Interactome Mapping Reveals Important Pathways in Skeletal Muscle Development of Pigs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
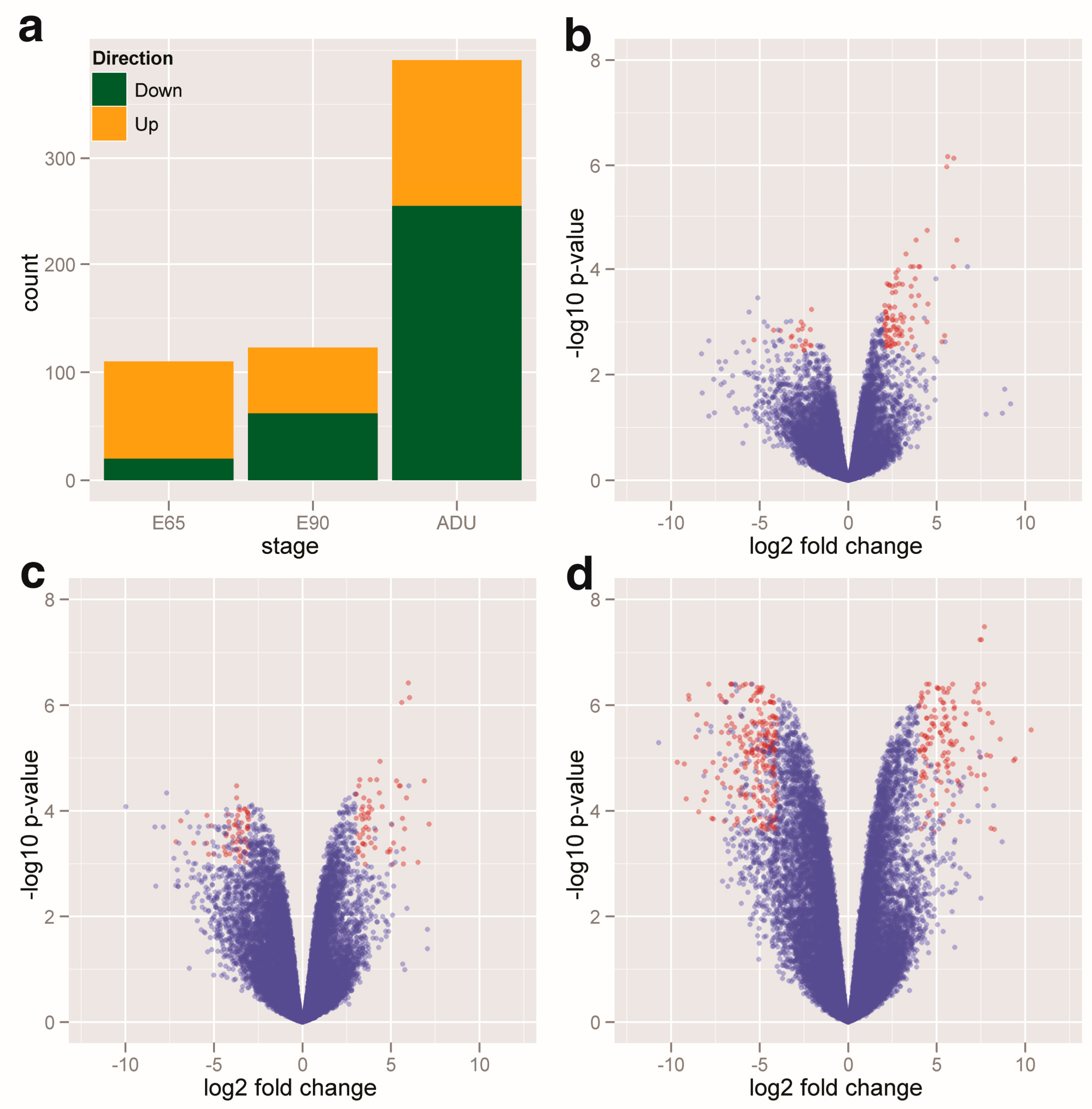
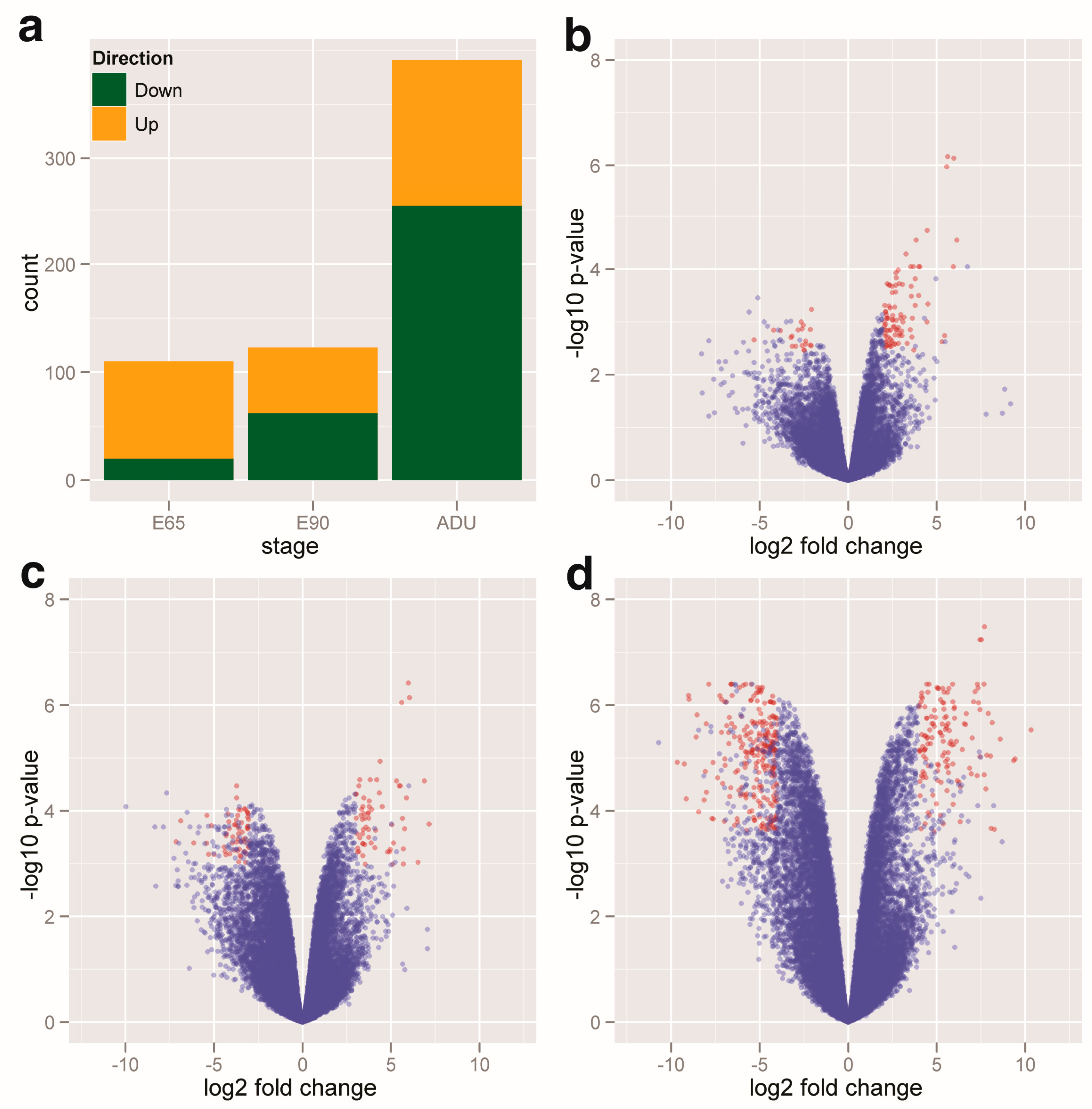
2.1. Profiling of the DEGs and DEMs
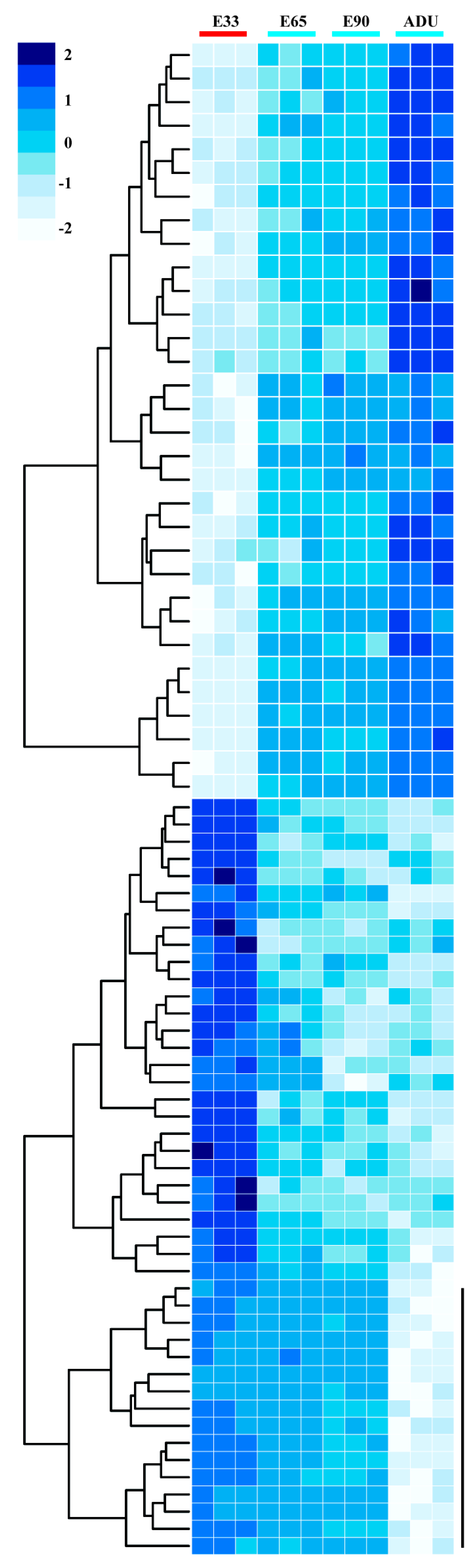
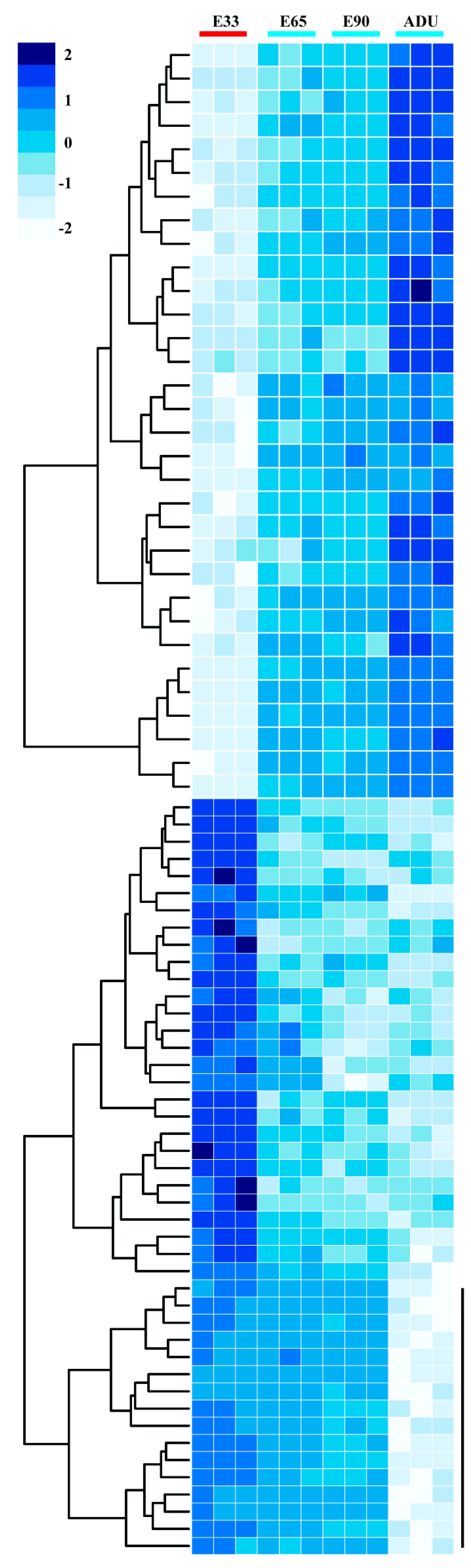
2.2. Linear Expression Patterns of DEGs
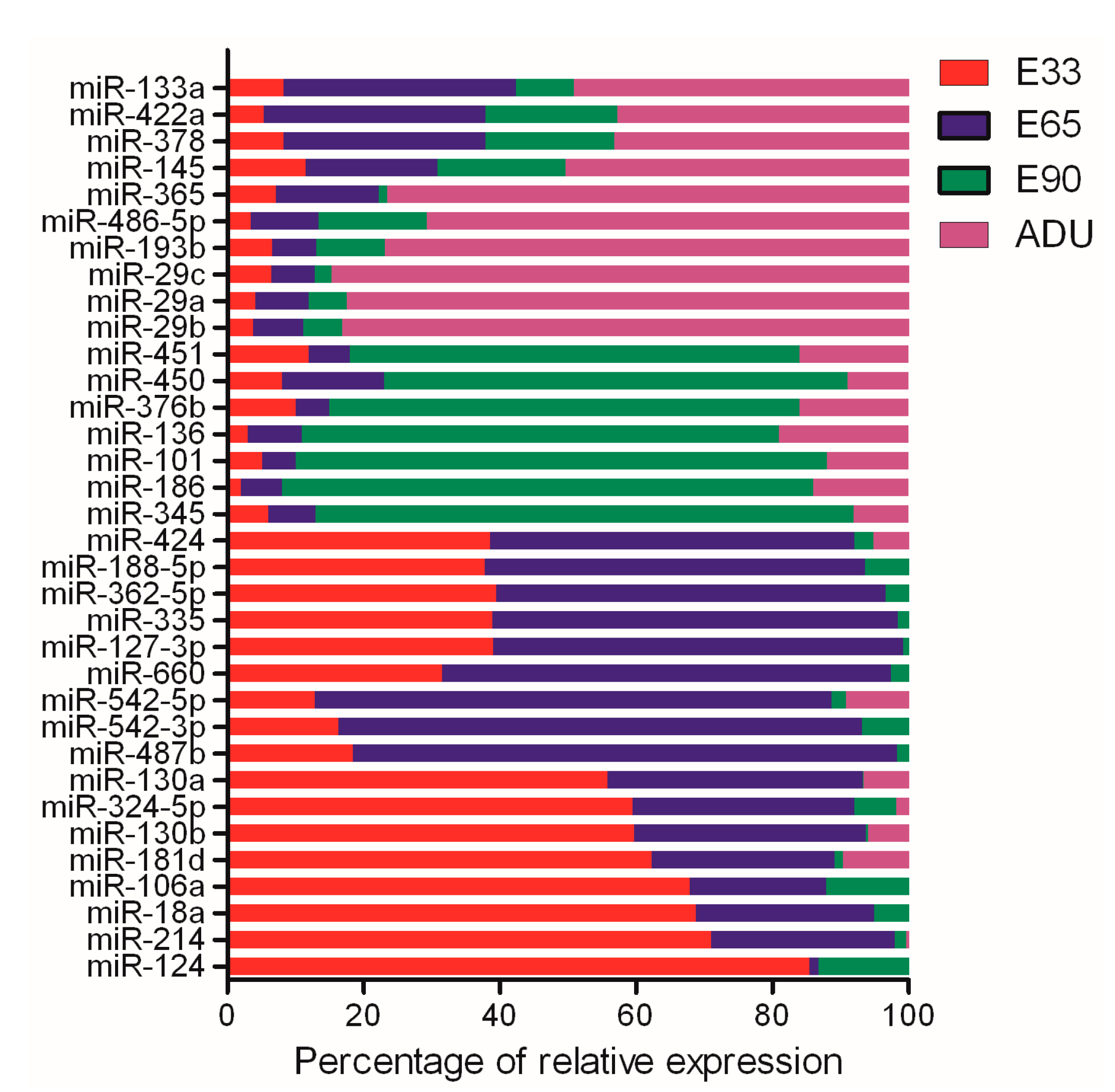
2.3. Characterization of the miRNA Patterns

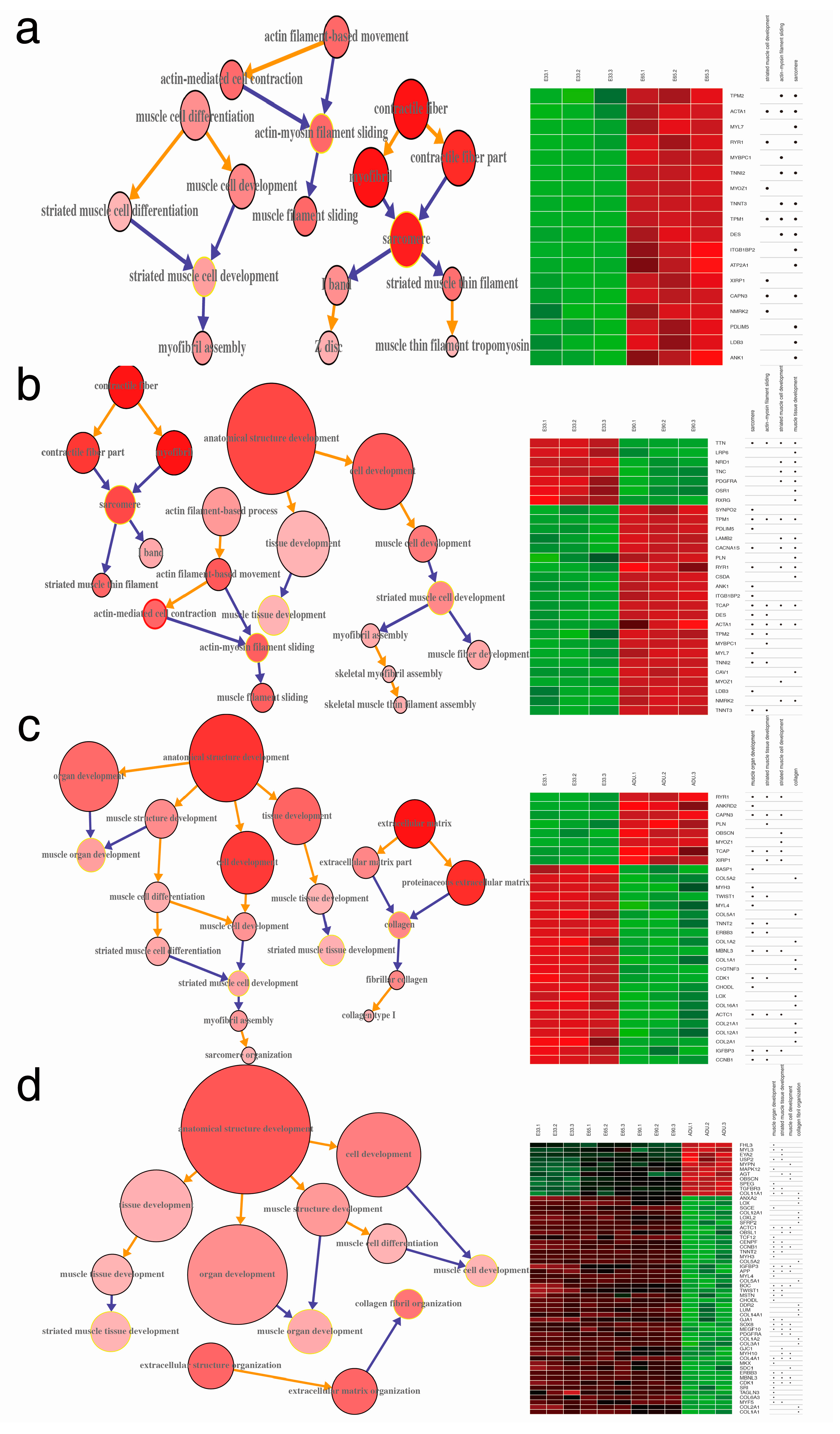
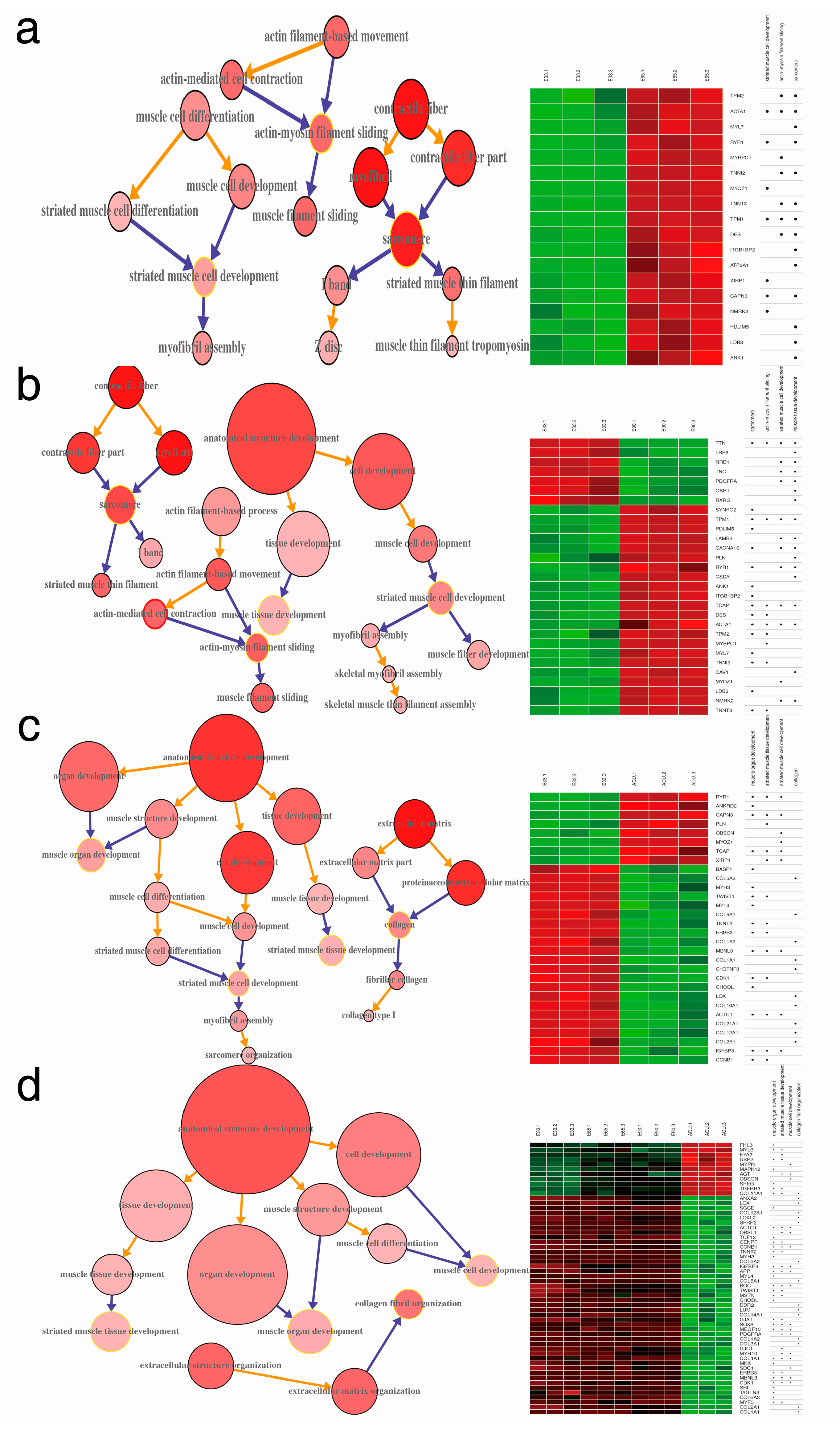
2.4. Gene Ontology Annotation (GOA) of DEGs
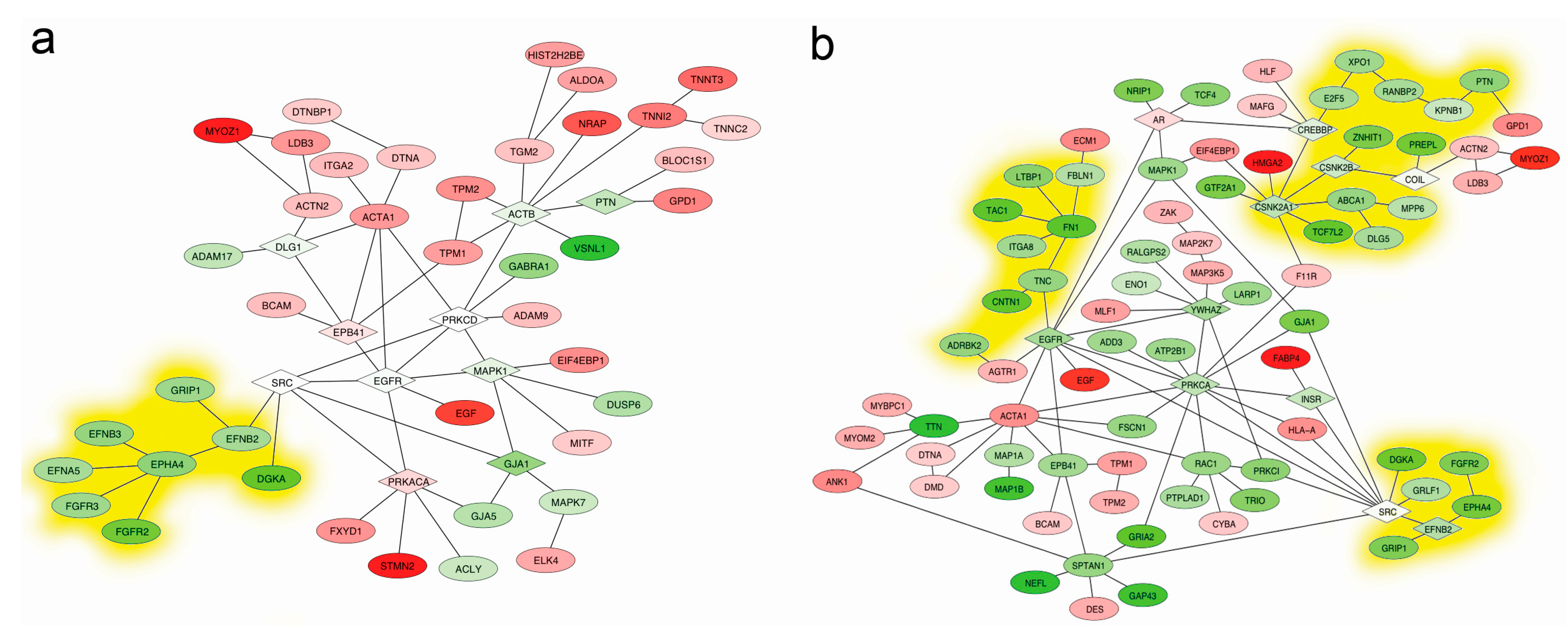
2.5. Mapping Interactome Networks of DEGs

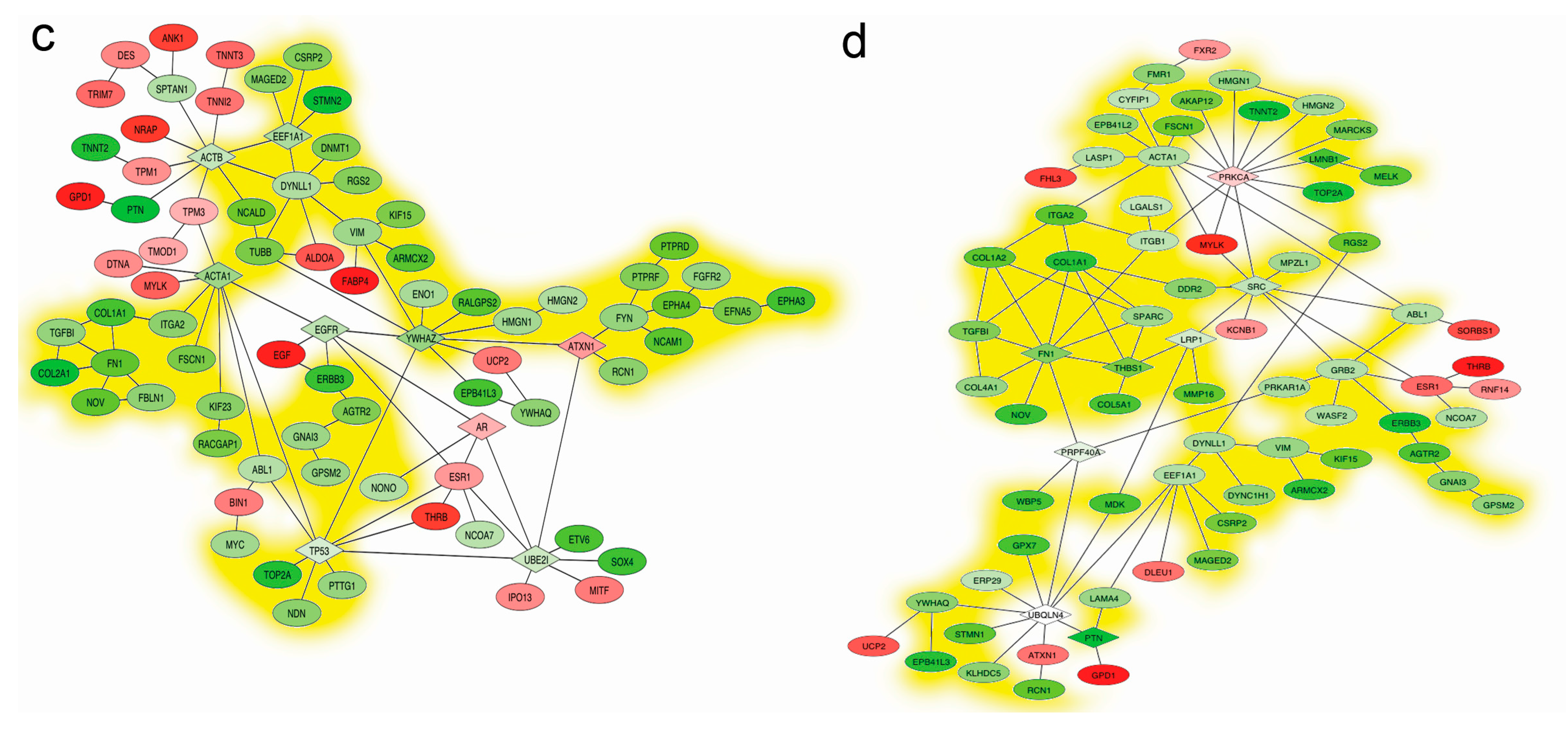
2.6. Discussion
3. Materials and Methods
3.1. Sample Collection and RNA Isolation
3.2. Porcine Gene Chip and Customized miRNA Array
3.3. Statistical Analysis of Gene Chip and miRNA Array
3.4. Databases and Sequences
3.5. Gene Ontology Annotation (GOA)
3.6. Interactome Mapping of the DEGs
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blandin, G.; Marchand, S.; Charton, K.; Daniele, N.; Gicquel, E.; Boucheteil, J.B.; Bentaib, A.; Barrault, L.; Stockholm, D.; Bartoli, M.; et al. A human skeletal muscle interactome centered on proteins involved in muscular dystrophies: LGMD interactome. Skelet. Muscle 2013, 3, 3. [Google Scholar]
- Grefte, S.; Kuijpers-Jagtman, A.M.; Torensma, R.; von den Hoff, J.W. Skeletal muscle development and regeneration. Stem Cells Dev. 2007, 16, 857–868. [Google Scholar]
- Zanou, N.; Gailly, P. Skeletal muscle hypertrophy and regeneration: Interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell. Mol. Life Sci. 2013, 70, 4117–4130. [Google Scholar]
- Yang, H.; Li, F.; Kong, X.; Yuan, X.; Wang, W.; Huang, R.; Li, T.; Geng, M.; Wu, G.; Yin, Y. Chemerin regulates proliferation and differentiation of myoblast cells via ERK1/2 and mTOR signaling pathways. Cytokine 2012, 60, 646–652. [Google Scholar]
- Yang, H.; Li, F.; Xiong, X.; Kong, X.; Zhang, B.; Yuan, X.; Fan, J.; Duan, Y.; Geng, M.; Li, L.; et al. Soy isoflavones modulate adipokines and myokines to regulate lipid metabolism in adipose tissue, skeletal muscle and liver of male Huanjiang mini-pigs. Mol. Cell. Endocrinol. 2013, 365, 44–51. [Google Scholar]
- Zhou, Y.; Tang, X.; Song, Q.; Ji, Y.; Wang, H.; Jiao, H.; Ouyang, H.; Pang, D. Identification and characterization of pig embryo microRNAs by Solexa sequencing. Reprod. Domest. Anim. 2013, 48, 112–120. [Google Scholar]
- Ropka-Molik, K.; Zukowski, K.; Eckert, R.; Gurgul, A.; Piorkowska, K.; Oczkowicz, M. Comprehensive analysis of the whole transcriptomes from two different pig breeds using RNA-Seq method. Anim. Genet. 2014, 45, 674–684. [Google Scholar]
- Townley-Tilson, W.H.; Callis, T.E.; Wang, D. MicroRNAs 1, 133, and 206: Critical factors of skeletal and cardiac muscle development, function, and disease. Int. J. Biochem. Cell Biol. 2010, 42, 1252–1255. [Google Scholar]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar]
- Drummond, M.J.; Glynn, E.L.; Fry, C.S.; Dhanani, S.; Volpi, E.; Rasmussen, B.B. Essential amino acids increase microRNA-499, -208b, and -23a and downregulate myostatin and myocyte enhancer factor 2C mRNA expression in human skeletal muscle. J. Nutr. 2009, 139, 2279–2284. [Google Scholar]
- Eisenberg, I.; Eran, A.; Nishino, I.; Moggio, M.; Lamperti, C.; Amato, A.A.; Lidov, H.G.; Kang, P.B.; North, K.N.; Mitrani-Rosenbaum, S.; et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 17016–17021. [Google Scholar]
- Pas, M.F.W.T.; Everts, M.E.; Haagsman, H. Muscle Development of Livestock Animals: Physiology, Genetics and Meat Quality; CABI Publishing: Cambridge, MA, USA, 2004; pp. 39–79. [Google Scholar]
- Berard, J.; Kalbe, C.; Losel, D.; Tuchscherer, A.; Rehfeldt, C. Potential sources of early-postnatal increase in myofibre number in pig skeletal muscle. Histochem. Cell Biol. 2011, 136, 217–225. [Google Scholar]
- Yang, H.; Fu, D.; Shao, H.; Kong, X.; Wang, W.; Yang, X.; Nyachoti, C.M.; Yin, Y. Impacts of birth weight on plasma, liver and skeletal muscle neutral amino acid profiles and intestinal amino acid transporters in suckling Huanjiang mini-piglets. PLoS One 2012, 7, e50921. [Google Scholar]
- Li, H.Y.; Xi, Q.Y.; Xiong, Y.Y.; Liu, X.L.; Cheng, X.; Shu, G.; Wang, S.B.; Wang, L.N.; Gao, P.; Zhu, X.T.; et al. Identification and comparison of microRNAs from skeletal muscle and adipose tissues from two porcine breeds. Anim. Genet. 2012, 43, 704–713. [Google Scholar]
- Xie, S.S.; Li, X.Y.; Liu, T.; Cao, J.H.; Zhong, Q.; Zhao, S.H. Discovery of porcine microRNAs in multiple tissues by a Solexa deep sequencing approach. PLoS One 2011, 6, e16235. [Google Scholar]
- Hitachi, K.; Tsuchida, K. Role of microRNAs in skeletal muscle hypertrophy. Front. Physiol. 2013, 4, 408. [Google Scholar]
- Huang, T.H.; Zhu, M.J.; Li, X.Y.; Zhao, S.H. Discovery of porcine microRNAs and profiling from skeletal muscle tissues during development. PLoS One 2008, 3, e3225. [Google Scholar]
- Costamagna, D.; Quattrocelli, M.; Duelen, R.; Sahakyan, V.; Perini, I.; Palazzolo, G.; Sampaolesi, M. Fate choice of post-natal mesoderm progenitors: Skeletal versus cardiac muscle plasticity. Cell. Mol. Life Sci. 2014, 71, 615–627. [Google Scholar]
- Kim, J.A.; Shon, Y.H.; Lim, J.O.; Yoo, J.J.; Shin, H.I.; Park, E.K. MYOD mediates skeletal myogenic differentiation of human amniotic fluid stem cells and regeneration of muscle injury. Stem Cell Res. Ther. 2013, 4, 147. [Google Scholar]
- Kanisicak, O.; Mendez, J.J.; Yamamoto, S.; Yamamoto, M.; Goldhamer, D.J. Progenitors of skeletal muscle satellite cells express the muscle determination gene, MyoD. Dev. Biol. 2009, 332, 131–141. [Google Scholar]
- Dobashi, Y.; Suzuki, S.; Sato, E.; Hamada, Y.; Yanagawa, T.; Ooi, A. EGFR-dependent and independent activation of Akt/mTOR cascade in bone and soft tissue tumors. Mod. Pathol. 2009, 22, 1328–1340. [Google Scholar]
- Haflidadottir, B.S.; Bergsteinsdottir, K.; Praetorius, C.; Steingrimsson, E. miR-148 regulates Mitf in melanoma cells. PLoS One 2010, 5, e11574. [Google Scholar]
- Maurer, B.; Stanczyk, J.; Jungel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheumatol. 2010, 62, 1733–1743. [Google Scholar]
- Ashmore, C.R.; Addis, P.B.; Doerr, L. Development of muscle fibers in the fetal pig. J. Anim. Sci. 1973, 36, 1088–1093. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Gentleman, R.; Carey, V.; Dudoit, S.; Irizarry, R.; Huber, W. Bioinformatics and Computational Biology Sulutions Using R and Bioconductor; Springer: New York, NY, USA, 2005; pp. 33–442. [Google Scholar]
- Wettenhall, J.M.; Smyth, G.K. limmaGUI: A graphical user interface for linear modeling of microarray data. Bioinformatics 2004, 20, 3705–3706. [Google Scholar]
- Ensembl Website. Available online: http://www.ensembl.org (accessed on 17 November 2014).
- NCBI Website. Available online: http://www.ncbi.nlm.nih.gov (accessed on 17 November 2014).
- miRBase Website. Available online: http://www.mirbase.org (accessed on 17 November 2014).
- Friedlander, M.R.; Chen, W.; Adamidi, C.; Maaskola, J.; Einspanier, R.; Knespel, S.; Rajewsky, N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008, 26, 407–415. [Google Scholar]
- TargetScan Website. Available online: http://www.targetscan.org (accessed on 17 November 2014).
- Gene Ontology Consortium. Available online: http://www.geneontology.org (accessed on 17 November 2014).
- AmiGO Website. Available online: http://amigo.geneontology.org (accessed on 17 November 2014).
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar]
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar]
- Carey, V.J.; Gentry, J.; Whalen, E.; Gentleman, R. Network structures and algorithms in Bioconductor. Bioinformatics 2005, 21, 135–136. [Google Scholar]
- Beisser, D.; Klau, G.W.; Dandekar, T.; Muller, T.; Dittrich, M.T. BioNet: An R-Package for the functional analysis of biological networks. Bioinformatics 2010, 26, 1129–1130. [Google Scholar]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, J.; Huang, T.; Li, X.; Zhao, S. Interactome Mapping Reveals Important Pathways in Skeletal Muscle Development of Pigs. Int. J. Mol. Sci. 2014, 15, 21788-21802. https://doi.org/10.3390/ijms151221788
Cao J, Huang T, Li X, Zhao S. Interactome Mapping Reveals Important Pathways in Skeletal Muscle Development of Pigs. International Journal of Molecular Sciences. 2014; 15(12):21788-21802. https://doi.org/10.3390/ijms151221788
Chicago/Turabian StyleCao, Jianhua, Tinghua Huang, Xinyun Li, and Shuhong Zhao. 2014. "Interactome Mapping Reveals Important Pathways in Skeletal Muscle Development of Pigs" International Journal of Molecular Sciences 15, no. 12: 21788-21802. https://doi.org/10.3390/ijms151221788