Modulation of Cyclins, p53 and Mitogen-Activated Protein Kinases Signaling in Breast Cancer Cell Lines by 4-(3,4,5-Trimethoxyphenoxy)benzoic Acid

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
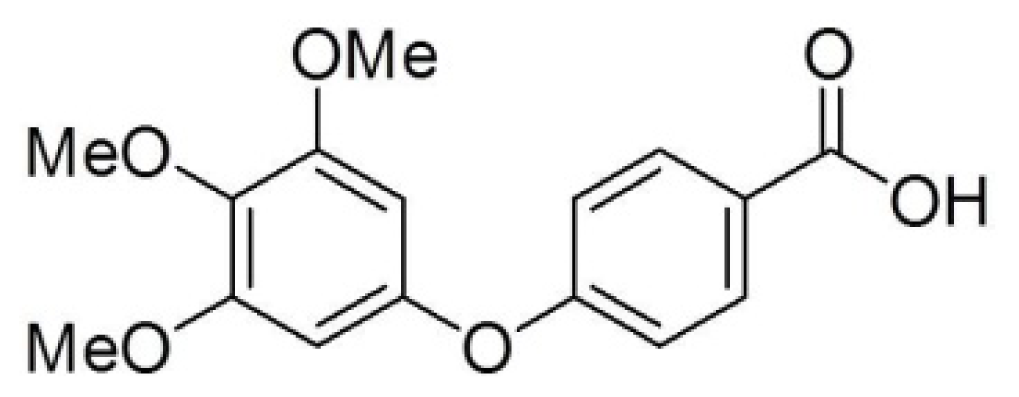
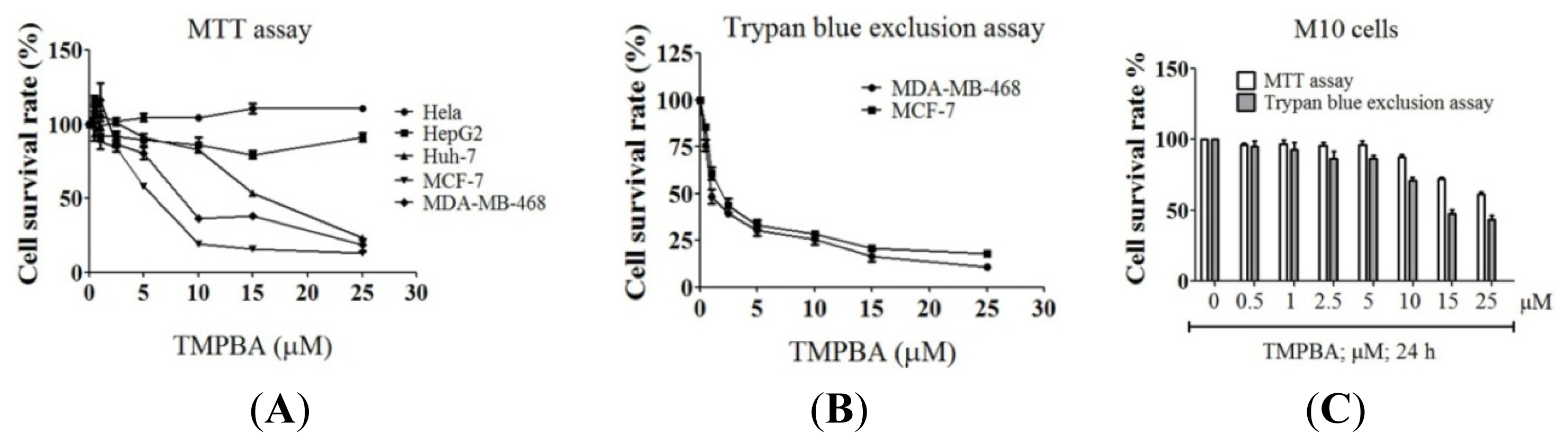
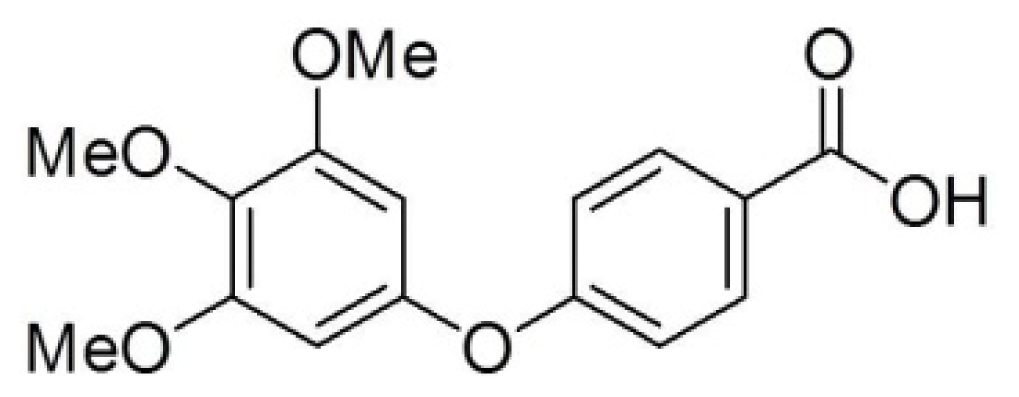
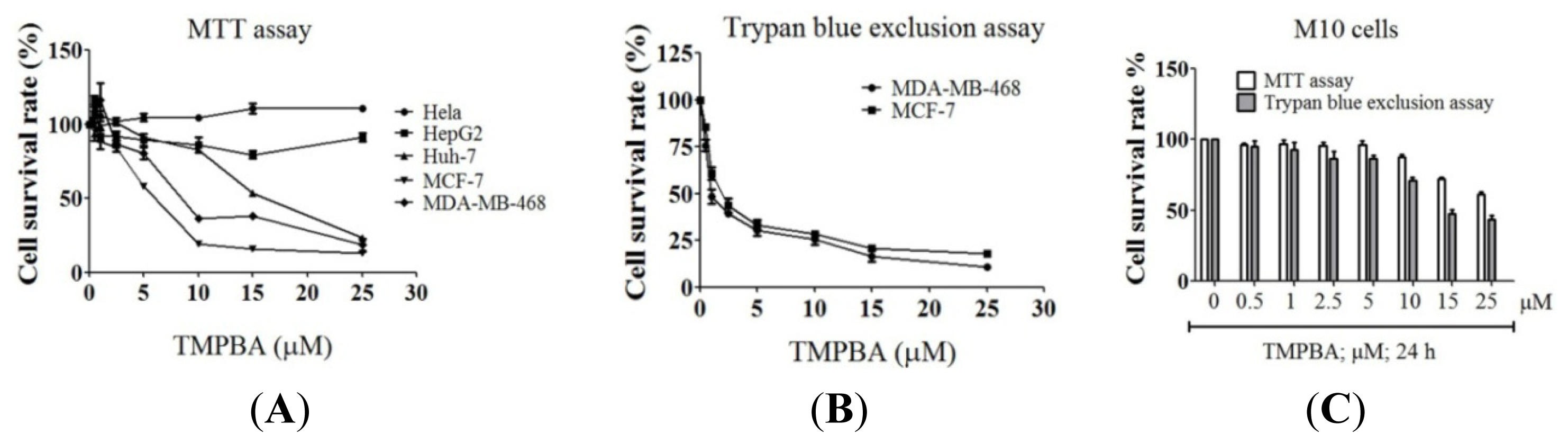
2.1. Differential Susceptibility of Cancer Cell Lines to TMPBA-Induced Cell Death
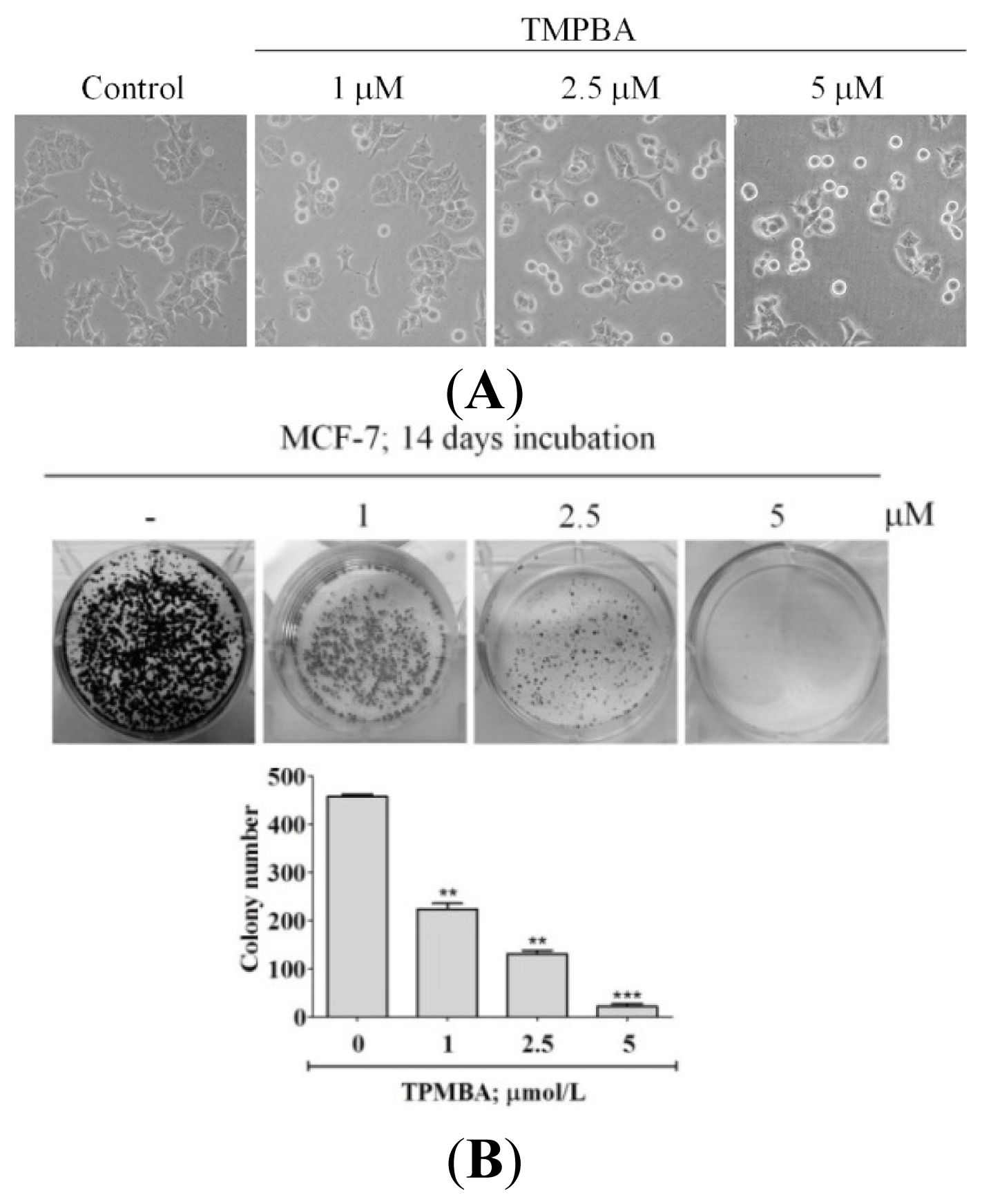
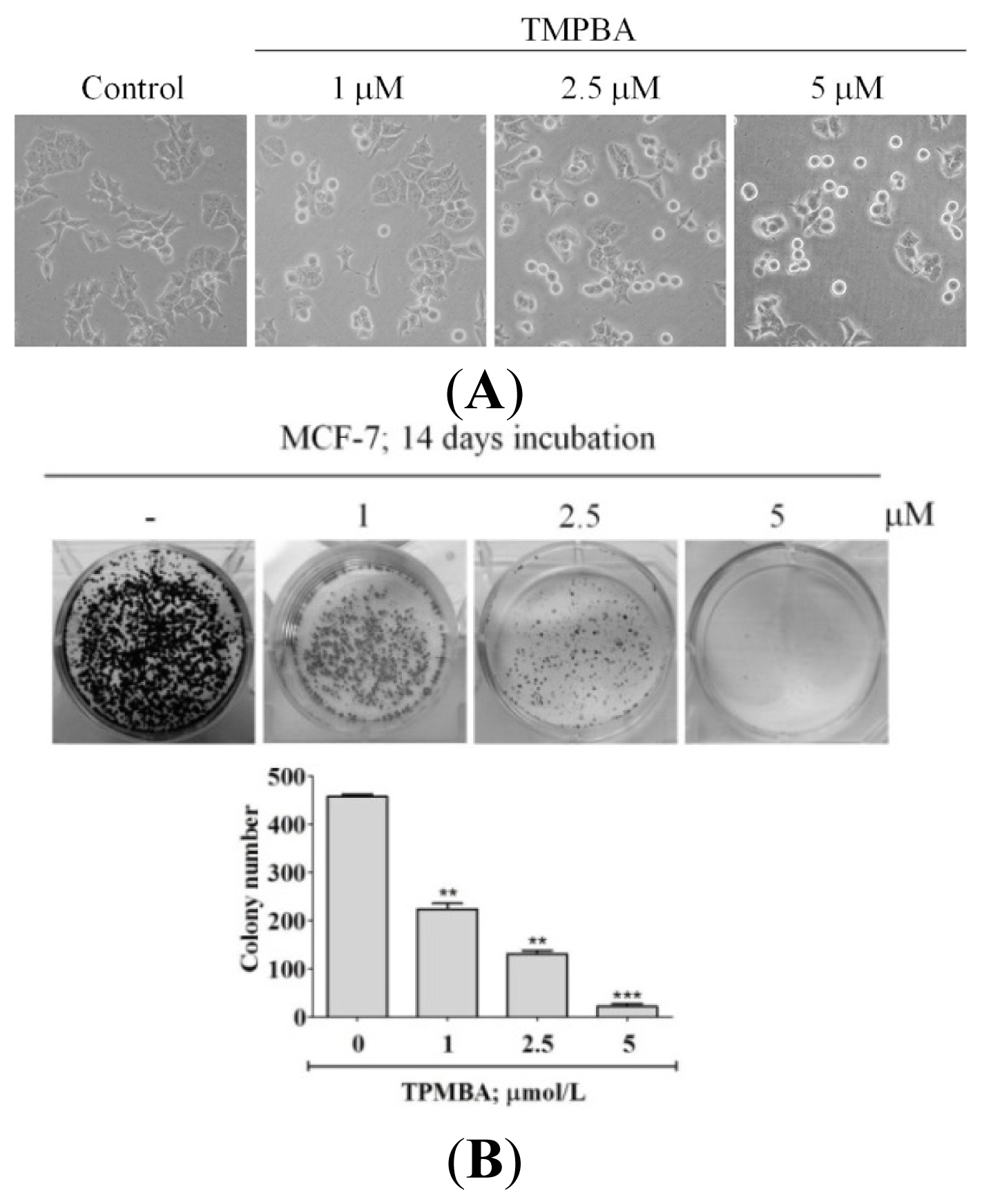
2.2. TMPBA Changed Cell Morphology and Decreased Colony Formation in Breast Cancer Cells
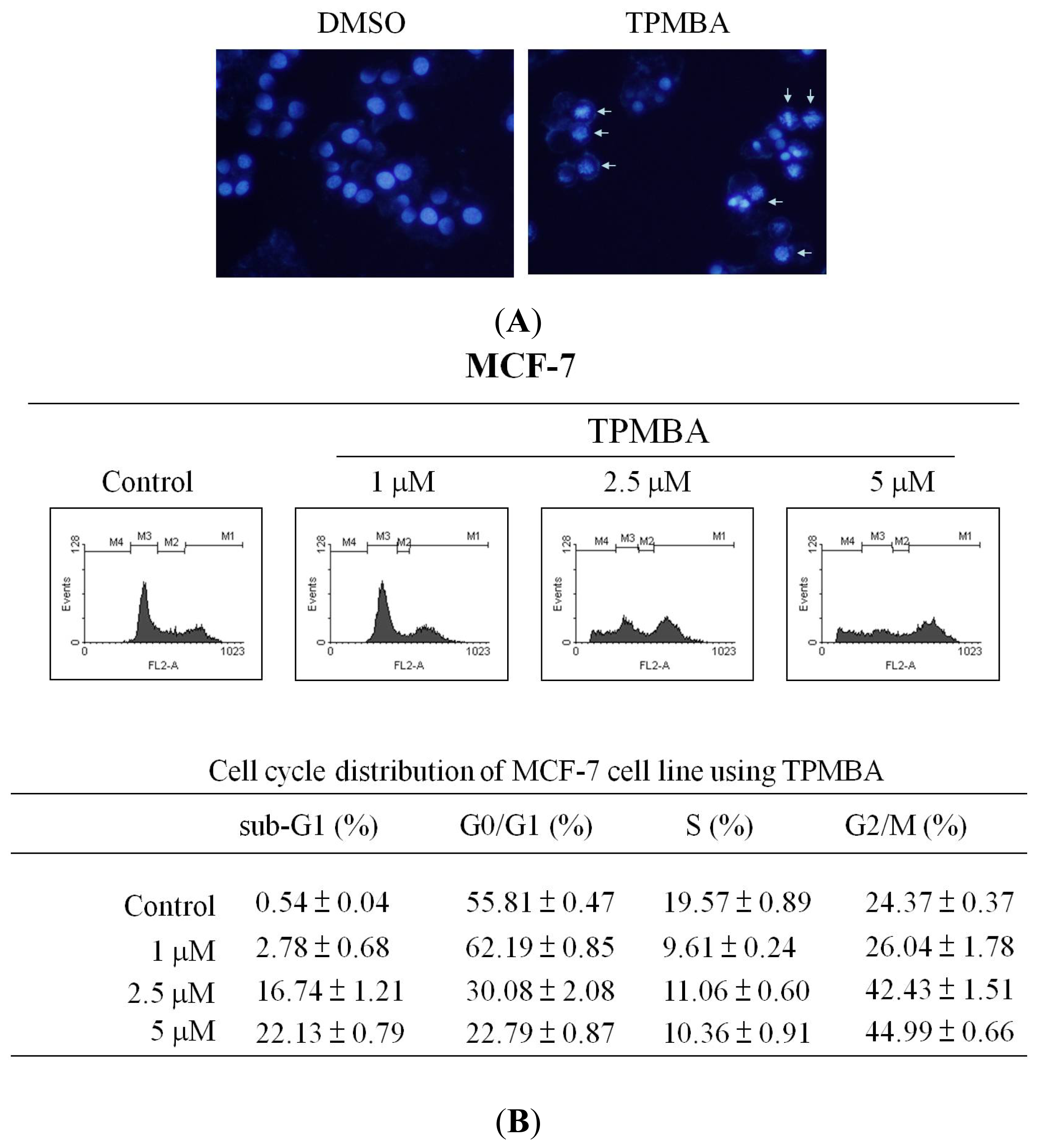
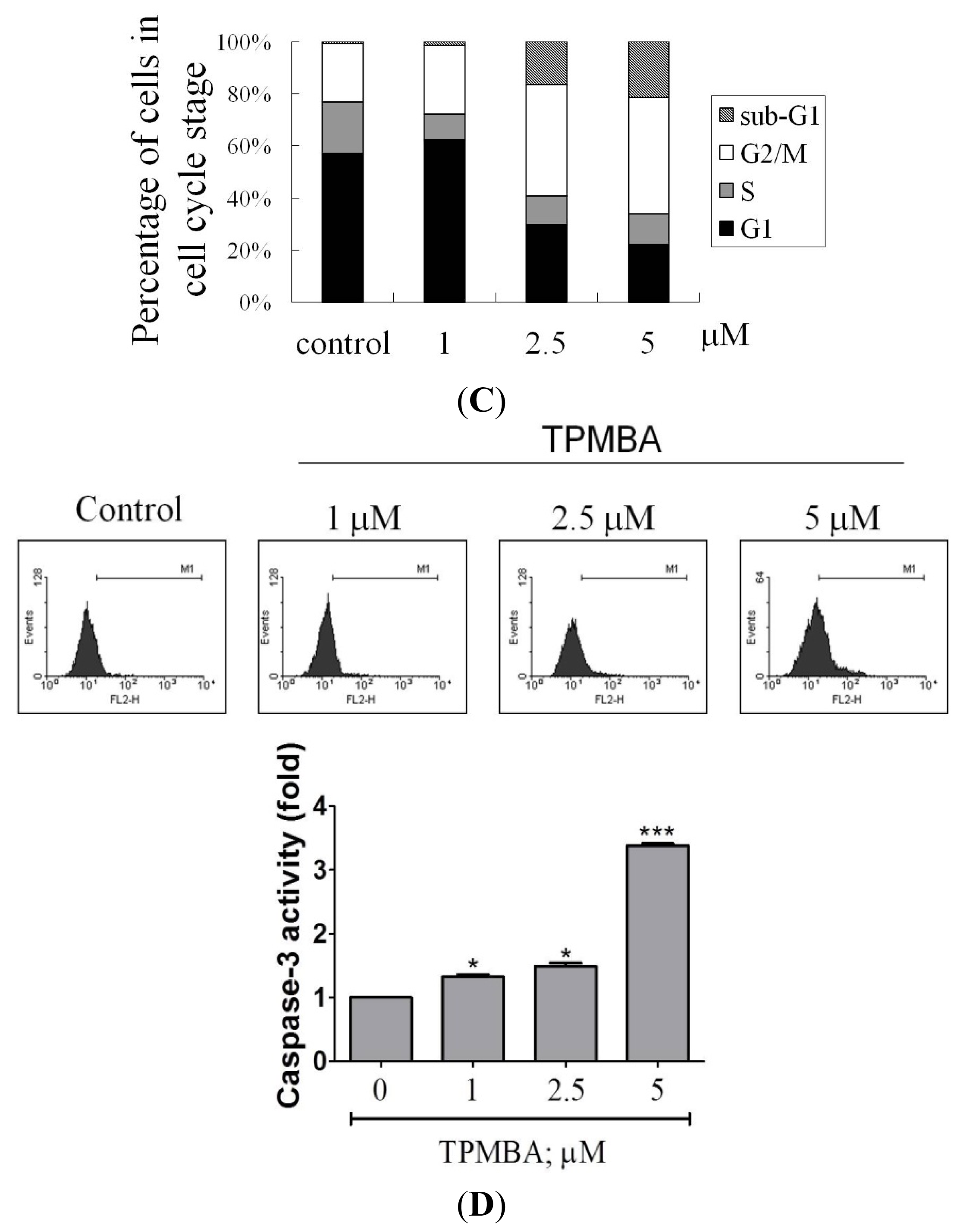
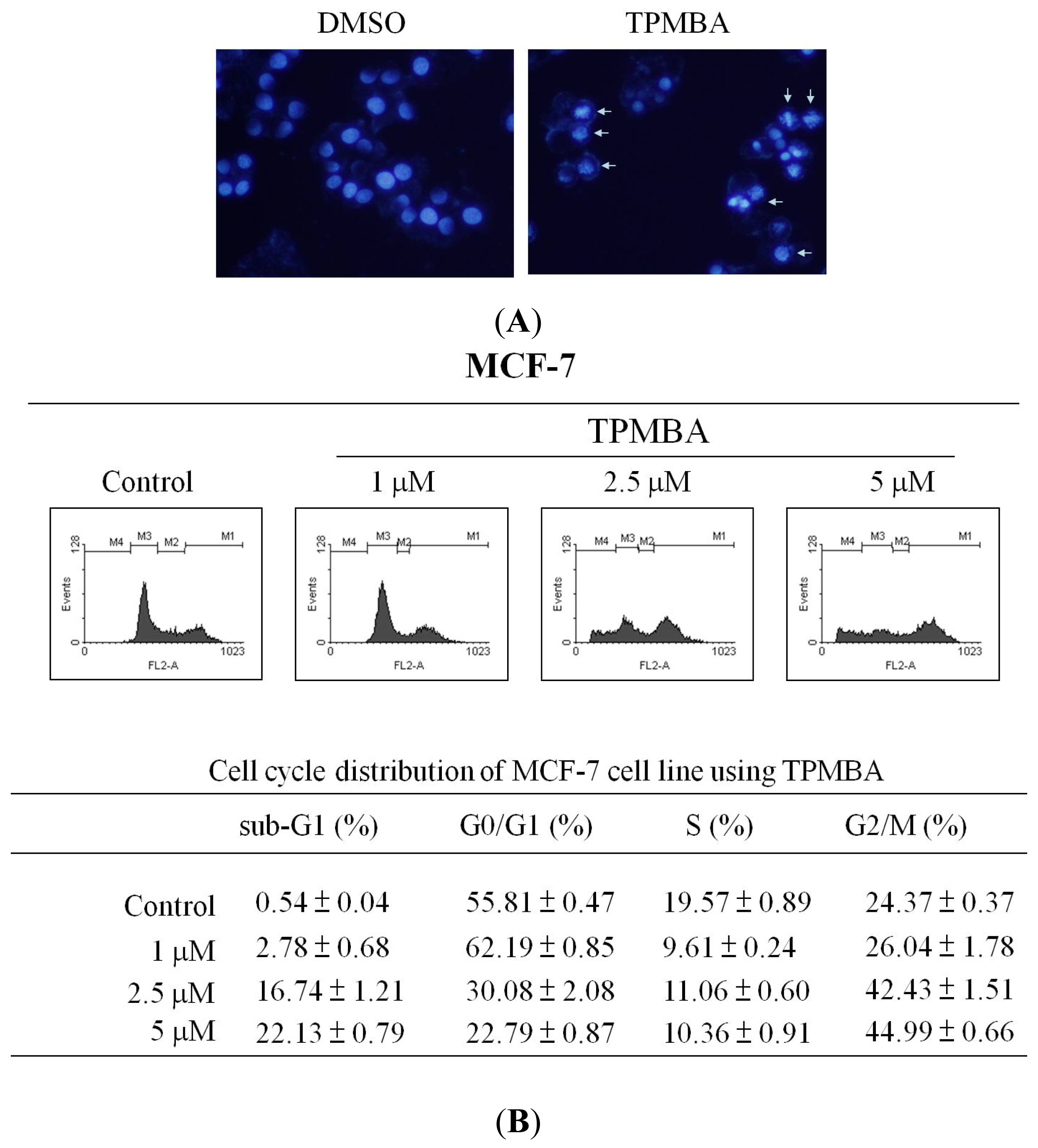
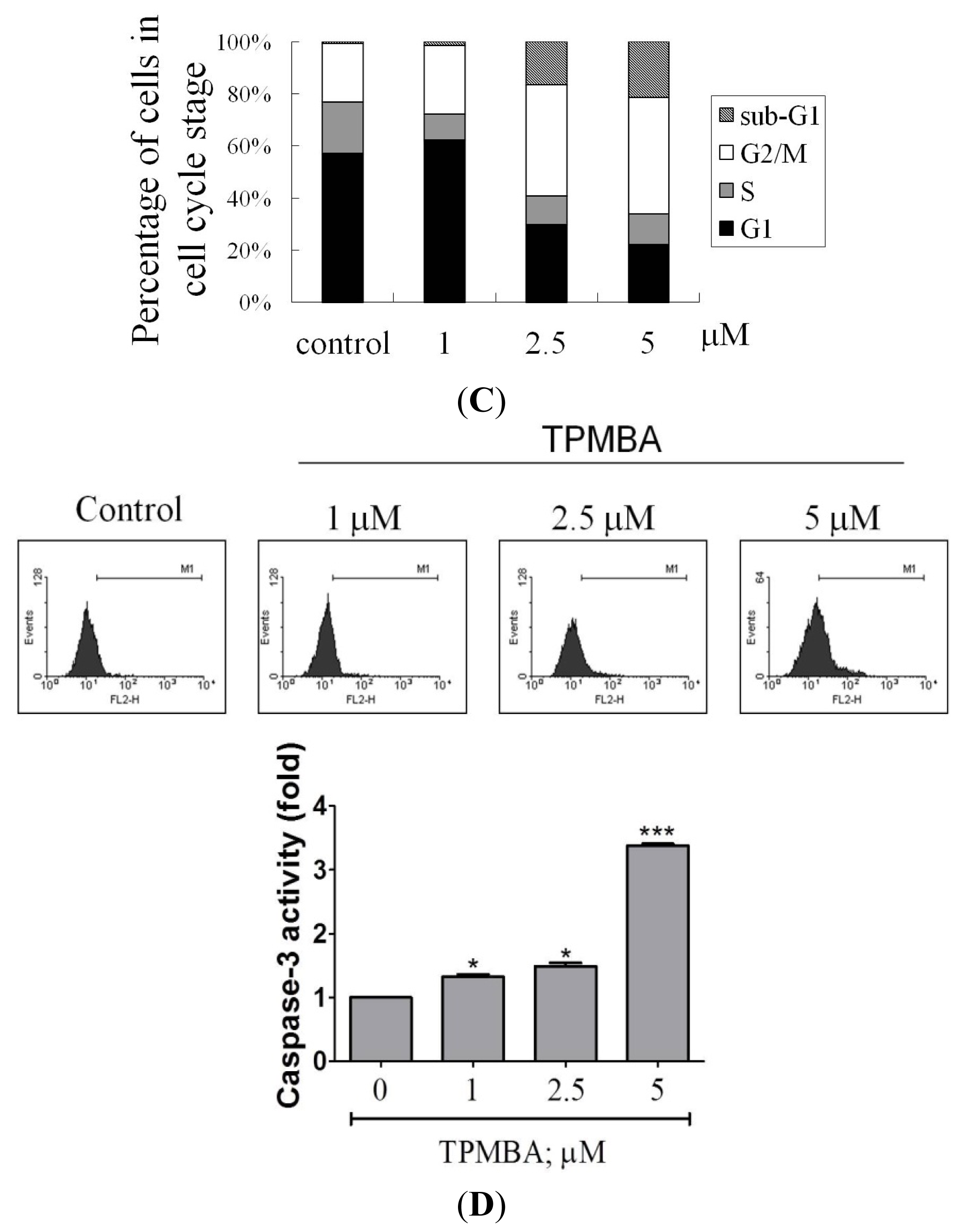
2.3. Induction of Cell Apoptosis and G2/M Cell Cycle Arrest by TMPBA Treatment in Breast Cancer Cells
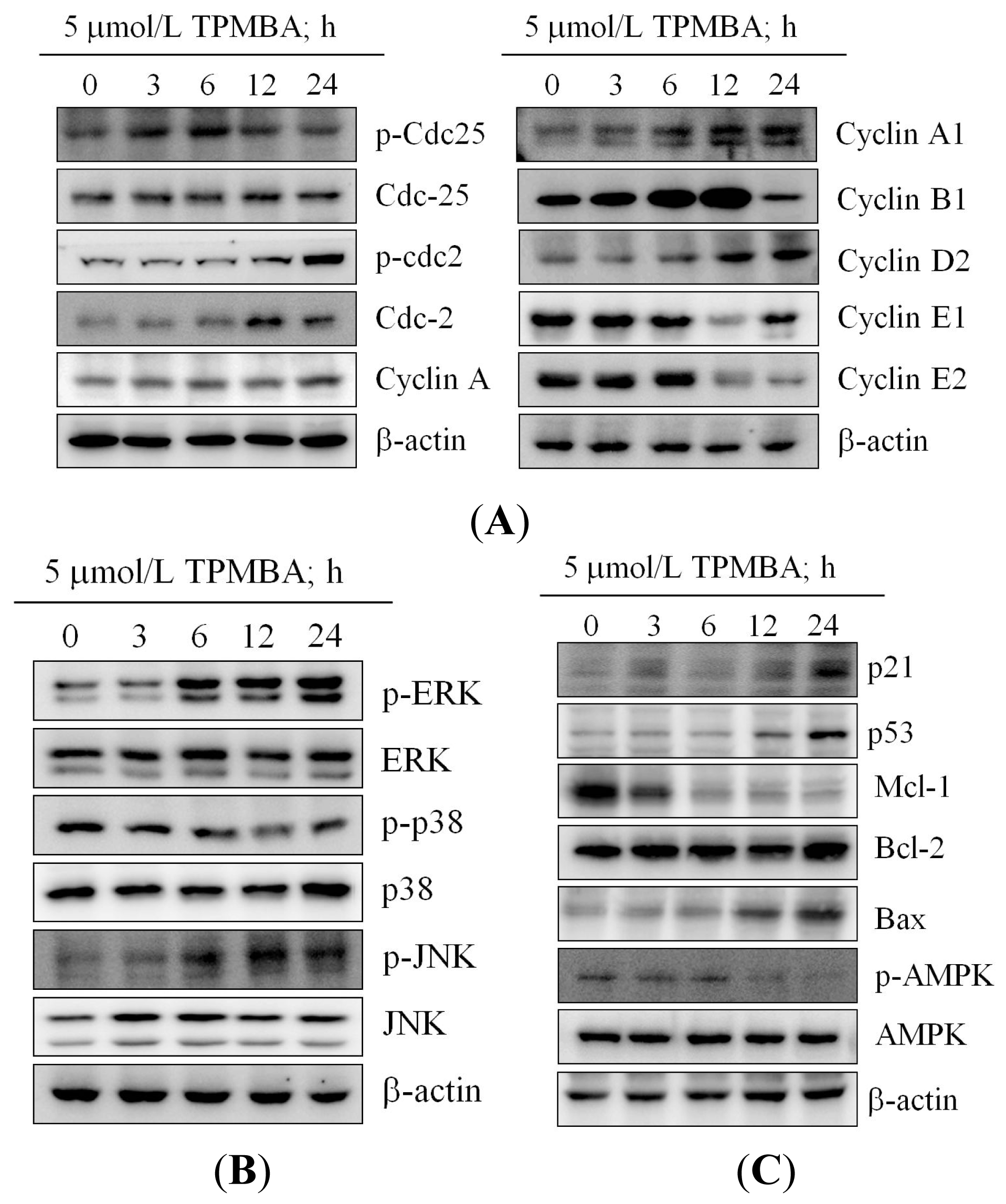
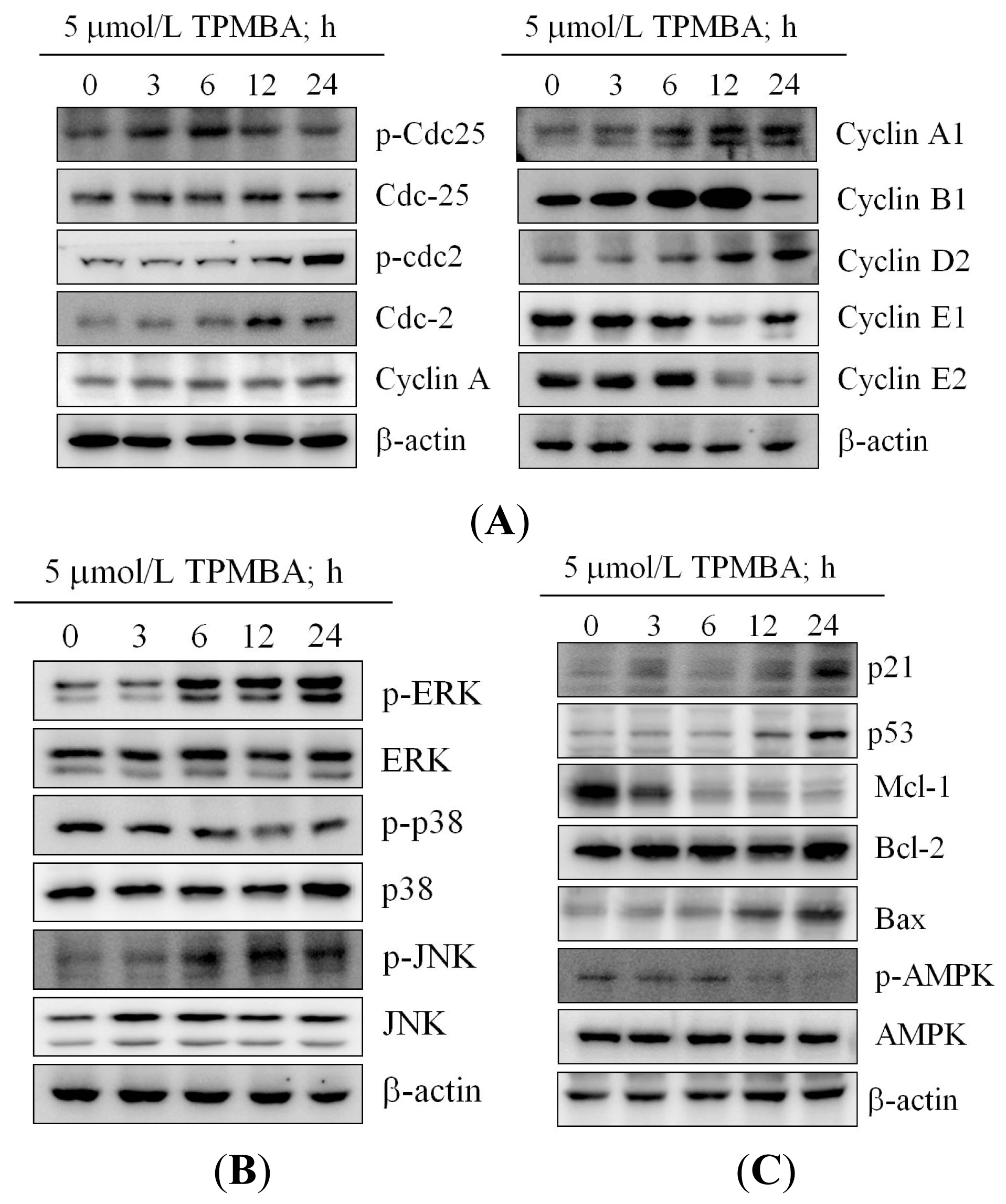
2.4. Modulation of the Expression of Cyclin B, Cyclin E, cdc2, and cdc25 in TMPBA-Treated Cells
2.5. Effect of TMPBA on MAPK Signaling Pathways
2.6. TMPBA Effect on the Expression of p21, p53, Bax, Bcl-2, MCL-1 and AMPK
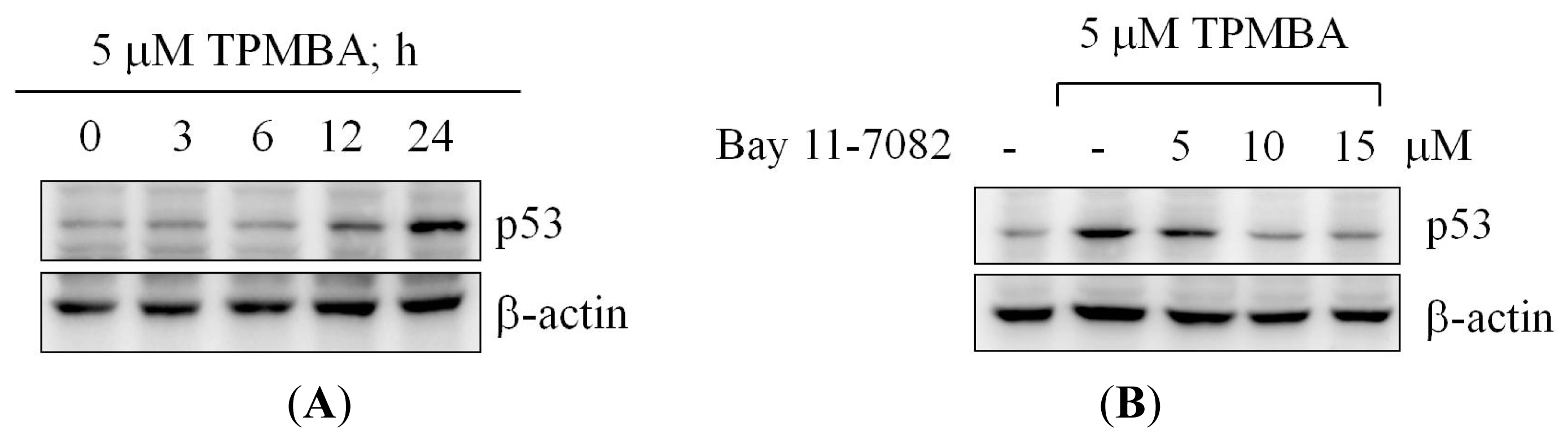
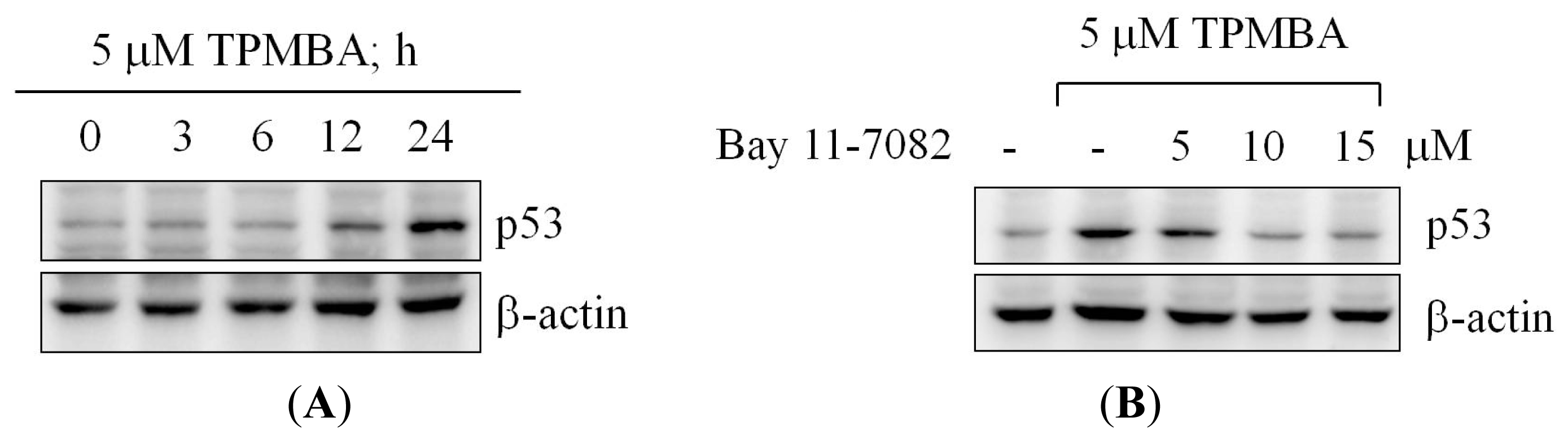
2.7. TMPBA-Induced p53 Expression via NF-κB Signaling Pathway
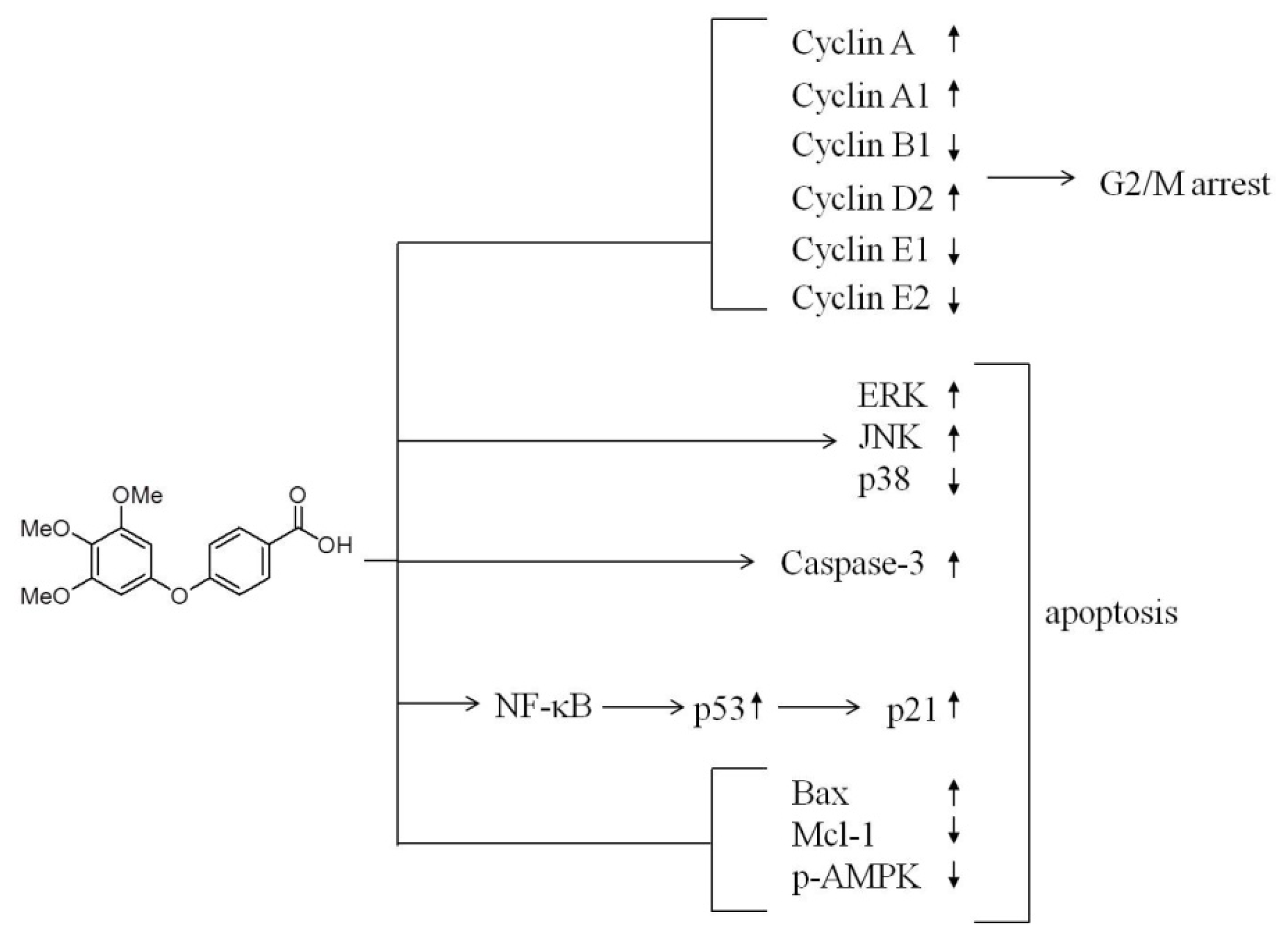
3. Discussion
4. Experimental Section
4.1. Cell Culture and Material
4.2. Cell Viability
4.3. Trypan Blue Exclusion Method
4.4. Colony Formation Assay
4.5. Morphological Observation of Nuclear Changes
4.6. Cell Cycle Analysis
4.7. Analysis of Caspase-3 Activity
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin 2011, 61, 69–90. [Google Scholar]
- Abdellatif, K.R.A.; Belal, A.; Omar, H.A. Design, synthesis and biological evaluation of novel triaryl (Z)-olefins as tamoxifen analogues. Bioorg. Med. Chem. Lett 2013, 23, 4960–4963. [Google Scholar]
- Dupont, W.D.; Page, D.L. Risk factors for breast cancer in women with proliferative breast disease. N. E. J. Med 1985, 312, 146–151. [Google Scholar]
- Peshkin, B.N.; Alabek, M.L.; Isaacs, C. BRCA1/2 mutations and triple negative breast cancers. Breast Dis 2010, 32, 25–33. [Google Scholar]
- Wu, A.H.; Pike, M.C.; Williams, L.D.; Spicer, D.; Tseng, C.C.; Churchwell, M.I.; Doerge, D.R. Tamoxifen, soy, and lifestyle factors in Asian American women with breast cancer. J. Clin. Oncol 2007, 25, 3024–3030. [Google Scholar]
- Newcomb, P.A.; Kampman, E.; Trentham-Dietz, A.; Egan, K.M.; Titus, L.J.; Baron, J.A.; Hampton, J.M.; Passarelli, M.N.; Willett, W.C. Alcohol consumption before and after breast cancer diagnosis: Associations with survival from breast cancer, cardiovascular disease, and other causes. J. Clin. Oncol 2013, 31, 1939–1946. [Google Scholar]
- Abdelgawad, M.A.; Belal, A.; Omar, H.A.; Hegazy, L.; Rateb, M.E. Synthesis, anti-breast cancer activity, and molecular modeling of some benzothiazole and benzoxazole derivatives. Arch. Pharm 2013, 346, 534–541. [Google Scholar]
- Weng, J.R.; Omar, H.A.; Kulp, S.K.; Chen, C.S. Pharmacological exploitation of indole-3-carbinol to develop potent antitumor agents. Mini Rev. Med. Chem 2010, 10, 398–404. [Google Scholar]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp. Cell Res 2000, 256, 58–66. [Google Scholar]
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430. [Google Scholar]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar]
- Degterev, A.; Boyce, M.; Yuan, J. A decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar]
- Takimoto, R.; El-Deiry, W.S. Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 2000, 19, 1735–1743. [Google Scholar]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar]
- Tokino, T.; Nakamura, Y. The role of p53-target genes in human cancer. Crit. Rev. Oncol. Hematol 2000, 33, 1–6. [Google Scholar]
- Cheng, W.L.; Lin, T.Y.; Tseng, Y.H.; Chu, F.H.; Chueh, P.J.; Kuo, Y.H.; Wang, S.Y. Inhibitory effect of human breast cancer cell proliferation via p21-mediated G1 cell cycle arrest by araliadiol isolated from Aralia cordata Thunb. Planta Med 2011, 77, 164–168. [Google Scholar]
- Yoshida, K.; Miki, Y. The cell death machinery governed by the p53 tumor suppressor in response to DNA damage. Cancer Sci 2010, 101, 831–835. [Google Scholar]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar]
- Shah, M.A.; Schwartz, G.K. Cyclin-dependent kinases as targets for cancer therapy. Cancer Chemother. Biol. Response Modif 2003, 21, 145–170. [Google Scholar]
- Gillett, C.; Fantl, V.; Smith, R.; Fisher, C.; Bartek, J.; Dickson, C.; Barnes, D.; Peters, G. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res 1994, 54, 1812–1817. [Google Scholar]
- Keyomarsi, K.; Conte, D., Jr.; Toyofuku, W.; Fox, M.P. Deregulation of cyclin E in breast cancer. Oncogene 1995, 11, 941–950. [Google Scholar]
- Shapiro, G.I.; Harper, J.W. Anticancer drug targets: Cell cycle and checkpoint control. J. Clin. Investig 1999, 104, 1645–1653. [Google Scholar]
- Gan, F.F.; Nagle, A.A.; Ang, X.; Ho, O.H.; Tan, S.H.; Yang, H.; Chui, W.K.; Chew, E.H. Shogaols at proapoptotic concentrations induce G(2)/M arrest and aberrant mitotic cell death associated with tubulin aggregation. Apoptosis 2011, 16, 856–867. [Google Scholar]
- Wadhwa, R.; Singh, R.; Gao, R.; Shah, N.; Widodo, N.; Nakamoto, T.; Ishida, Y.; Terao, K.; Kaul, S.C. Water extract of ashwagandha leaves has anticancer activity: Identification of an active component and its mechanism of action. PLoS One 2013, 8, e77189. [Google Scholar]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 2002, 12, 9–18. [Google Scholar]
- Furlong, E.E.; Rein, T.; Martin, F. YY1 and NF1 both activate the human p53 promoter by alternatively binding to a composite element, and YY1 and E1A cooperate to amplify p53 promoter activity. Mol. Cell. Biol 1996, 16, 5933–5945. [Google Scholar]
- Omar, H.A.; Sargeant, A.M.; Weng, J.R.; Wang, D.; Kulp, S.K.; Patel, T.; Chen, C.S. Targeting of the Akt-nuclear factor-κB signaling network by [1-(4-chloro-3-nitrobenzenesulfonyl)-1H-indol- 3-yl]-methanol (OSU-A9), a novel indole-3-carbinol derivative, in a mouse model of hepatocellular carcinoma. Mol. Pharmacol 2009, 76, 957–968. [Google Scholar]
- Rao, C.V.; Patlolla, J.M.; Qian, L.; Zhang, Y.; Brewer, M.; Mohammed, A.; Desai, D.; Amin, S.; Lightfoot, S.; Kopelovich, L. Chemopreventive effects of the p53-modulating agents CP-31398 and Prima-1 in tobacco carcinogen-induced lung tumorigenesis in A/J mice. Neoplasia 2013, 15, 1018–1027. [Google Scholar]
- Weng, J.R.; Tsai, C.H.; Omar, H.A.; Sargeant, A.M.; Wang, D.; Kulp, S.K.; Shapiro, C.L.; Chen, C.S. OSU-A9, a potent indole-3-carbinol derivative, suppresses breast tumor growth by targeting the Akt-NF-κB pathway and stress response signaling. Carcinogenesis 2009, 30, 1702–1709. [Google Scholar]
- Hung, J.H.; Teng, Y.N.; Wang, L.H.; Su, I.J.; Wang, C.C.; Huang, W.; Lee, K.H.; Lu, K.Y. Induction of Bcl-2 expression by hepatitis B virus pre-S2 mutant large surface protein resistance to 5-fluorouracil treatment in Huh-7 cells. PLoS One 2011, 6, e28977. [Google Scholar]
- Omar, H.A.; Arafa El, S.A.; Salama, S.A.; Arab, H.H.; Wu, C.H.; Weng, J.R. OSU-A9 inhibits angiogenesis in human umbilical vein endothelial cells via disrupting Akt-NF-κB and MAPK signaling pathways. Toxicol. Appl. Pharmacol 2013, 272, 616–624. [Google Scholar]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol 2013, 6, 41–47. [Google Scholar]
- Ryan, K.M.; Phillips, A.C.; Vousden, K.H. Regulation and function of the p53 tumor suppressor protein. Curr. Opin. Cell Biol 2001, 13, 332–337. [Google Scholar]
- Chandel, N.S.; vander Heiden, M.G.; Thompson, C.B.; Schumacker, P.T. Redox regulation of p53 during hypoxia. Oncogene 2000, 19, 3840–3848. [Google Scholar]
- Chang, B.D.; Xuan, Y.; Broude, E.V.; Zhu, H.; Schott, B.; Fang, J.; Roninson, I.B. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999, 18, 4808–4818. [Google Scholar]
- Lin, W.C.; Chuang, Y.C.; Chang, Y.S.; Lai, M.D.; Teng, Y.N.; Su, I.J.; Wang, C.C.; Lee, K.H.; Hung, J.H. Endoplasmic reticulum stress stimulates p53 expression through NF-κB activation. PLoS One 2012, 7, e39120. [Google Scholar]
- Wu, H.; Lozano, G. NF-kappa B activation of p53. A potential mechanism for suppressing cell growth in response to stress. J. Biol. Chem 1994, 269, 20067–20074. [Google Scholar]
- Ryan, K.M.; Ernst, M.K.; Rice, N.R.; Vousden, K.H. Role of NF-κB in p53-mediated programmed cell death. Nature 2000, 404, 892–897. [Google Scholar]
- Meisse, D.; van de Casteele, M.; Beauloye, C.; Hainault, I.; Kefas, B.A.; Rider, M.H.; Foufelle, F.; Hue, L. Sustained activation of AMP-activated protein kinase induces c-Jun N-terminal kinase activation and apoptosis in liver cells. FEBS Lett 2002, 526, 38–42. [Google Scholar]
- Kefas, B.A.; Cai, Y.; Ling, Z.; Heimberg, H.; Hue, L.; Pipeleers, D.; van de Casteele, M. AMP-activated protein kinase can induce apoptosis of insulin-producing MIN6 cells through stimulation of c-Jun-N-terminal kinase. J. Mol. Endocrinol 2003, 30, 151–161. [Google Scholar]
- Igata, M.; Motoshima, H.; Tsuruzoe, K.; Kojima, K.; Matsumura, T.; Kondo, T.; Taguchi, T.; Nakamaru, K.; Yano, M.; Kukidome, D.; et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circulation Res 2005, 97, 837–844. [Google Scholar]
- Hickson-Bick, D.L.; Buja, L.M.; McMillin, J.B. Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. J. Mol. Cell. Cardiol 2000, 32, 511–519. [Google Scholar]
- Weng, J.R.; Bai, L.Y.; Omar, H.A.; Sargeant, A.M.; Yeh, C.T.; Chen, Y.Y.; Tsai, M.H.; Chiu, C.F. A novel indole-3-carbinol derivative inhibits the growth of human oral squamous cell carcinoma in vitro. Oral Oncol. 2010, 46, 748–754. [Google Scholar]

© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, K.-H.; Ho, W.-Y.; Wu, S.-J.; Omar, H.A.; Huang, P.-J.; Wang, C.C.C.; Hung, J.-H. Modulation of Cyclins, p53 and Mitogen-Activated Protein Kinases Signaling in Breast Cancer Cell Lines by 4-(3,4,5-Trimethoxyphenoxy)benzoic Acid. Int. J. Mol. Sci. 2014, 15, 743-757. https://doi.org/10.3390/ijms15010743
Lee K-H, Ho W-Y, Wu S-J, Omar HA, Huang P-J, Wang CCC, Hung J-H. Modulation of Cyclins, p53 and Mitogen-Activated Protein Kinases Signaling in Breast Cancer Cell Lines by 4-(3,4,5-Trimethoxyphenoxy)benzoic Acid. International Journal of Molecular Sciences. 2014; 15(1):743-757. https://doi.org/10.3390/ijms15010743
Chicago/Turabian StyleLee, Kuan-Han, Wen-Yueh Ho, Shu-Jing Wu, Hany A. Omar, Po-Jui Huang, Clay C. C. Wang, and Jui-Hsiang Hung. 2014. "Modulation of Cyclins, p53 and Mitogen-Activated Protein Kinases Signaling in Breast Cancer Cell Lines by 4-(3,4,5-Trimethoxyphenoxy)benzoic Acid" International Journal of Molecular Sciences 15, no. 1: 743-757. https://doi.org/10.3390/ijms15010743