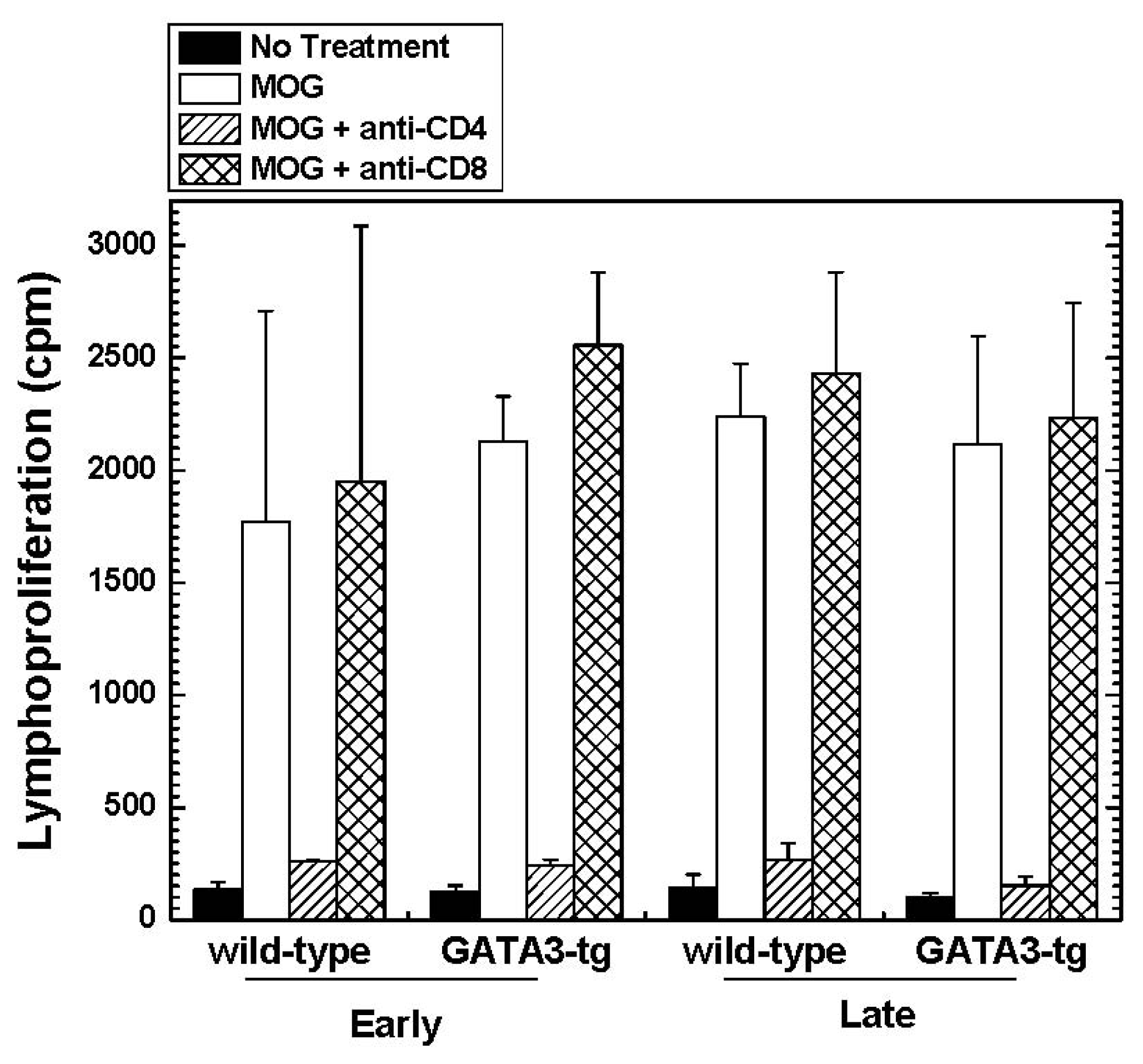
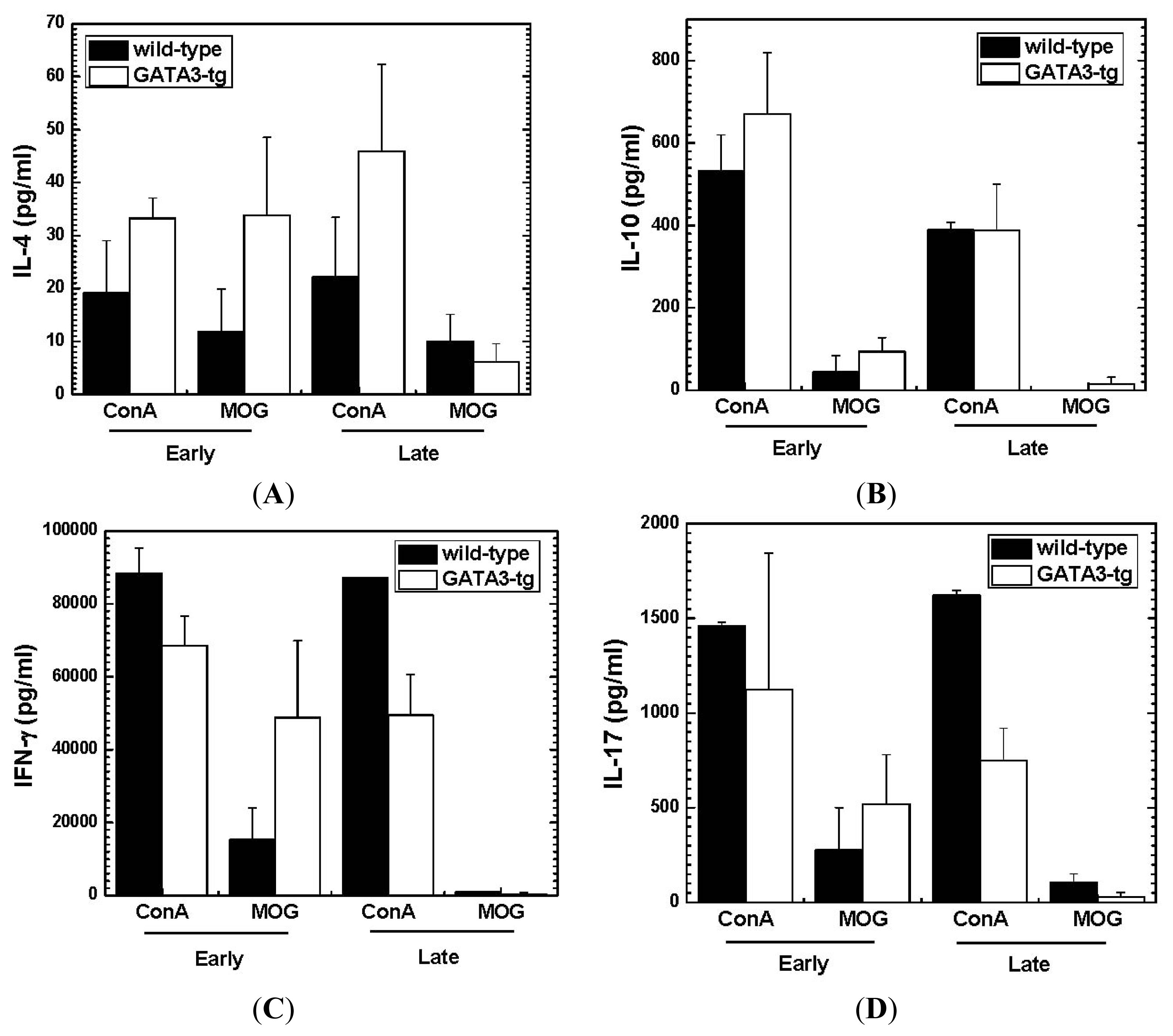
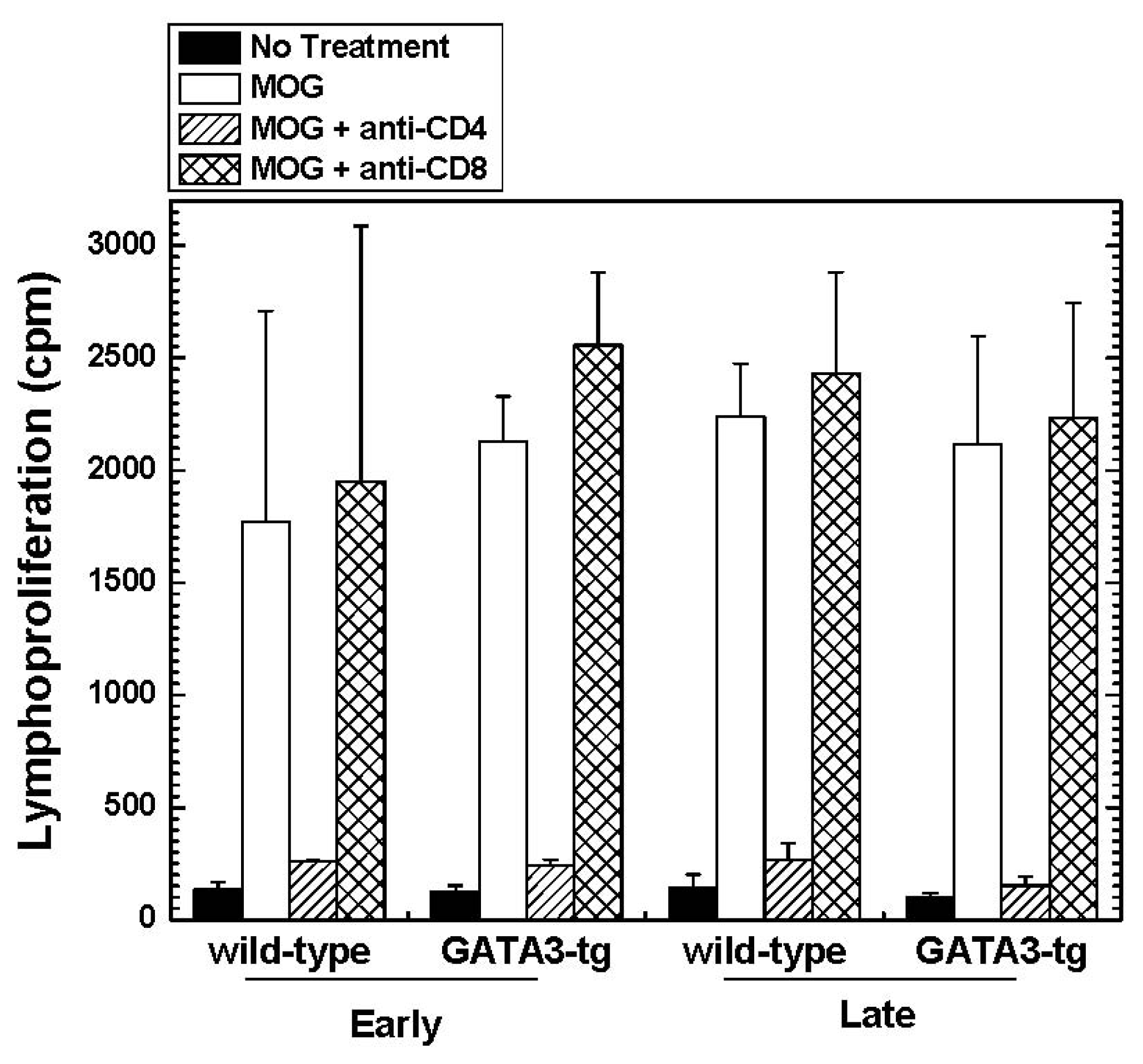
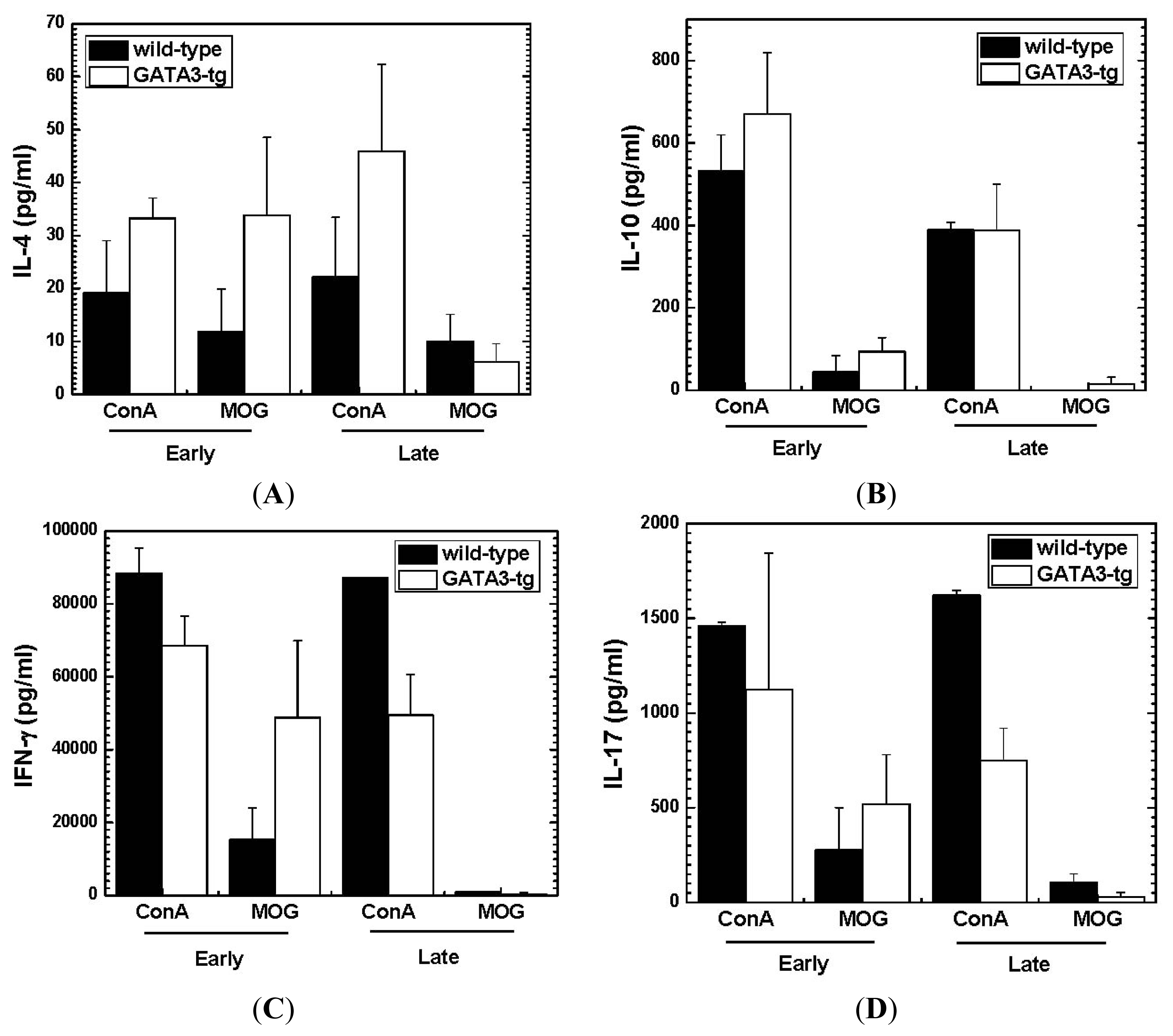
2.1.4. Altered Cytokine Profiles without Changes in the Overall MOG-Specific Lymphoproliferation in GATA3-tg Mice
2.2. Discussion
3. Experimental Section
3.1. EAE Induction
3.2. Pathology
3.3. Immunohistochemistry
3.4. Lymphoproliferative Assay
3.5. Cytokine Assay
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sato, F.; Omura, S.; Martinez, N.E.; Tsunoda, I. Animal Models of Multiple Sclerosis. In Neuroinflammation, 1st ed.; Minagar, A., Ed.; Elsevier Publications: London, UK, 2011; pp. 55–79. [Google Scholar]
- Bielekova, B.; Goodwin, B.; Richert, N.; Cortese, I.; Kondo, T.; Afshar, G.; Gran, B.; Eaton, J.; Antel, J.; Frank, J.A.; et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99 in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat. Med 2000, 6, 1167–1175. [Google Scholar]
- Rizvi, S.A.; Kim, E.; Moodie, J. Glatiramer in the treatment of multiple sclerosis. Int. J. Nanomed 2006, 1, 283–289. [Google Scholar]
- Rudick, R.A.; Stuart, W.H.; Calabresi, P.A.; Confavreux, C.; Galetta, S.L.; Radue, E.W.; Lublin, F.D.; Weinstock-Guttman, B.; Wynn, D.R.; Lynn, F.; et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N. Engl. J. Med 2006, 354, 911–923. [Google Scholar]
- Tsunoda, I.; Libbey, J.E.; Kuang, L.-Q.; Terry, E.J.; Fujinami, R.S. Massive apoptosis in lymphoid organs in animal models for primary and secondary progressive multiple sclerosis. Am. J. Pathol 2005, 167, 1631–1646. [Google Scholar]
- Martinez, N.E.; Sato, F.; Omura, S.; Minagar, A.; Alexander, J.S.; Tsunoda, I. Immunopathological patterns from EAE and Theiler’s virus infection: Is multiple sclerosis a homogenous 1-stage or heterogenous 2-stage disease? Pathophysiology 2013, 20, 71–84. [Google Scholar]
- Tsunoda, I.; Fujinami, R.S. Two models for multiple sclerosis: Experimental allergic encephalomyelitis and Theiler’s murine encephalomyelitis virus. J. Neuropathol. Exp. Neurol 1996, 55, 673–686. [Google Scholar]
- Tuohy, V.K.; Sobel, R.A.; Lees, M.B. Myelin proteolipid protein-induced experimental allergic encephalomyelitis. Variations of disease expression in different strains of mice. J. Immunol 1988, 140, 1868–1873. [Google Scholar]
- Dyment, D.A.; Herrera, B.M.; Cader, M.Z.; Willer, C.J.; Lincoln, M.R.; Sadovnick, A.D.; Risch, N.; Ebers, G.C. Complex interactions among MHC haplotypes in multiple sclerosis: Susceptibility and resistance. Hum. Mol. Genet 2005, 14, 2019–2026. [Google Scholar]
- Kasper, L.H.; Shoemaker, J. Multiple sclerosis immunology: The healthy immune system vs. the MS immune system. Neurology 2010, 74, S2–S8. [Google Scholar]
- Balabanov, R.; Strand, K.; Goswami, R.; McMahon, E.; Begolka, W.; Miller, S.D.; Popko, B. Interferon-γ-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J. Neurosci 2007, 27, 2013–2024. [Google Scholar]
- Sato, W.; Tomita, A.; Ichikawa, D.; Lin, Y.; Kishida, H.; Miyake, S.; Ogawa, M.; Okamoto, T.; Murata, M.; Kuroiwa, Y.; et al. CCR2+CCR5+ T cells produce matrix metalloproteinase-9 and osteopontin in the pathogenesis of multiple sclerosis. J. Immunol 2012, 189, 5057–5065. [Google Scholar]
- McGeachy, M.J.; Anderton, S.M. Cytokines in the induction and resolution of experimental autoimmune encephalomyelitis. Cytokine 2005, 32, 81–84. [Google Scholar]
- Kennedy, M.K.; Torrance, D.S.; Picha, K.S.; Mohler, K.M. Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J. Immunol 1992, 149, 2496–2505. [Google Scholar]
- Falcone, M.; Rajan, A.J.; Bloom, B.R.; Brosnan, C.F. A critical role for IL-4 in regulating disease severity in experimental allergic encephalomyelitis as demonstrated in IL-4-deficient C57BL/6 mice and BALB/c mice. J. Immunol 1998, 160, 4822–4830. [Google Scholar]
- Betteli, E.; Das, M.P.; Howard, E.D.; Weiner, H.L.; Sobel, R.A.; Kuchroo, V.K. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10 and IL-4 deficient and transgenic mice. J. Immunol 1998, 161, 3299–3306. [Google Scholar]
- Cua, D.J.; Groux, H.; Hinton, D.R.; Stohlman, S.A.; Coffman, R.L. Transgenic interleukin 10 prevents induction of experimental autoimmune encephalomyelitis. J. Exp. Med 1999, 189, 1005–1010. [Google Scholar]
- Croxford, J.L.; Feldmann, M.; Chernajovsky, Y.; Baker, D. Different therapeutic outcomes in experimental allergic encephalomyelitis dependent upon the mode of delivery of IL-10: A comparison of the effects of protein, adenoviral or retroviral IL-10 delivery into the central nervous system. J. Immunol 2001, 166, 4124–4130. [Google Scholar]
- Tsunoda, I.; Kuang, L.-Q.; Theil, D.J.; Fujinami, R.S. Antibody association with a novel model for primary progressive multiple sclerosis: Induction of relapsing-remitting and progressive forms of EAE in H2s mouse strains. Brain Pathol 2000, 10, 402–418. [Google Scholar]
- Tsunoda, I.; Kuang, L.-Q.; Igenge, I.Z.M.; Fujinami, R.S. Converting relapsing remitting to secondary progressive experimental allergic encephalomyelitis (EAE) by ultraviolet B irradiation. J. Neuroimmunol 2005, 160, 122–134. [Google Scholar]
- Lafaille, J.J.; Keere, F.V.; Hsu, A.L.; Baron, J.L.; Haas, W.; Raine, C.S.; Tonegawa, S. Myelin basic protein-specific T helper 2 (Th2) cells cause experimental autoimmune encephalomyelitis in immunodeficient hosts rather than protect them from the disease. J. Exp. Med 1997, 186, 307–312. [Google Scholar]
- Krakowski, M.; Owens, T. Interferon-γ confers resistance to experimental allergic encephalomyelitis. Eur. J. Immunol 1996, 26, 1641–1646. [Google Scholar]
- Ferber, I.A.; Brocke, S.; Taylor-Edwards, C.; Ridgway, W.; Dinisco, C.; Steinman, L.; Dalton, D.; Fathman, C.G. Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol 1996, 156, 5–7. [Google Scholar]
- Voorthuis, J.A.C.; Uitdehaag, B.M.J.; de Groot, C.J.A.; Goede, P.H.; van der Meide, P.H.; Dijkstra, C.D. Suppression of experimental allergic encephalomyelitis by intraventricular administration of interferon-gamma in Lewis rats. Clin. Exp. Immunol 1990, 81, 183–188. [Google Scholar]
- Lees, J.R.; Golumbek, P.T.; Sim, J.; Dorsey, D.; Russell, J.H. Regional CNS responses to IFN-γ determine lesion localization patterns during EAE pathogenesis. J. Exp. Med 2008, 205, 2633–2642. [Google Scholar]
- Willenborg, D.O.; Fordham, S.; Bernard, C.C.; Cowden, W.B.; Ramshaw, I.A. IFN-γ plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J. Immunol 1996, 157, 3223–3227. [Google Scholar]
- Willenborg, D.O.; Fordham, S.; Staykova, M.A.; Ramshaw, I.A.; Cowden, W.B. IFN-γ is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: A possible role for nitric oxide. J. Immunol 1999, 163, 5278–5286. [Google Scholar]
- Liu, L.; Okada, S.; Kong, X.-F.; Kreins, A.Y.; Cypowyj, S.; Abhyankar, A.; Toubiana, J.; Itan, Y.; Audry, M.; Nitschke, P.; et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med 2011, 208, 1635–1648. [Google Scholar]
- Masters, S.L.; Simon, A.; Aksentijevich, I.; Kastner, D.L. Horror autoinflammaticus: The molecular pathophysiology of autoinflammatory disease. Annu. Rev. Immunol 2009, 27, 621–668. [Google Scholar]
- Kimura, T.; Ishii, Y.; Yoh, K.; Morishima, Y.; Iizuka, T.; Kiwamoto, T.; Matsuno, Y.; Homma, S.; Nomura, A.; Sakamoto, T.; et al. Overexpression of the transcription factor GATA-3 enhances the development of pulmonary fibrosis. Am. J. Pathol 2006, 169, 96–104. [Google Scholar]
- Huber, J.P.; Ramos, H.J.; Gill, M.A.; Farrar, J.D. Cutting edge: Type I IFN reverses human Th2 commitment and stability by suppressing GATA3. J. Immunol 2010, 185, 813–817. [Google Scholar]
- Zheng, W.; Flavell, R.A. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997, 89, 587–596. [Google Scholar]
- Ferber, I.A.; Lee, H.-J.; Zonin, F.; Heath, V.; Mui, A.; Arai, N.; O’Garra, A. GATA-3 significantly downregulates IFN-γ production from developing Th1 cells in addition to inducing IL-4 and IL-5 levels. Clin. Immunol 1999, 91, 134–144. [Google Scholar]
- Ouyang, W.; Ranganath, S.H.; Weindel, K.; Bhattacharya, D.; Murphy, T.L.; Sha, W.C.; Murphy, K.M. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity 1998, 9, 745–755. [Google Scholar]
- Ho, I.; Pai, S. GATA-3-not just for Th2 cells anymore. Cell Mol. Immunol 2007, 4, 15–29. [Google Scholar]
- Yoh, K.; Shibuya, K.; Morito, N.; Nakano, T.; Ishizaki, K.; Shimohata, H.; Nose, M.; Izui, S.; Shibuya, A.; Koyama, A.; et al. Transgenic overexpression of GATA-3 in T lymphocytes improves autoimmune glomerulonephritis in mice with a BXSB/MpJ-Yaa genetic background. J. Am. Soc. Nephrol 2003, 14, 2494–2502. [Google Scholar]
- Kiwamoto, T.; Ishii, Y.; Morishima, Y.; Yoh, K.; Maeda, A.; Ishizaki, K.; Iizuka, T.; Hegab, A.E.; Matsuno, Y.; Homma, S.; et al. Transcription factors T-bet and GATA-3 regulate development of airway remodeling. Am. J. Respir. Crit. Care Med 2006, 174, 142–151. [Google Scholar]
- Kiwamoto, T.; Ishii, Y.; Morishima, Y.; Yoh, K.; Kikuchi, N.; Haraguchi, N.; Masuko, H.; Kawaguchi, M.; Nomura, A.; Sakamoto, T.; et al. Blockade of cysteinyl leukotriene-1 receptors suppresses airway remodelling in mice overexpressing GATA-3. Clin. Exp. Allergy 2011, 41, 116–128. [Google Scholar]
- Tsunoda, I.; Tanaka, T.; Terry, E.J.; Fujinami, R.S. Contrasting roles for axonal degeneration in an autoimmune versus viral model of multiple sclerosis: When can axonal injury be beneficial? Am. J. Pathol 2007, 170, 214–226. [Google Scholar]
- Tsunoda, I.; Kuang, L.-Q.; Libbey, J.E.; Fujinami, R.S. Axonal injury heralds virus-induced demyelination. Am. J. Pathol 2003, 162, 1259–1269. [Google Scholar]
- Tsunoda, I.; Kuang, L.-Q.; Tolley, N.D.; Whitton, J.L.; Fujinami, R.S. Enhancement of experimental allergic encephalomyelitis (EAE) by DNA immunization with myelin proteolipid protein (PLP) plasmid DNA. J. Neuropathol. Exp. Neurol 1998, 57, 758–767. [Google Scholar]
- Tsunoda, I.; Lane, T.E.; Blackett, J.; Fujinami, R.S. Distinct roles for IP-10/CXCL10 in three animal models, Theiler’s virus infection, EAE, and MHV infection, for multiple sclerosis: Implication of differing roles for IP-10. Mult. Scler 2004, 10, 26–34. [Google Scholar]
- Yoshimoto, T.; Bendelac, A.; Hu-Li, J.; Paul, W.E. Defective IgE production by SJL mice is linked to the absence of CD4+, NK1.1+ T cells that promptly produce interleukin 4. Proc. Natl. Acad. Sci. USA 1995, 92, 11931–11934. [Google Scholar]
- Yoshimoto, T.; Bendelac, A.; Watson, C.; Hu-Li, J.; Paul, W.E. Role of NK1.1+ T cells in a TH2 response and in immunoglobulin E production. Science 1995, 270, 1845–1847. [Google Scholar]
- Tsunoda, I.; Tanaka, T.; Taniguchi, M.; Fujinami, R.S. Contrasting roles for Vα14+ natural killer T cells in a viral model for multiple sclerosis. J. Neurovirol 2009, 15, 90–98. [Google Scholar]
- Tsunoda, I.; Tanaka, T.; Fujinami, R.S. Regulatory role of CD1d in neurotropic virus infection. J. Virol 2008, 82, 10279–10289. [Google Scholar]
- Segal, B.M.; Shevach, E.M. IL-12 unmasks latent autoimmune disease in resistant mice. J. Exp. Med 1996, 184, 771–775. [Google Scholar]
- Szabo, S.J.; Jacobson, N.G.; Dighe, A.S.; Gubler, U.; Murphy, K.M. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity 1995, 2, 665–675. [Google Scholar]
- Jäger, A.; Dardalhon, V.; Sobel, R.A.; Bettelli, E.; Kuchroo, V.K. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol 2009, 183, 7169–7177. [Google Scholar]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol 2006, 177, 566–573. [Google Scholar]
- Karlsson, F.; Martinez, N.E.; Gray, L.; Zhang, S.; Tsunoda, I.; Grisham, M.B. Therapeutic evaluation of ex vivo-generated versus natural regulatory T-cells in a mouse model of chronic gut inflammation. Inflamm. Bowel Dis 2013, 19, 2282–2294. [Google Scholar]
- Martinez, N.E.; Sato, F.; Kawai, E.; Omura, S.; Chervenak, R.P.; Tsunoda, I. Regulatory T cells and Th17 cells in viral infections: Implications for multiple sclerosis and myocarditis. Future Virol 2012, 7, 593–608. [Google Scholar]
- Hofstetter, H.H.; Ibrahim, S.M.; Koczan, D.; Kruse, N.; Weishaupt, A.; Toyka, K.V.; Gold, R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell. Immunol 2005, 237, 123–130. [Google Scholar]
- Chakravarti, S.; Sabatos, C.A.; Xiao, S.; Illes, Z.; Cha, E.K.; Sobel, R.A.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. Tim-2 regulates T helper type 2 responses and autoimmunity. J. Exp. Med 2005, 202, 437–444. [Google Scholar]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol 2005, 6, 1123–1132. [Google Scholar]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol 2005, 6, 1133–1141. [Google Scholar]
- Tsunoda, I.; Libbey, J.E.; Fujinami, R.S. TGF-β1 suppresses T cell infiltration and VP2 puff B mutation enhances apoptosis in acute polioencephalitis induced by Theiler’s virus. J. Neuroimmunol 2007, 190, 80–89. [Google Scholar]
- Isobe, N.; Kanamori, Y.; Yonekawa, T.; Matsushita, T.; Shigeto, H.; Kawamura, N.; Kira, J. First diagnostic criteria for atopic myelitis with special reference to discrimination from myelitis-onset multiple sclerosis. J. Neurol. Sci 2012, 316, 30–35. [Google Scholar]
- Nakamura, Y.; Ghaffar, O.; Olivenstein, R.; Taha, R.A.; Soussi-Gounni, A.; Zhang, D.H.; Ray, A.; Hamid, Q. Gene expression of the GATA-3 transcription factor is increased in atopic asthma. J. Allergy Clin. Immunol 1999, 103, 215–222. [Google Scholar]
- Ano, S.; Morishima, Y.; Ishii, Y.; Yoh, K.; Yageta, Y.; Ohtsuka, S.; Matsuyama, M.; Kawaguchi, M.; Takahashi, S.; Hizawa, N. Transcription factors GATA-3 and RORγt are important for determining the phenotype of allergic airway inflammation in a murine model of asthma. J. Immunol 2013, 190, 1056–1065. [Google Scholar]
- Keegan, M.; König, F.; McClelland, R.; Brück, W.; Morales, Y.; Bitsch, A.; Panitch, H.; Lassmann, H.; Weinshenker, B.; Rodriguez, M.; et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 2005, 366, 579–582. [Google Scholar]
- Zhumabekov, T.; Corbella, P.; Tolaini, M.; Kioussis, D. Improved version of a human CD2 minigene based vector for T cell-specific expression in transgenic mice. J. Immunol. Methods 1995, 185, 133–140. [Google Scholar]
- Tsunoda, I.; Terry, E.J.; Marble, B.J.; Lazarides, E.; Woods, C.; Fujinami, R.S. Modulation of experimental autoimmune encephalomyelitis by VLA-2 blockade. Brain Pathol 2007, 17, 45–55. [Google Scholar]
- Sato, F.; Martinez, N.E.; Shahid, M.; Rose, J.W.; Carlson, N.G.; Tsunoda, I. Resveratrol exacerbates both autoimmune and viral models for multiple sclerosis. Am. J. Pathol 2013, 183, 1390–1396. [Google Scholar]
- Peterson, L.K.; Tsunoda, I.; Fujinami, R.S. Role of CD5+ B-1 cells in EAE pathogenesis. Autoimmunity 2008, 41, 353–362. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Strain | N | Onset Days | Maximum Clinical Score | Cumulative Score | Incidence | ||
|---|---|---|---|---|---|---|---|
| Early b | Late c | Early b | Late c | ||||
| wild-type | 10 | 13.1 ± 0.3 | 3.4 ± 0.3 | 23.9 ± 5.0 | 16.8 ± 7.9 | 9/10 | 6/10 |
| GATA3-tg | 12 | 16.1 ± 0.3 * | 2.1 ± 0.4 * | 12.8 ± 3.9 | 8.9 ± 6.0 ** | 11/12 | 3/12 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fernando, V.; Omura, S.; Sato, F.; Kawai, E.; Martinez, N.E.; Elliott, S.F.; Yoh, K.; Takahashi, S.; Tsunoda, I. Regulation of an Autoimmune Model for Multiple Sclerosis in Th2-Biased GATA3 Transgenic Mice. Int. J. Mol. Sci. 2014, 15, 1700-1718. https://doi.org/10.3390/ijms15021700
Fernando V, Omura S, Sato F, Kawai E, Martinez NE, Elliott SF, Yoh K, Takahashi S, Tsunoda I. Regulation of an Autoimmune Model for Multiple Sclerosis in Th2-Biased GATA3 Transgenic Mice. International Journal of Molecular Sciences. 2014; 15(2):1700-1718. https://doi.org/10.3390/ijms15021700
Chicago/Turabian StyleFernando, Viromi, Seiichi Omura, Fumitaka Sato, Eiichiro Kawai, Nicholas E. Martinez, Sadie Faith Elliott, Keigyou Yoh, Satoru Takahashi, and Ikuo Tsunoda. 2014. "Regulation of an Autoimmune Model for Multiple Sclerosis in Th2-Biased GATA3 Transgenic Mice" International Journal of Molecular Sciences 15, no. 2: 1700-1718. https://doi.org/10.3390/ijms15021700
APA StyleFernando, V., Omura, S., Sato, F., Kawai, E., Martinez, N. E., Elliott, S. F., Yoh, K., Takahashi, S., & Tsunoda, I. (2014). Regulation of an Autoimmune Model for Multiple Sclerosis in Th2-Biased GATA3 Transgenic Mice. International Journal of Molecular Sciences, 15(2), 1700-1718. https://doi.org/10.3390/ijms15021700