Pluripotent State Induction in Mouse Embryonic Fibroblast Using mRNAs of Reprogramming Factors

Abstract
:1. Introduction
2. Results
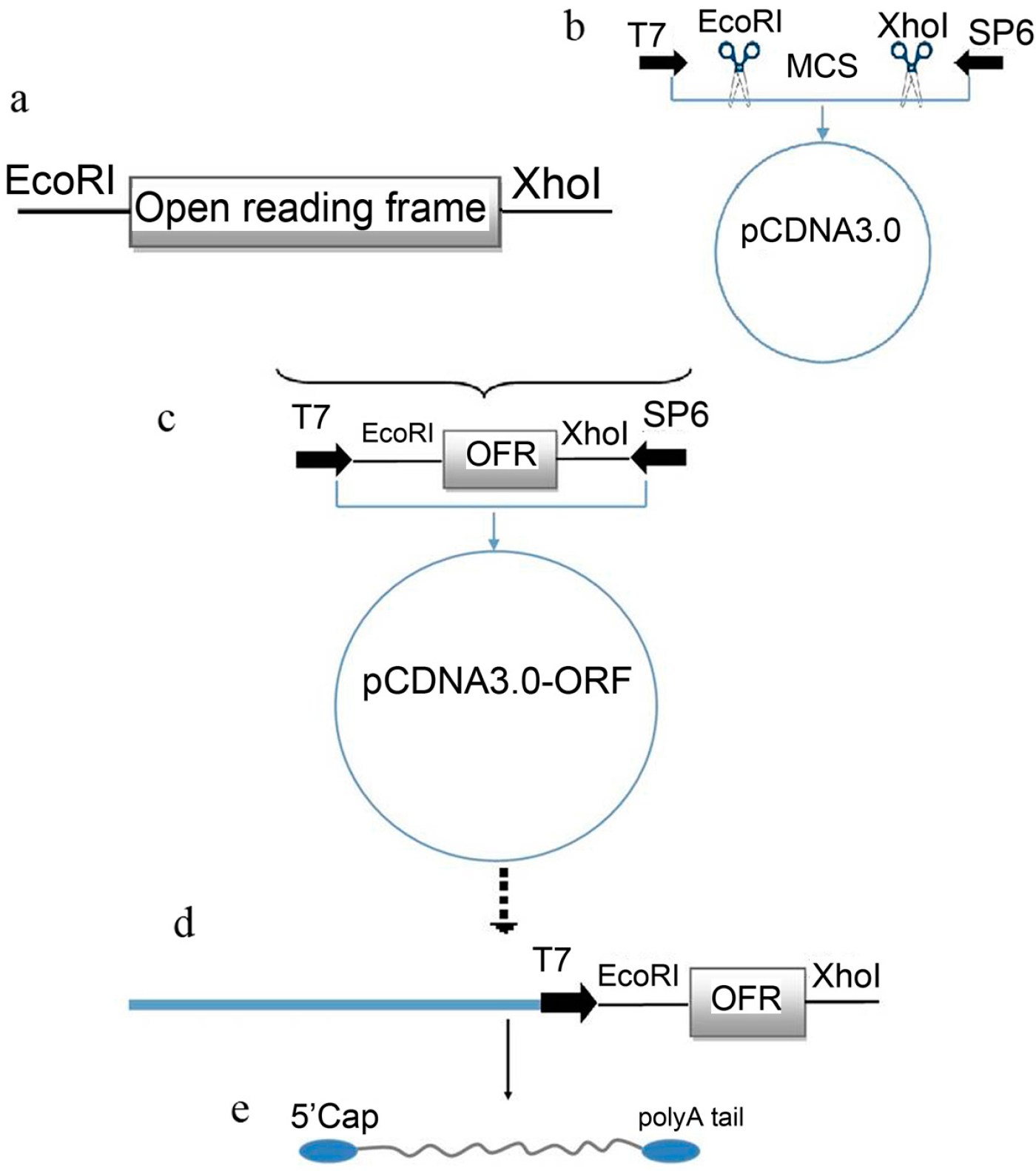
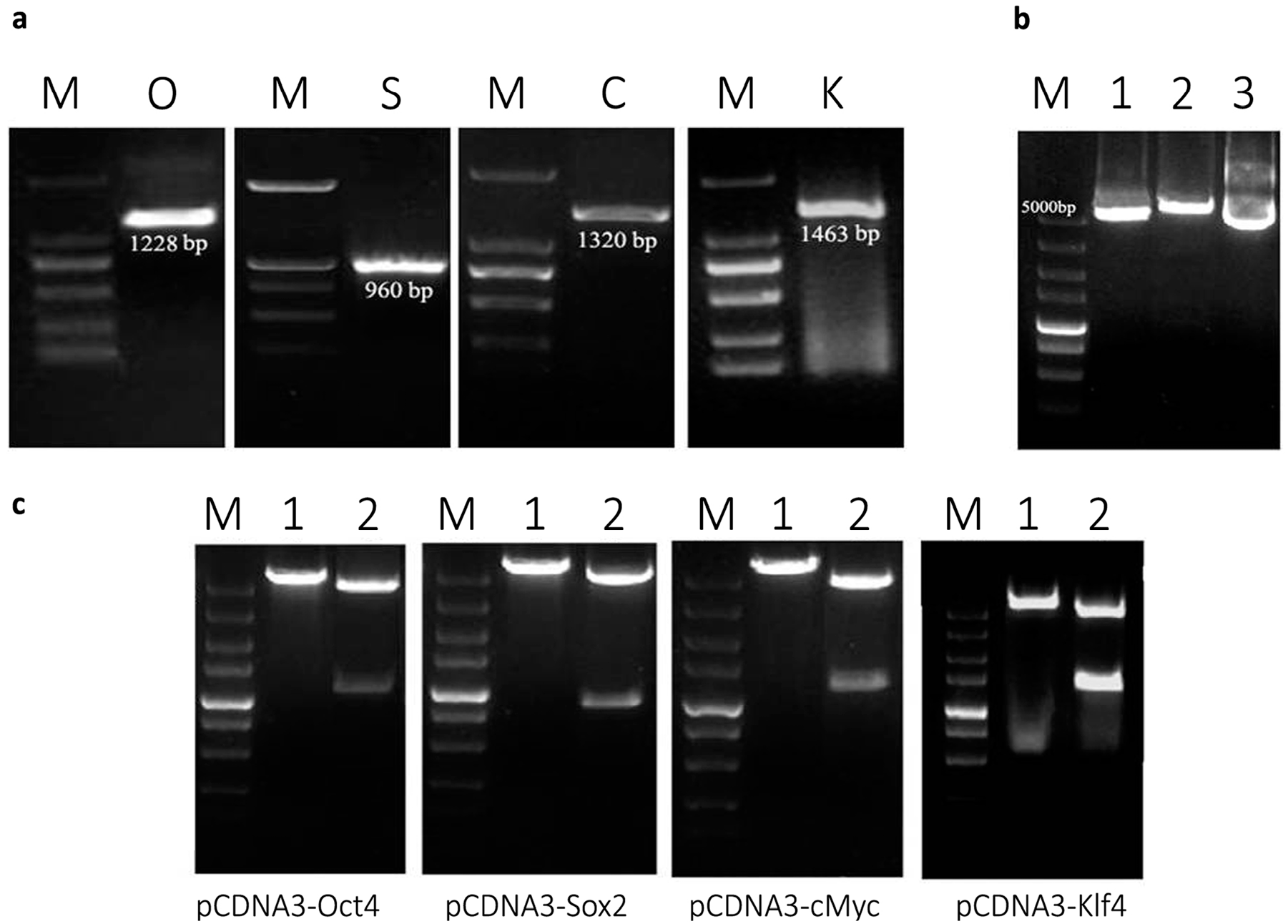
2.1. Plasmid Construction and mRNA Synthesis
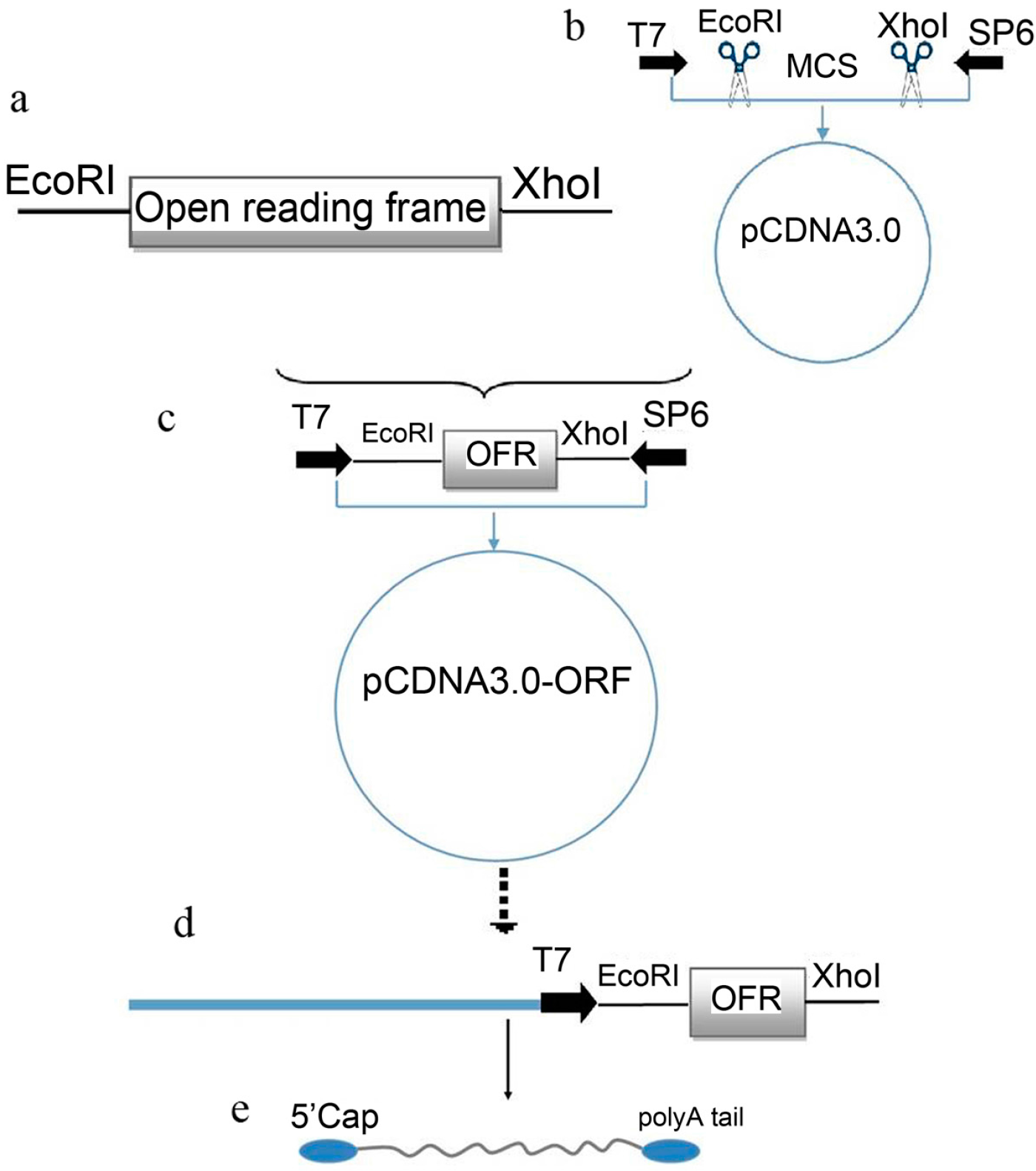
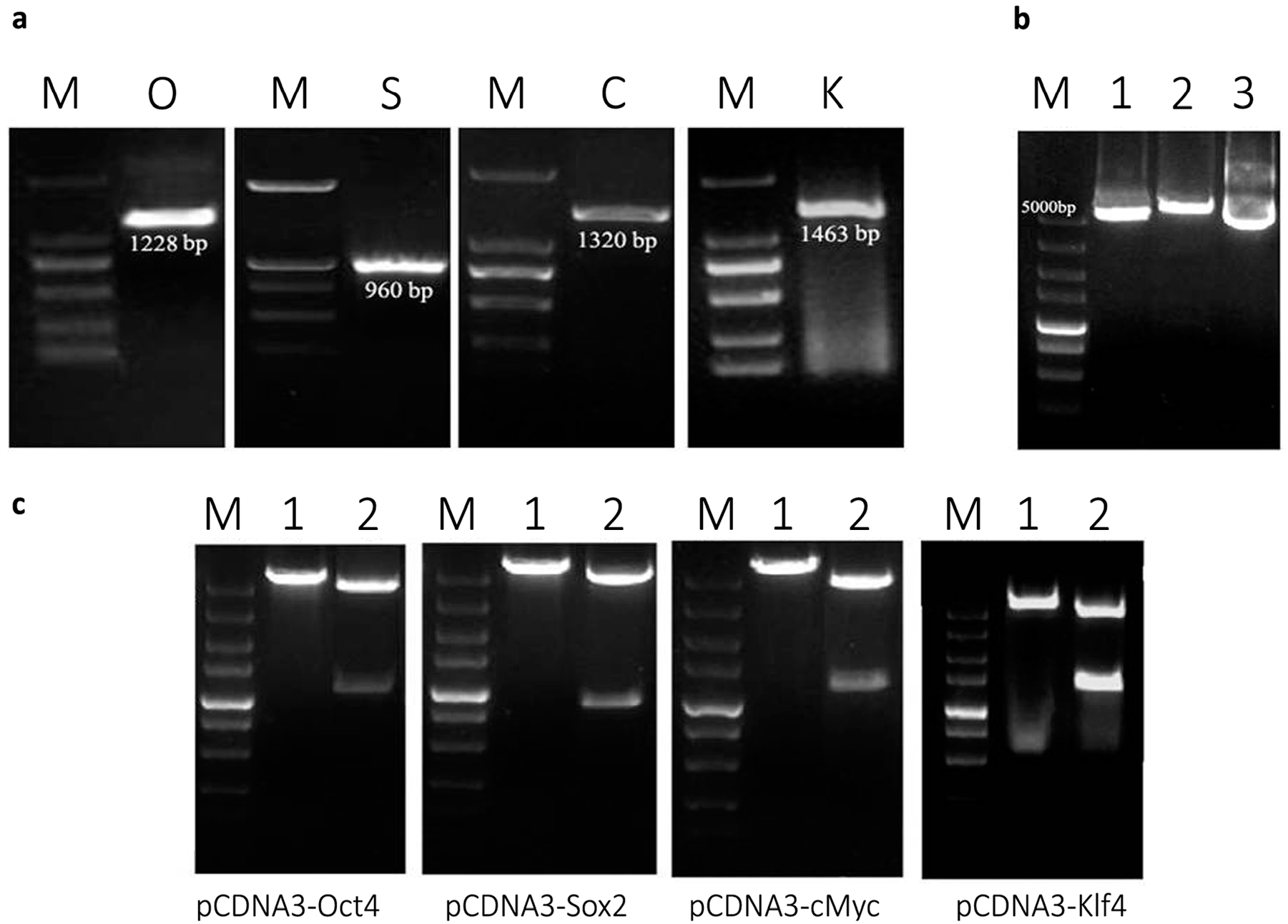
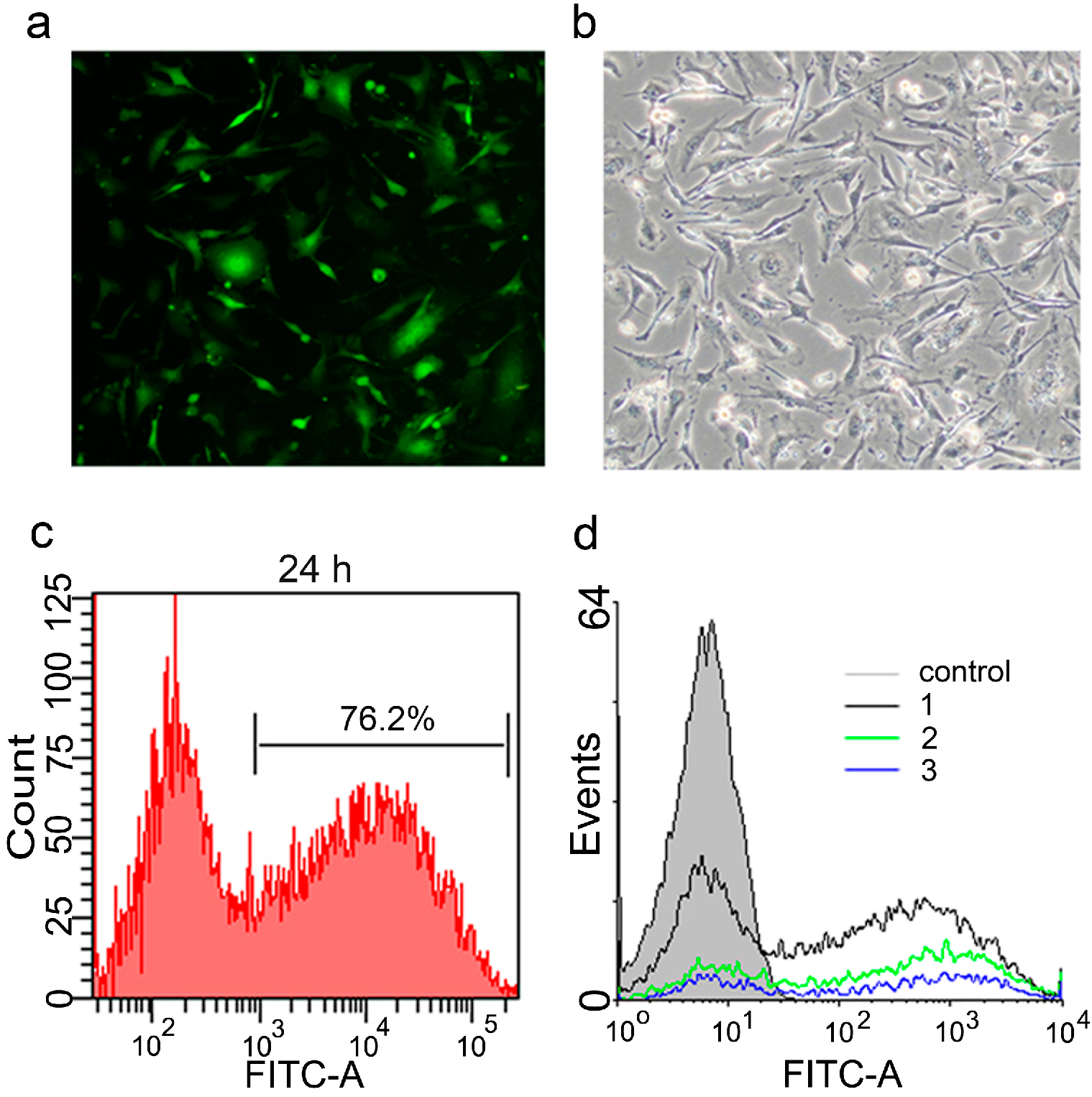
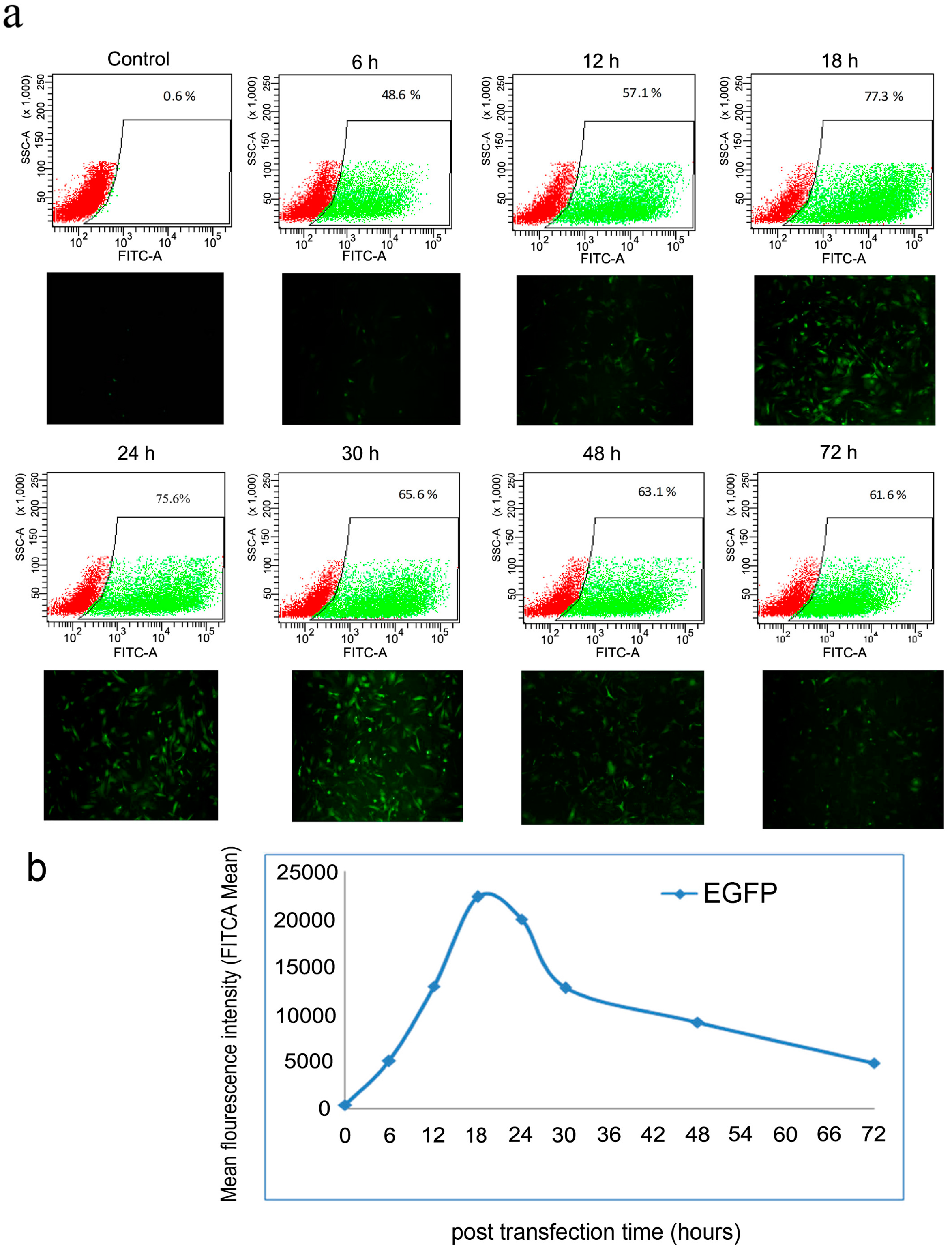
2.2. Optimization of Transfection Conditions
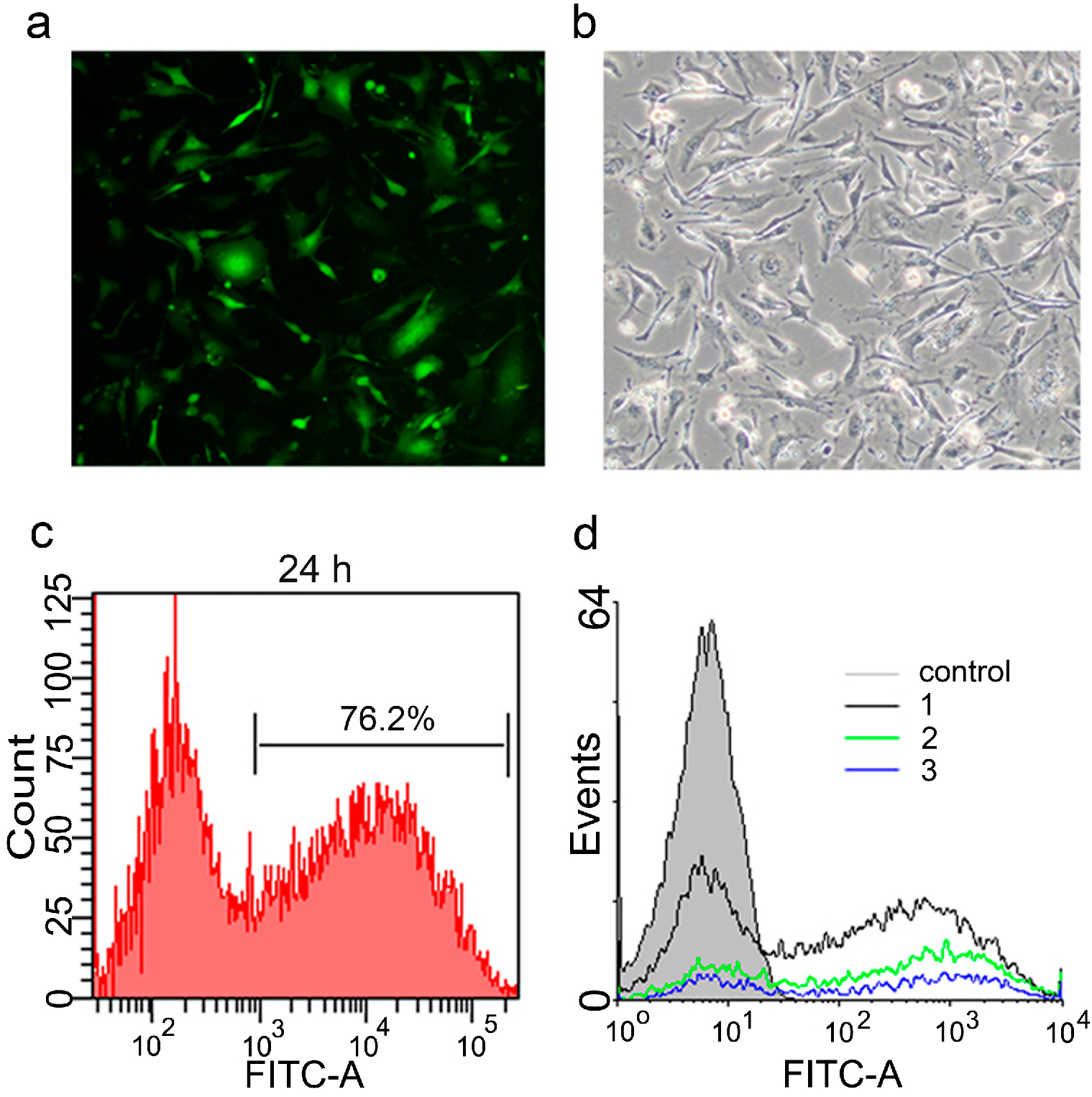
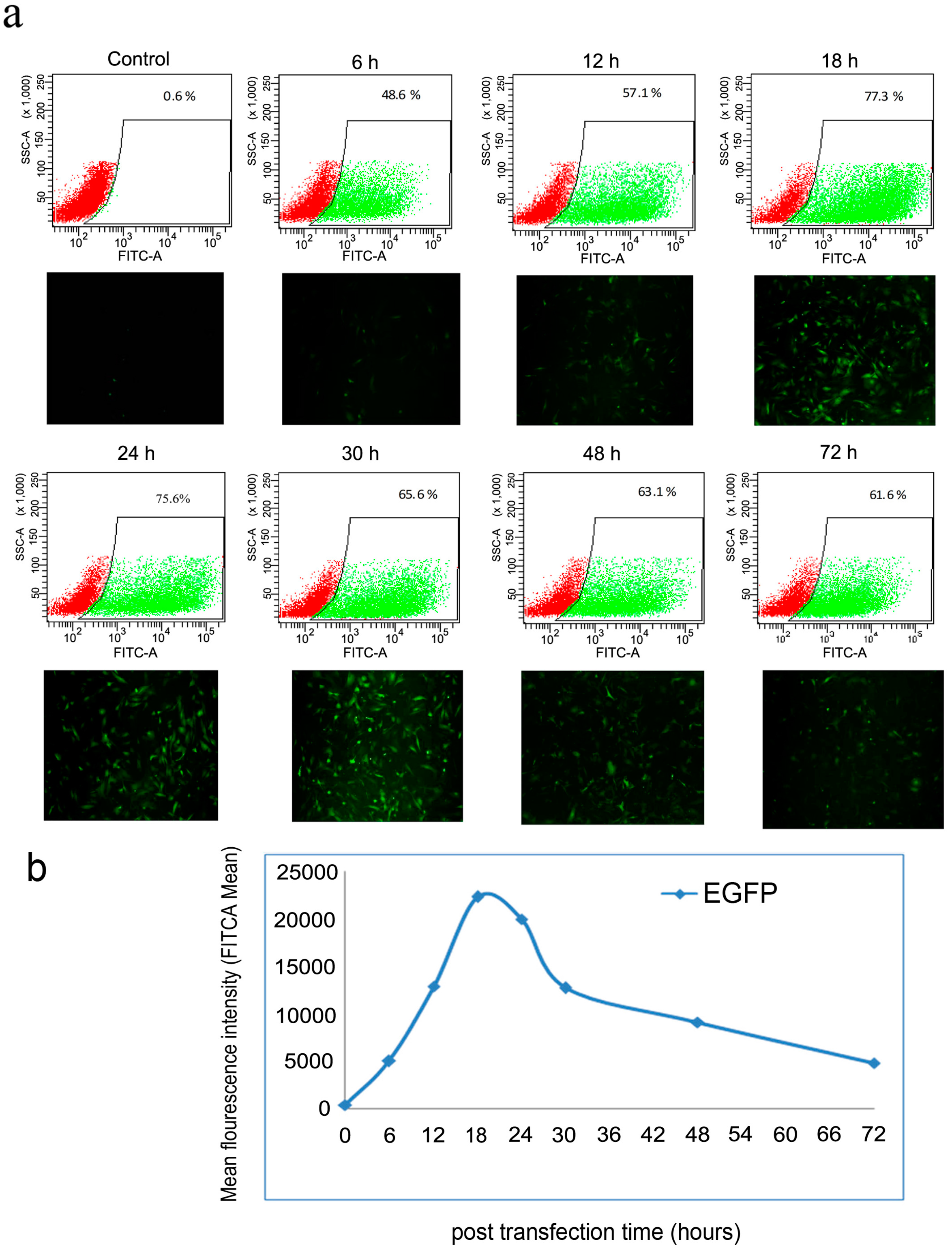
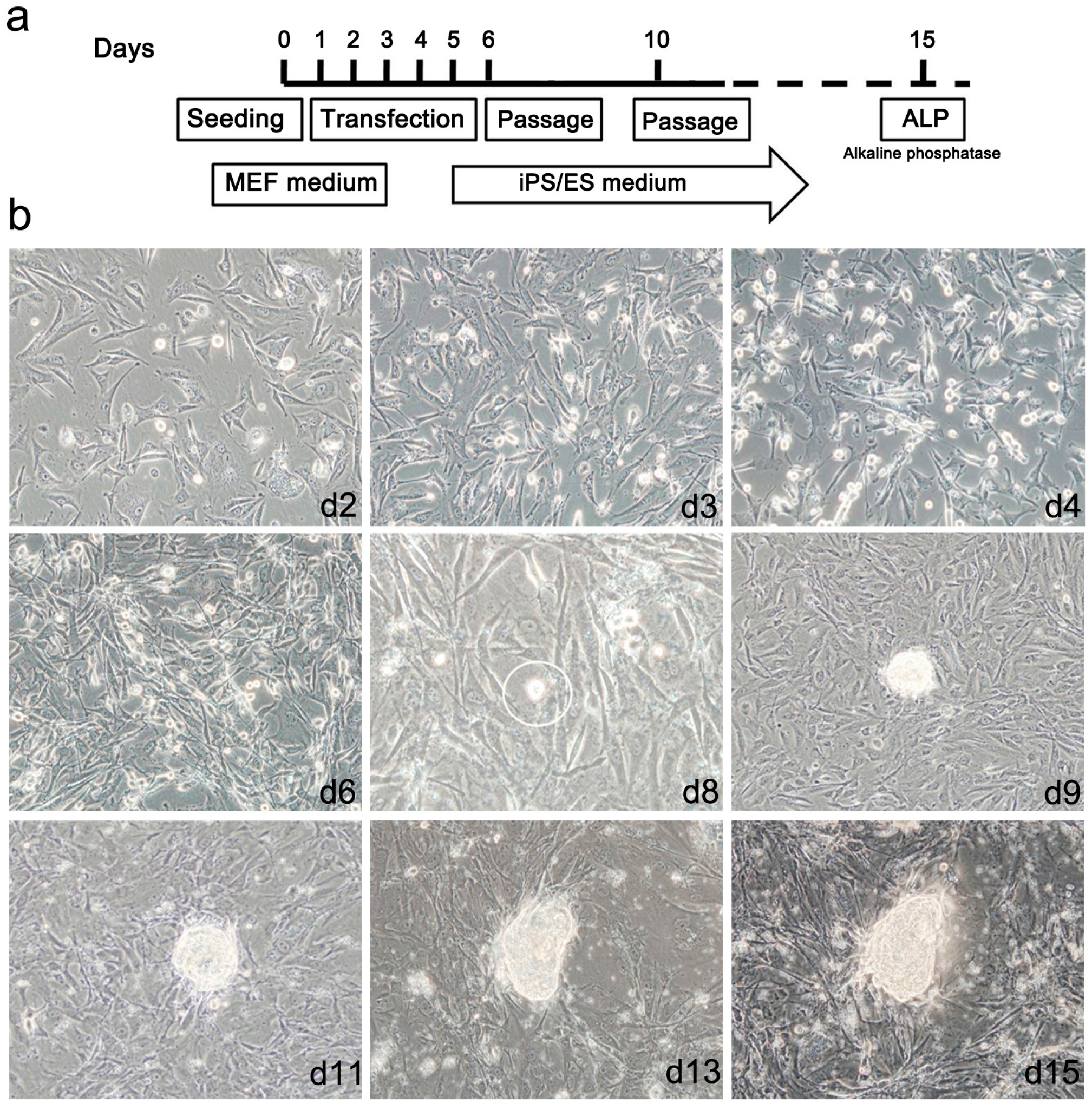
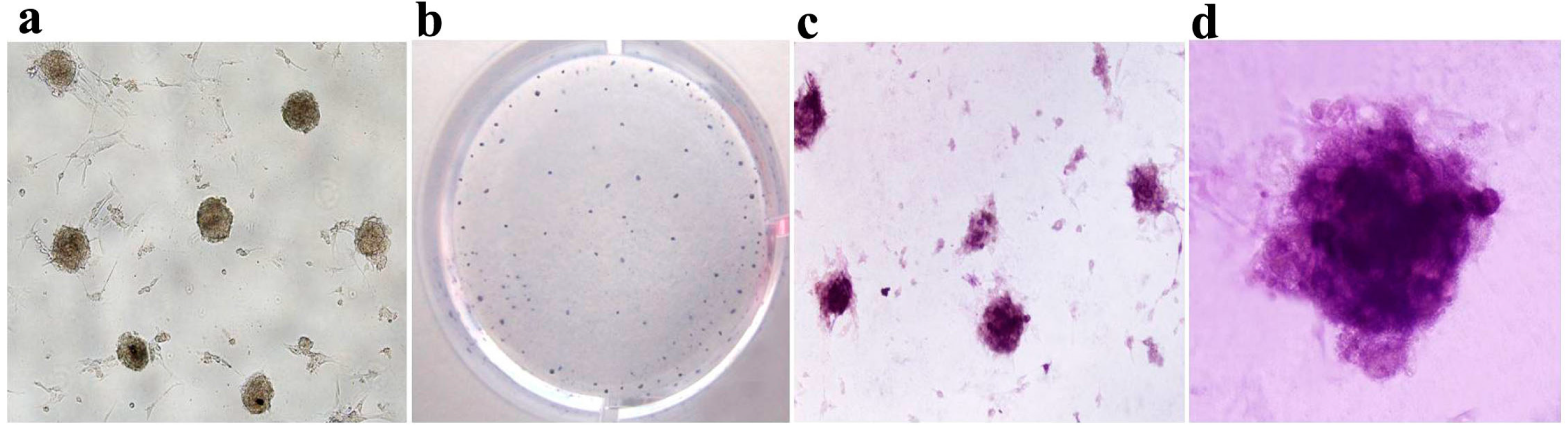
2.3. Generation of mRNA Induced Pluripotent Stem Cells (mRNA iPS)


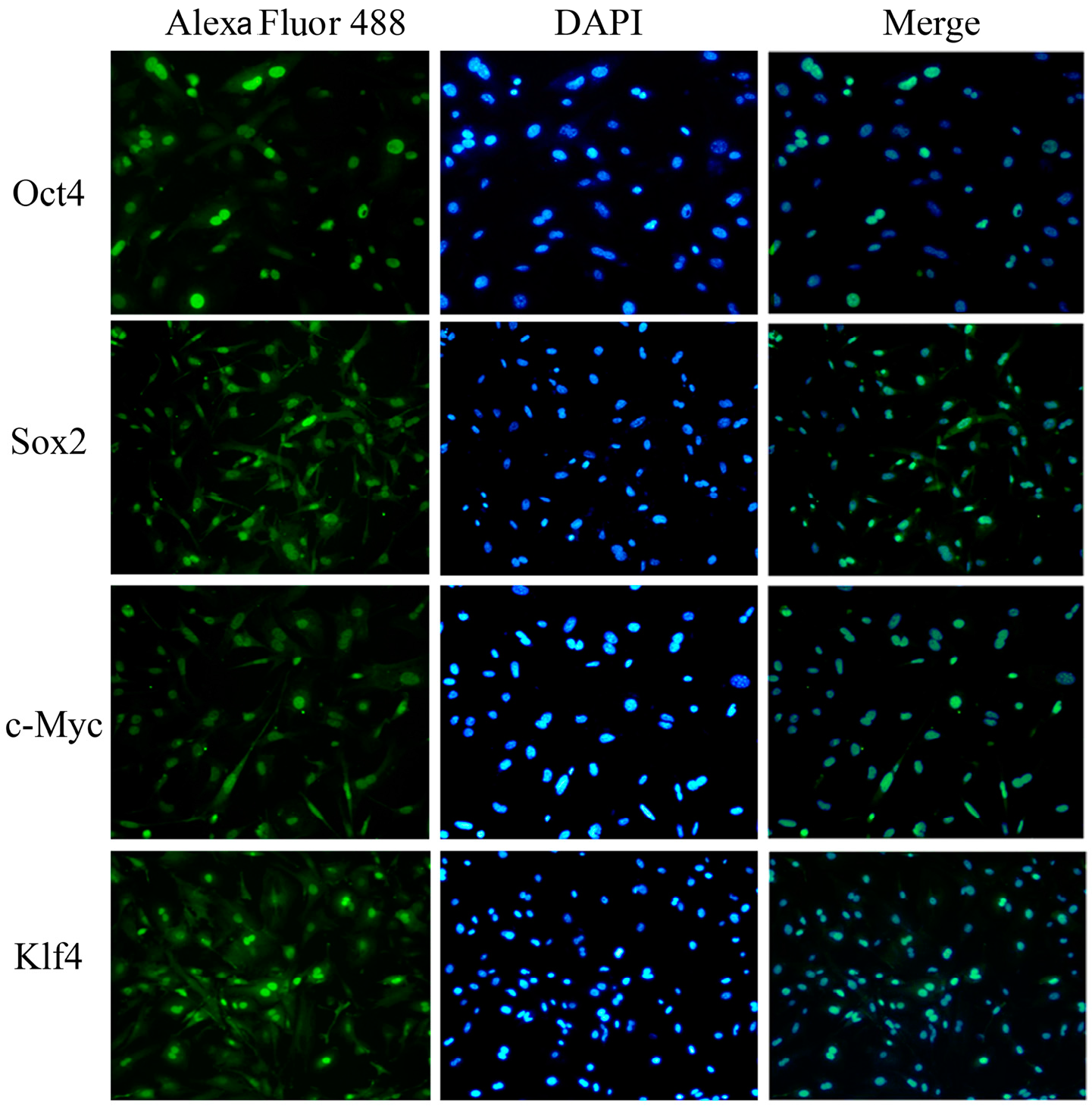
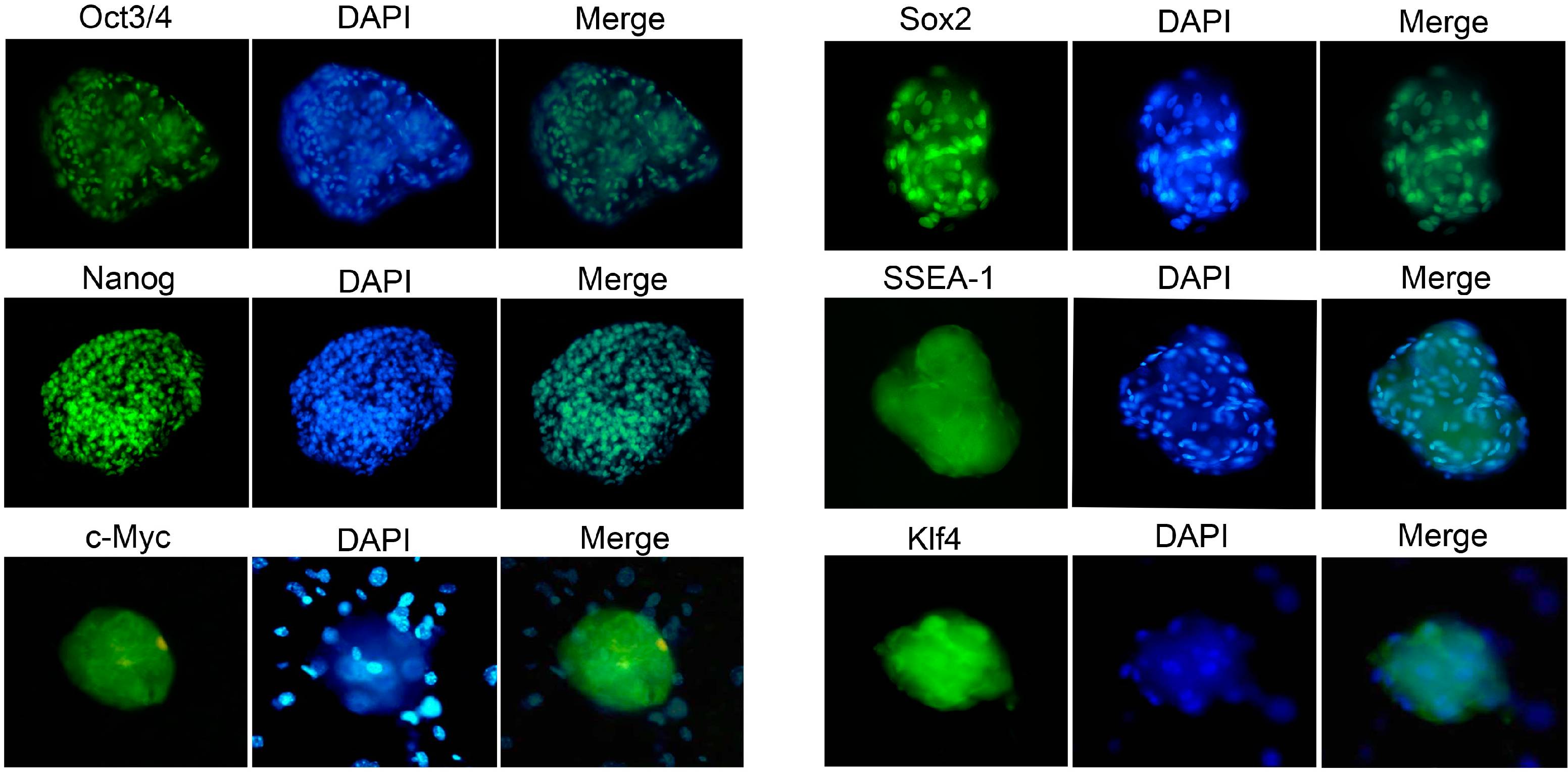
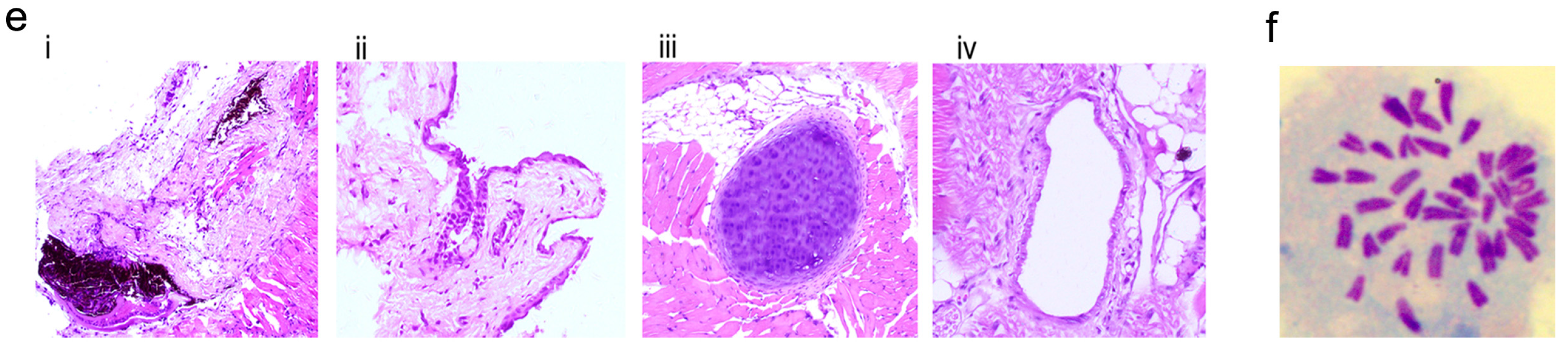
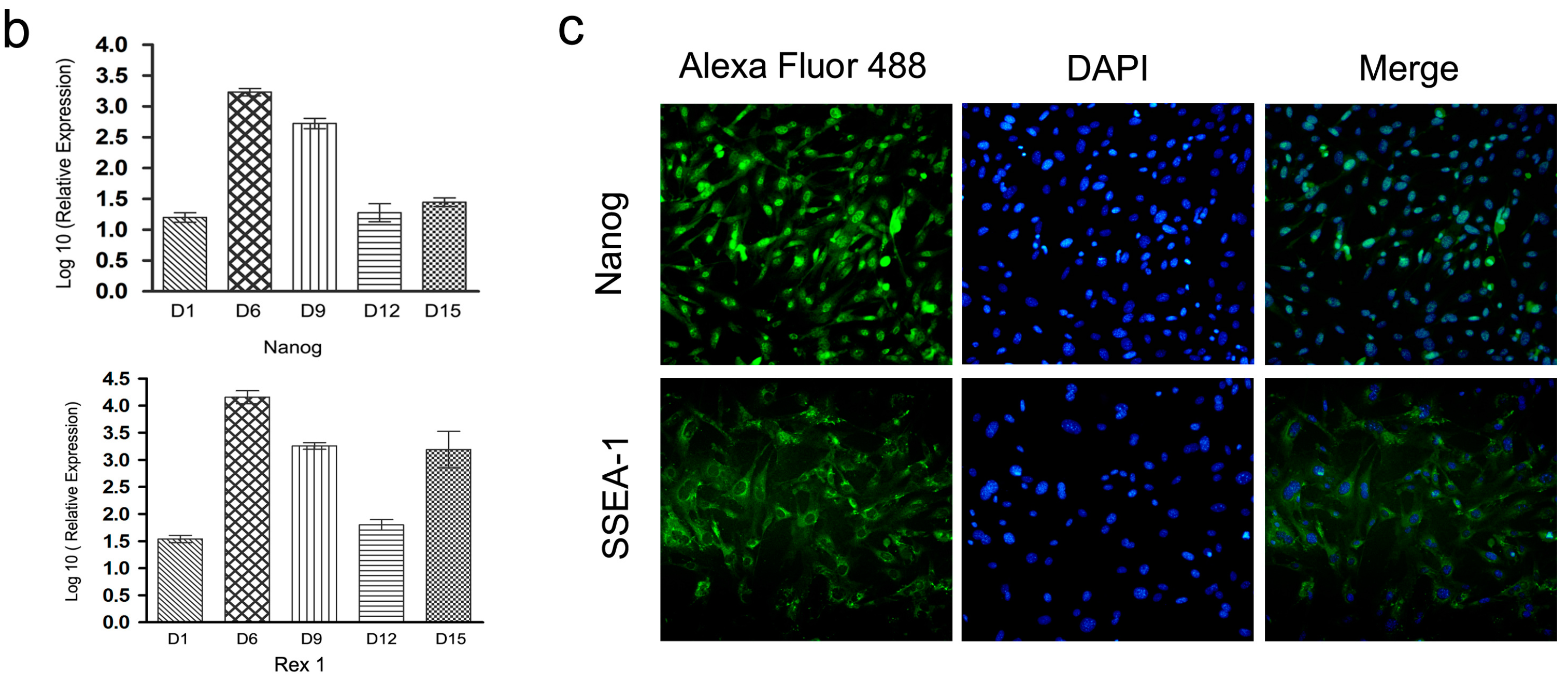
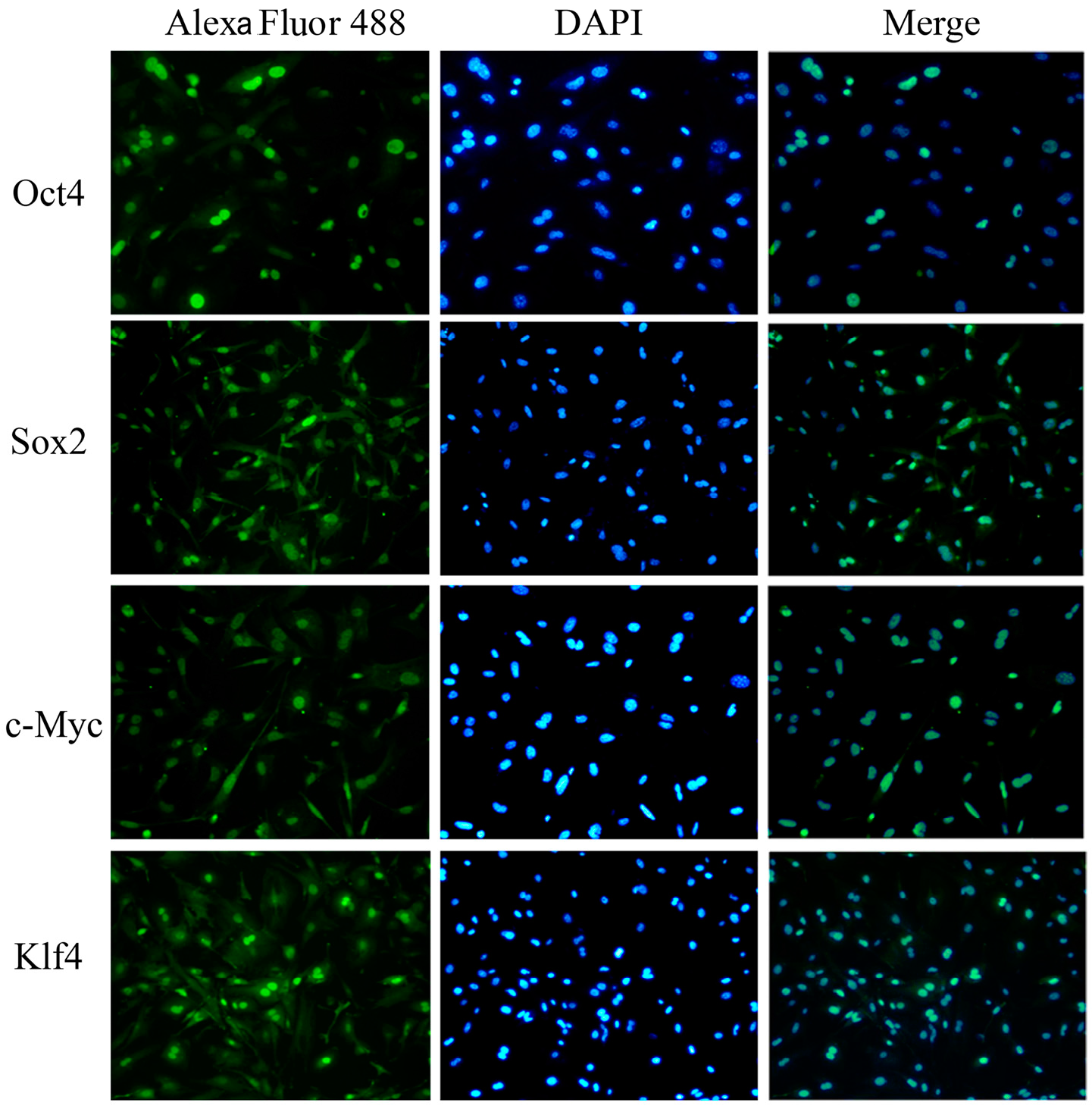
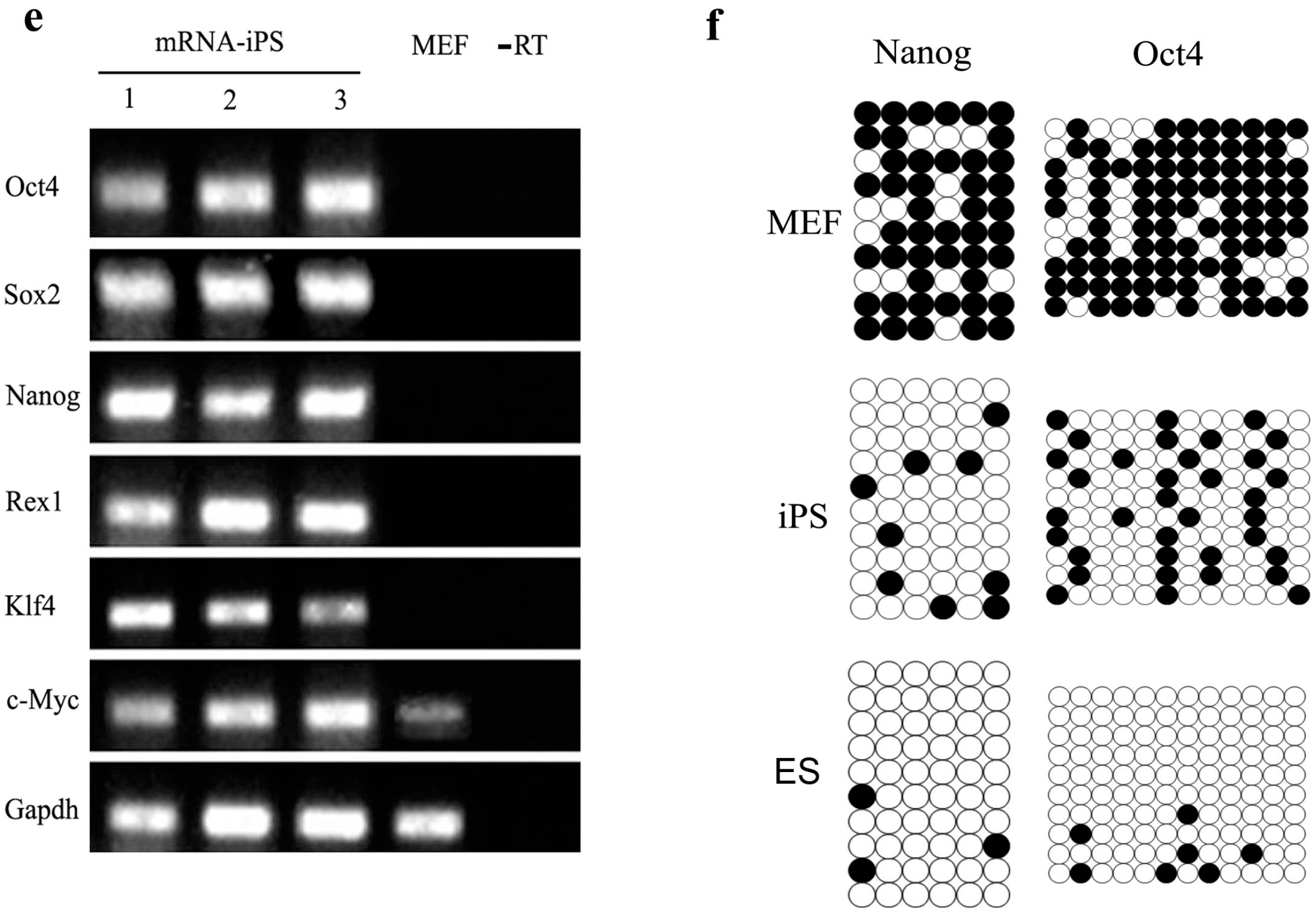
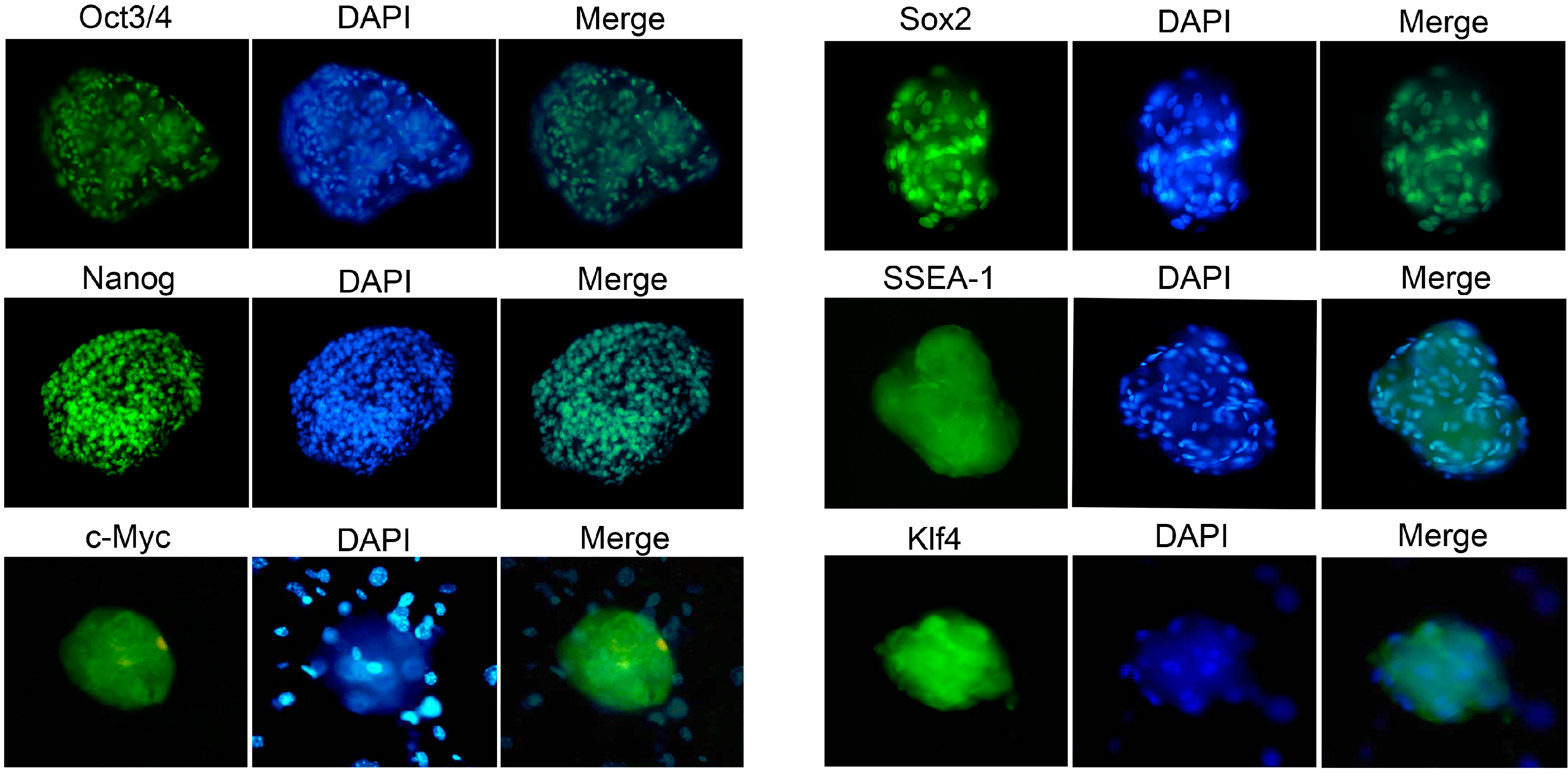
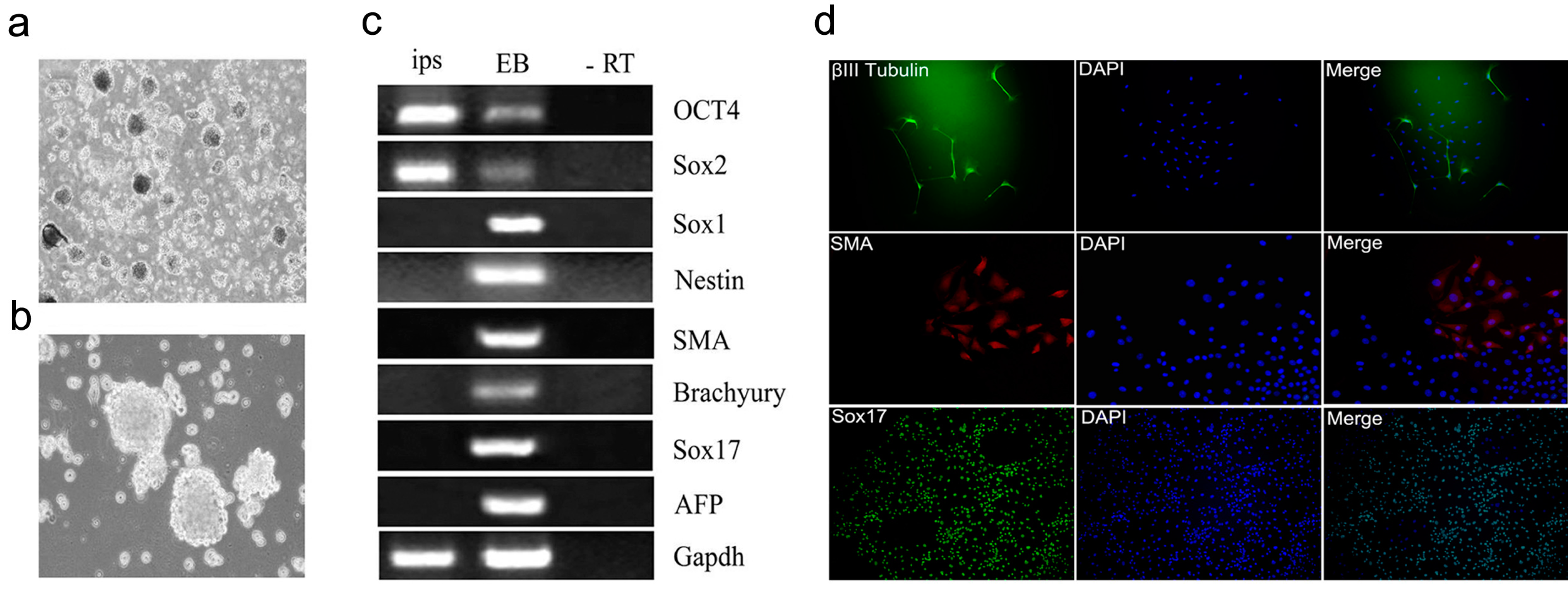
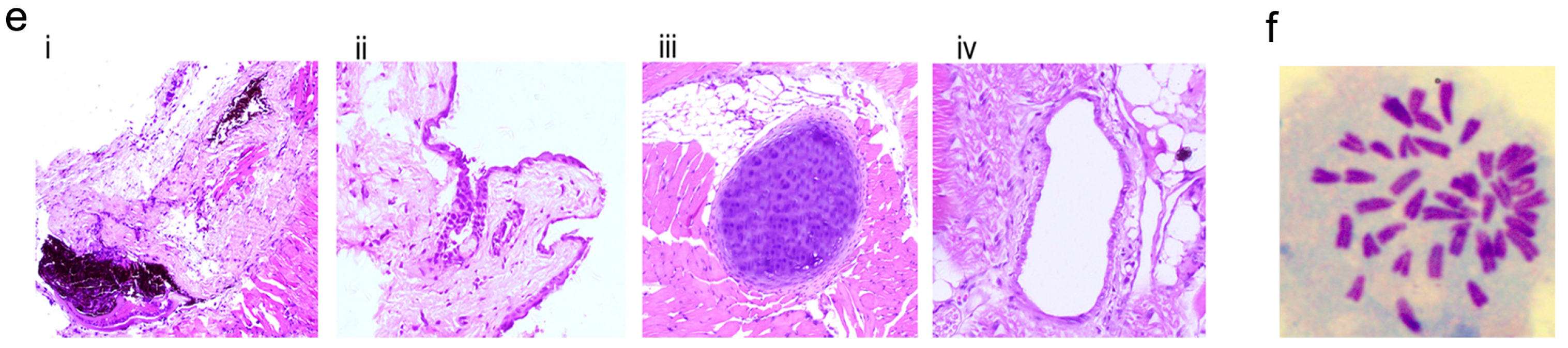
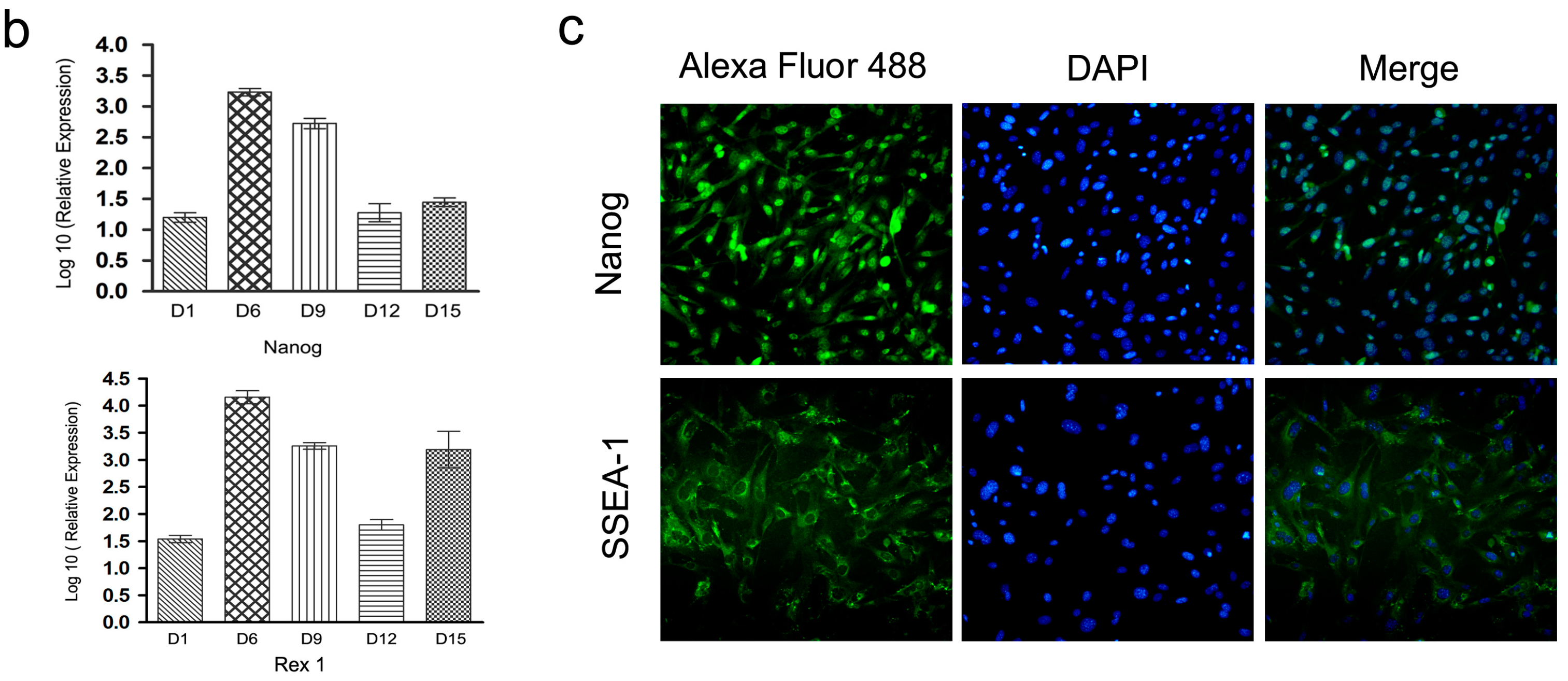
2.4. Characterization and Identification of the Generated mRNA iPSCs

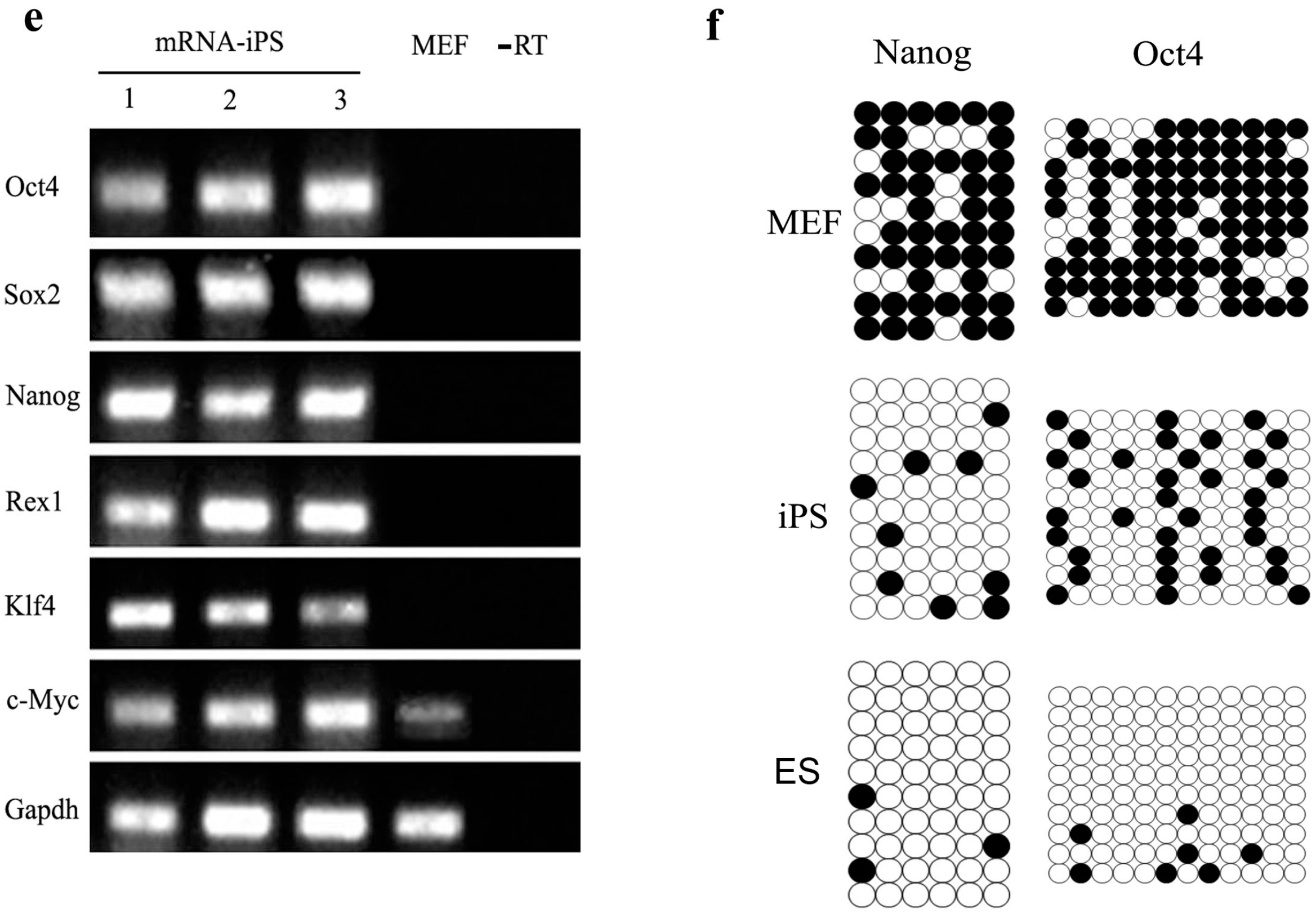
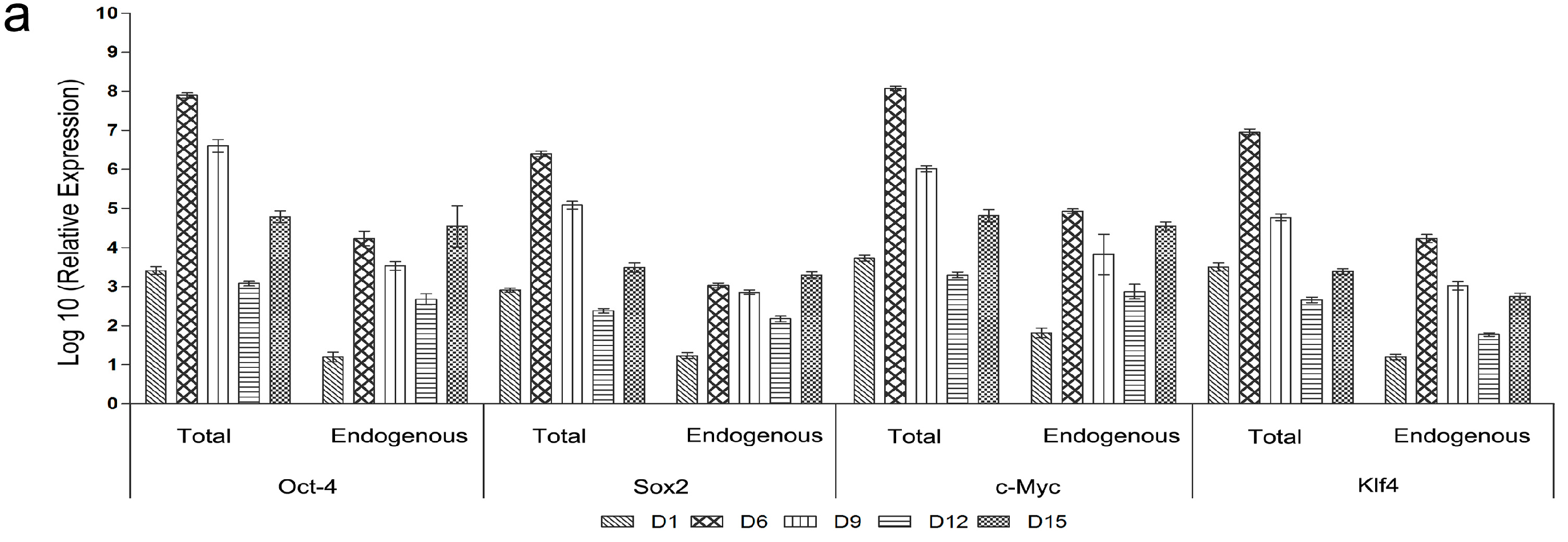
2.5. Genetic and Epigenetic Changes of the Reprogramming Factors and Pluripotency Markers during mRNA iPS Generation


3. Discussion
4. Materials and Methods
4.1. RNA Extraction and cDNA Amplification
4.2. Plasmid Construction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession No. | ORF Primers |
|---|---|---|
| Oct4 | NM_013633 | F: 5'CGGAATTCCGCCACCTTCCCCATGGCTGGACACC3'
R: 5'CCCTCGAGGGTGATCAACAGCATCACTGAGCTTC3' |
| Sox2 | NM_011443 | F: 5'CGGAATTCCGATGTATAACATGATGGAGACGGAGCT3'
R: 5'CCCTCGAGGGTCACATGTGCGACAGGGGCAGT3' |
| c-Myc | NM_001177352 | F: 5'CGGAATTCCGATGCCCCTCAACGTGAACTTCACC3'
R: 5'CCCTCGAGGGTTATGCACCAGAGTTTCGAAGC3' |
| Klf4 | NM_010637 | F: 5'CGGAATTCCGATGAGGCAGCCACCTGGCGAGT3' R: 5'CCCTCGAGGGCTACGTGGGATTTAAAAGTGCCTC3' |
4.3. In Vitro Transcription of mRNA
4.4. Mouse Embryonic Fibroblast (MEF) Isolation
4.5. Cell Culture
4.6. Cell Transfection
4.7. Quantitative Real-Time PCR (qPCR)
| Gene | Forward Primer | Reverse Primer | References | |
|---|---|---|---|---|
| Oct4 | Total | 5'CAGACCACCATCTGTCGCTTC3' | 5'AGACTCCACCTCACACGGTTCTC3' | This study |
| Endogenous | 5'TCTTTCCACCAGGCCCCCGGCTC3' | 5'TGCGGGCGGACATGGGGAGATCC3' | [11] | |
| Sox2 | Total | 5'GGTTACCTCTTCCTCCCACTCCAG3' | 5'TCACATGTGCGACAGGGGCAG3' | |
| Endogenous | 5'TAGAGCTAGACTCCGGGCGATGA3' | 5'TTGCCTTAAACAAGACCACGAAA3' | ||
| c-Myc | Total | 5'CCTAGTGCTGCATGAGGAGACAC3' | 5'TCCACAGACACCACATCAATTTCTT3' | This study |
| Endogenous | 5'TGACCTAACTCGAGGAGGAGCTGGAATC3' | 5'AAGTTTGAGGCAGTTAAAATTATGGCTGAAGC3' | [11] | |
| Klf4 | Total | 5'ACAGCCACCCACACTTGTGACTA3' | 5'GGCGAATTTCCACCCACAG3' | This study |
| Endogenous | 5'GCGAACTCACACAGGCGAGAAACC3' | 5'TCGCTTCCTCTTCCTCCGACACA3' | [11] | |
| Gapdh | – | 5'TGTGTCCGTCGTGGATCTGA3' | 5'TTGCTGTTGAAGTCGCAGGAG3' | This study |
4.8. Immunofluorescence
4.9. Fluorescence Activated Cell Sorting (FACS)
4.10. Alkaline Phosphatase Staining
4.11. Bisulfite Genomic Sequencing
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Nanog | 5'GATTTTGTAGGTGGGATTAATTGTGAATTT3' | 5'ACCAAAAAAACCCACACTCATATCAATATA3' |
| Oct4 | F1: 5'GTTGTTTTGTTTTGGTTTTGGATAT3' F2: 5'ATGGGTTGAAATATTGGGTTTATTTA3' | 5'CCACCCTCTAACCTTAACCTCTAAC3' |
4.12. In Vitro Differentiation of mRNA iPSCs
| Gene | Accession No. | Primers |
|---|---|---|
| Sox1 | NM_009233 | F: 5'GGATCTCTGGTCAAGTCGGAG3'
R: 5'CTGGCGCTCGGCTCTCCAGAG3' |
| Nestin | NM_016701 | F: 5' TCTGGAAGTCAACAGAGGTGG3'
R: 5'ACGGAGTCTTGTTCACCTGC3' |
| α-SMA | NM_007392 | F: 5'GAGAAGAGCTACGAACTGCCTGAC3'
R: 5'CACATCTGCTGGAAGGTAGACAG3' |
| Bra | NM_009309 | F: 5'GTTCCTGGTGCTGGCACCCTCTGC3'
R: 5'CAGACCAGAGACTGGGATACTG3' |
| Sox17 | NM_011441 | F: 5'CACAGCAGAACCCAGATCTGCA3'
R: 5'CATGTGCGGAGACATCAGCGGAG3' |
| AFP | NM_007423 | F: 5'GTGAGCATTGCCTCCACGTGCTG3'
R: 5'GTGACAGCCGCCAGCTGCTCCTC3' |
4.13. Teratoma Formation and Histological Analysis
4.14. Karyotyping Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H. Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Gurdon, J.B.; Wilmut, I. Nuclear transfer to eggs and oocytes. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Jullien, J.; Pasque, V.; Halley-Stott, R.P.; Miyamoto, K.; Gurdon, J.B. Mechanisms of nuclear reprogramming by eggs and oocytes: A deterministic process? Nat. Rev. Mol. Cell Biol. 2011, 12, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Takahama, Y.; Abe, K.; Nakatsuji, N.; Tada, T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr. Biol. 2001, 11, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.A.; Atienza, J.; Melton, D.A.; Eggan, K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science 2005, 309, 1369–1373. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S.; Blau, H.M. Nuclear reprogramming to a pluripotent state by three approaches. Nature 2010, 465, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Hansis, C.; Barreto, G.; Maltry, N.; Niehrs, C. Nuclear reprogramming of human somatic cells by xenopus egg extract requires BRG1. Curr. Biol. 2004, 14, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Freberg, C.T.; Dahl, J.A.; Timoskainen, S.; Collas, P. Epigenetic reprogramming of Oct4 and Nanog regulatory regions by embryonal carcinoma cell extract. Mol. Biol. Cell 2007, 18, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Taranger, C.K.; Noer, A.; Sorensen, A.L.; Hakelien, A.M.; Boquest, A.C.; Collas, P. Induction of dedifferentiation, genomewide transcriptional programming, and epigenetic reprogramming by extracts of carcinoma and embryonic stem cells. Mol. Biol. Cell 2005, 16, 5719–5735. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, T.; Hayashi, Y.; Ogura, A. Participation of the female pronucleus derived from the second polar body in full embryonic development of mice. J. Reprod. Fertil. 1997, 110, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hamalainen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. Piggybac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Brennand, K.; Hochedlinger, K. Reprogramming of pancreatic beta cells into induced pluripotent stem cells. Curr. Biol. 2008, 18, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Norrby, K.; Paca, A.; Mileikovsky, M.; Mohseni, P.; Woltjen, K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 2009, 458, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Maherali, N.; Sridharan, R.; Xie, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachechko, R.; Tchieu, J.; Jaenisch, R.; et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007, 448, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Brambrink, T.; Foreman, R.; Welstead, G.G.; Lengner, C.J.; Wernig, M.; Suh, H.; Jaenisch, R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2008, 2, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Maherali, N.; Breault, D.T.; Hochedlinger, K. Defining molecular cornerstones during fibroblast to ips cell reprogramming in mouse. Cell Stem Cell 2008, 2, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Sommer, A.G.; Longmire, T.A.; Christodoulou, C.; Thomas, D.D.; Gostissa, M.; Alt, F.W.; Murphy, G.J.; Kotton, D.N.; Mostoslavsky, G. Excision of reprogramming transgenes improves the differentiation potential of iPS cells generated with a single excisable vector. Stem Cells 2010, 28, 64–74. [Google Scholar] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Jia, F.; Wilson, K.D.; Sun, N.; Gupta, D.M.; Huang, M.; Li, Z.; Panetta, N.J.; Chen, Z.Y.; Robbins, R.C.; Kay, M.A.; et al. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods 2010, 7, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggybac transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chang, D.C.; Chang-Lin, S.; Lin, C.H.; Wu, D.T.; Chen, D.T.; Ying, S.Y. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA 2008, 14, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [PubMed]
- Miyoshi, N.; Ishii, H.; Nagano, H.; Haraguchi, N.; Dewi, D.L.; Kano, Y.; Nishikawa, S.; Tanemura, M.; Mimori, K.; Tanaka, F.; et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011, 8, 633–638. [Google Scholar] [CrossRef]
- Smits, E.; Ponsaerts, P.; Lenjou, M.; Nijs, G.; van Bockstaele, D.R.; Berneman, Z.N.; van Tendeloo, V.F. RNA-based gene transfer for adult stem cells and T cells. Leukemia 2004, 18, 1898–1902. [Google Scholar] [CrossRef]
- Wiehe, J.M.; Ponsaerts, P.; Rojewski, M.T.; Homann, J.M.; Greiner, J.; Kronawitter, D.; Schrezenmeier, H.; Hombach, V.; Wiesneth, M.; Zimmermann, O.; et al. mRNA-mediated gene delivery into human progenitor cells promotes highly efficient protein expression. J. Cell. Mol. Med. 2007, 11, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Boczkowski, D.; Lee, J.; Pruitt, S.; Nair, S. Dendritic cells engineered to secrete anti-gitr antibodies are effective adjuvants to dendritic cell-based immunotherapy. Cancer Gene Ther. 2009, 16, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Sul, J.Y.; Wu, C.W.; Zeng, F.; Jochems, J.; Lee, M.T.; Kim, T.K.; Peritz, T.; Buckley, P.; Cappelleri, D.J.; Maronski, M.; et al. Transcriptome transfer produces a predictable cellular phenotype. Proc. Natl. Acad. Sci. USA 2009, 106, 7624–7629. [Google Scholar] [CrossRef] [PubMed]
- Melhem, N.M.; Liu, X.D.; Boczkowski, D.; Gilboa, E.; Barratt-Boyes, S.M. Robust CD4+ and CD8+ T cell responses to siv using mRNA-transfected DC expressing autologous viral Ag. Eur. J. Immunol. 2007, 37, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Dollins, C.M.; Boczkowski, D.; Sullenger, B.A.; Nair, S. Activated b cells modified by electroporation of multiple mRNAs encoding immune stimulatory molecules are comparable to mature dendritic cells in inducing in vitro antigen-specific T-cell responses. Immunology 2008, 125, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Sul, J.Y.; Peternko, N.B.; Lee, J.H.; Lee, M.; Patel, V.V.; Kim, J.; Eberwine, J.H. Transcriptome transfer provides a model for understanding the phenotype of cardiomyocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 11918–11923. [Google Scholar] [CrossRef] [PubMed]
- Plews, J.R.; Li, J.; Jones, M.; Moore, H.D.; Mason, C.; Andrews, P.W.; Na, J. Activation of pluripotency genes in human fibroblast cells by a novel mRNA based approach. PLoS One 2010, 5, e14397. [Google Scholar] [PubMed]
- Yakubov, E.; Rechavi, G.; Rozenblatt, S.; Givol, D. Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem. Biophys. Res. Commun. 2010, 394, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.; Naaldijk, Y.M.; Fabian, C.; Wirth, H.; Binder, H.; Nikkhah, G.; Armstrong, L.; Stolzing, A. Reprogramming of human huntington fibroblasts using mRNA. ISRN Cell Biol. 2012, 2012, 12. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Rossi, D.J. Reprogramming human fibroblasts to pluripotency using modified mRNA. Nat. Protoc. 2013, 8, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Durruthy-Durruthy, J.; Briggs, S.F.; Awe, J.; Ramathal, C.Y.; Karumbayaram, S.; Lee, P.C.; Heidmann, J.D.; Clark, A.; Karakikes, I.; Loh, K.M.; et al. Rapid and efficient conversion of integration-free human induced pluripotent stem cells to gmp-grade culture conditions. PLoS One 2014, 9, e94231. [Google Scholar] [CrossRef]
- Tavernier, G.; Wolfrum, K.; Demeester, J.; De Smedt, S.C.; Adjaye, J.; Rejman, J. Activation of pluripotency-associated genes in mouse embryonic fibroblasts by non-viral transfection with in vitro-derived mRNAs encoding Oct4, Sox2, Klf4 and cMyc. Biomaterials 2012, 33, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Sullenger, B.A.; Gilboa, E. Emerging clinical applications of RNA. Nature 2002, 418, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Ponsaerts, P.; van Tendeloo, V.F.; Berneman, Z.N. Cancer immunotherapy using RNA-loaded dendritic cells. Clin. Exp. Immunol. 2003, 134, 378–384. [Google Scholar] [PubMed]
- Saeboe-Larssen, S.; Fossberg, E.; Gaudernack, G. mRNA-based electrotransfection of human dendritic cells and induction of cytotoxic T lymphocyte responses against the telomerase catalytic subunit (hTERT). J. Immunol. Methods 2002, 259, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Ponsaerts, P.; van Tendeloo, V.F.; Cools, N.; van Driessche, A.; Lardon, F.; Nijs, G.; Lenjou, M.; Mertens, G.; van Broeckhoven, C.; van Bockstaele, D.R.; et al. mRNA-electroporated mature dendritic cells retain transgene expression, phenotypical properties and stimulatory capacity after cryopreservation. Leukemia 2002, 16, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.R.; Tsakou, G.; Grunebach, F.; Schmidt, S.M.; Brossart, P. Induction of chronic lymphocytic leukemia (CLL)-specific CD4- and CD8-mediated T-cell responses using RNA-transfected dendritic cells. Blood 2004, 103, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Van Tendeloo, V.F.; van Broeckhoven, C.; Berneman, Z.N. Gene therapy: Principles and applications to hematopoietic cells. Leukemia 2001, 15, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Yisraeli, J.K.; Melton, D.A. Synthesis of long, capped transcripts in vitro by SP6 and T7 RNA polymerases. Methods Enzymol. 1989, 180, 42–50. [Google Scholar] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5'-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Antiviral signaling through pattern recognition receptors. J. Biochem. 2007, 141, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors and type I interferons. J. Biol. Chem. 2007, 282, 15319–15323. [Google Scholar] [CrossRef] [PubMed]
- Angel, M.; Yanik, M.F. Innate immune suppression enables frequent transfection with RNA encoding reprogramming proteins. PLoS One 2010, 5, e11756. [Google Scholar] [PubMed]
- Drews, K.; Tavernier, G.; Demeester, J.; Lehrach, H.; de Smedt, S.C.; Rejman, J.; Adjaye, J. The cytotoxic and immunogenic hurdles associated with non-viral mRNA-mediated reprogramming of human fibroblasts. Biomaterials 2012, 33, 4059–4068. [Google Scholar] [CrossRef] [PubMed]
- Van Tendeloo, V.F.; Willems, R.; Ponsaerts, P.; Lenjou, M.; Nijs, G.; Vanhove, M.; Muylaert, P.; van Cauwelaert, P.; van Broeckhoven, C.; van Bockstaele, D.R.; et al. High-level transgene expression in primary human t lymphocytes and adult bone marrow CD34+ cells via electroporation-mediated gene delivery. Gene Ther. 2000, 7, 1431–1437. [Google Scholar]
- Jin, J.; Kwon, Y.W.; Paek, J.S.; Cho, H.J.; Yu, J.; Lee, J.Y.; Chu, I.S.; Park, I.H.; Park, Y.B.; Kim, H.S.; et al. Analysis of differential proteomes of induced pluripotent stem cells by protein-based reprogramming of fibroblasts. J. Proteome Res. 2011, 10, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Ratanasirintrawoot, S.; Park, I.H.; Manos, P.D.; Loh, Y.H.; Huo, H.; Miller, J.D.; Hartung, O.; Rho, J.; Ince, T.A.; et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat. Biotechnol. 2009, 27, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Nachman, I.; Regev, A.; Meissner, A. Dynamic single-cell imaging of direct reprogramming reveals an early specifying event. Nat. Biotechnol. 2010, 28, 521–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsson, S.; Gurdon, J. DNA demethylation is necessary for the epigenetic reprogramming of somatic cell nuclei. Nat. Cell Biol. 2004, 6, 984–990. [Google Scholar] [PubMed]
- Robinton, D.A.; Daley, G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature 2012, 481, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Hochedlinger, K. Induced pluripotency: History, mechanisms, and applications. Genes Dev. 2010, 24, 2239–2263. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K.; Maruyama, M.; Maeda, M.; Yamanaka, S. The homeoprotein nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003, 113, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Gertsenstein, M.; Vintersten, K.; Behringer, R. Karyotyping mouse cells. CSH Protoc. 2008, 2008. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Sayed, A.K.; Zhang, Z.; Zhang, L.; Liu, Z.; Abbott, L.C.; Zhang, Y.; Li, B. Pluripotent State Induction in Mouse Embryonic Fibroblast Using mRNAs of Reprogramming Factors. Int. J. Mol. Sci. 2014, 15, 21840-21864. https://doi.org/10.3390/ijms151221840
El-Sayed AK, Zhang Z, Zhang L, Liu Z, Abbott LC, Zhang Y, Li B. Pluripotent State Induction in Mouse Embryonic Fibroblast Using mRNAs of Reprogramming Factors. International Journal of Molecular Sciences. 2014; 15(12):21840-21864. https://doi.org/10.3390/ijms151221840
Chicago/Turabian StyleEl-Sayed, Ahmed Kamel, Zhentao Zhang, Lei Zhang, Zhiyong Liu, Louise C. Abbott, Yani Zhang, and Bichun Li. 2014. "Pluripotent State Induction in Mouse Embryonic Fibroblast Using mRNAs of Reprogramming Factors" International Journal of Molecular Sciences 15, no. 12: 21840-21864. https://doi.org/10.3390/ijms151221840
APA StyleEl-Sayed, A. K., Zhang, Z., Zhang, L., Liu, Z., Abbott, L. C., Zhang, Y., & Li, B. (2014). Pluripotent State Induction in Mouse Embryonic Fibroblast Using mRNAs of Reprogramming Factors. International Journal of Molecular Sciences, 15(12), 21840-21864. https://doi.org/10.3390/ijms151221840