Hypoxic Conditioned Medium from Human Amniotic Fluid-Derived Mesenchymal Stem Cells Accelerates Skin Wound Healing through TGF-β/SMAD2 and PI3K/Akt Pathways


Abstract
:1. Introduction
2. Results
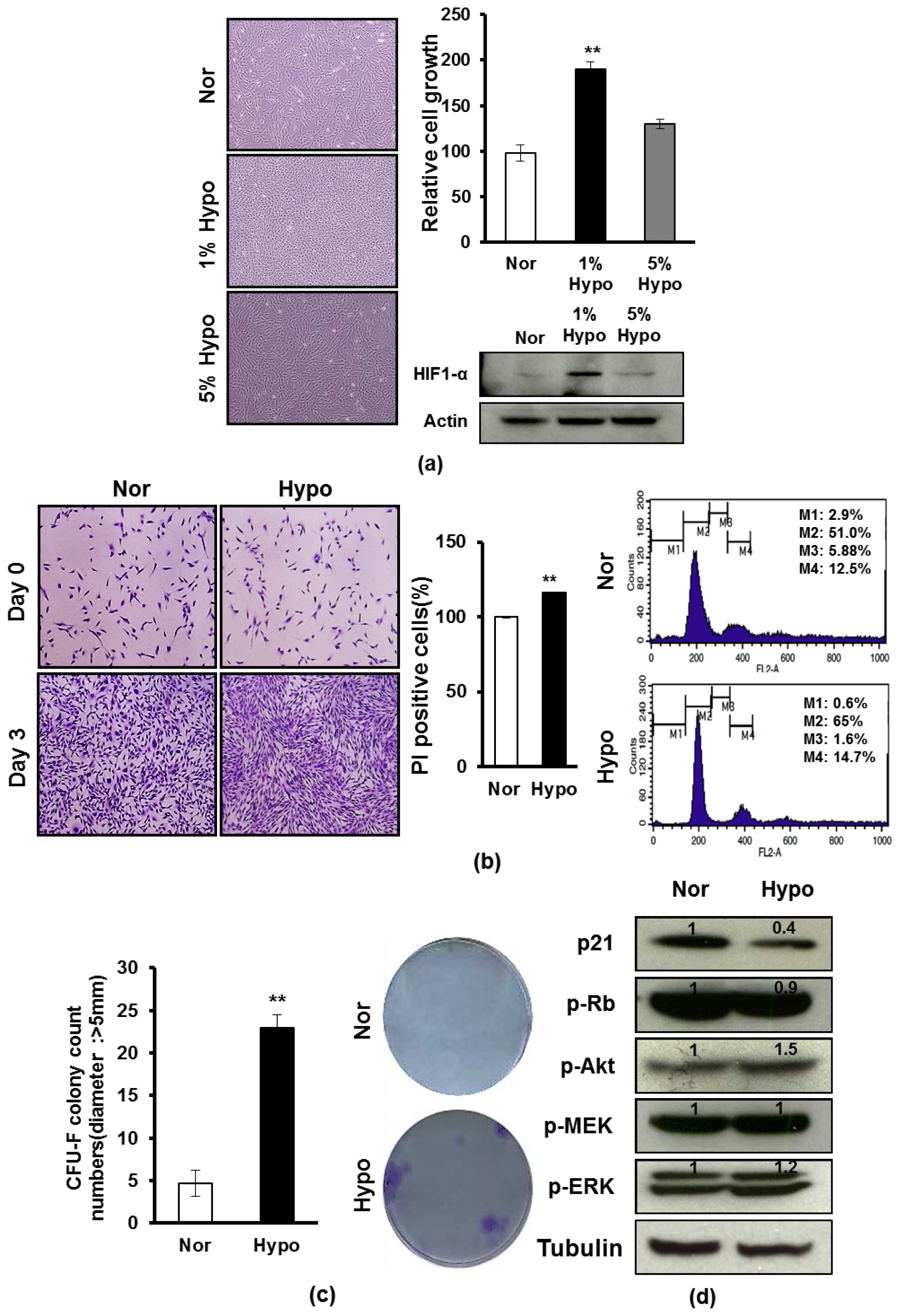
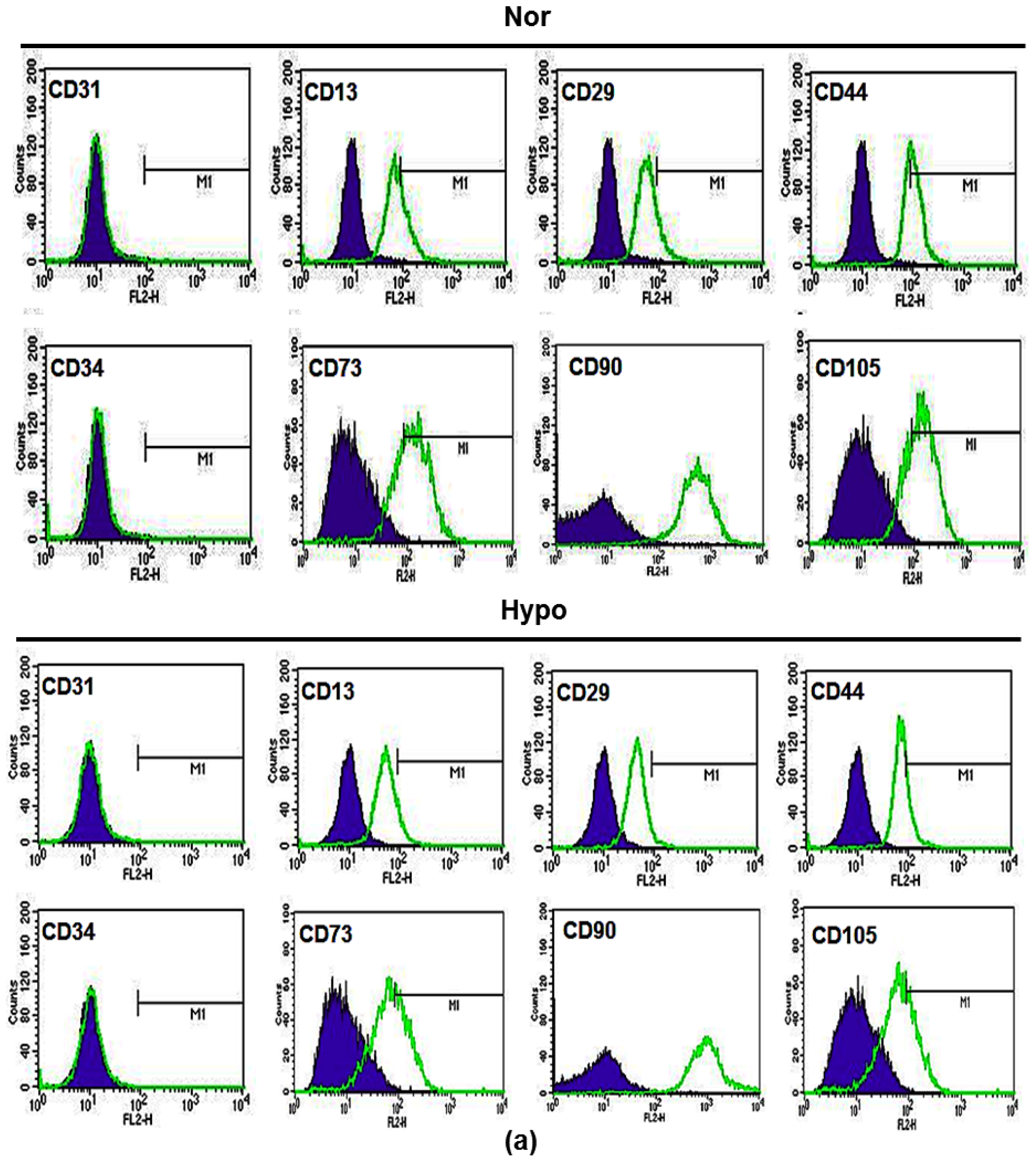
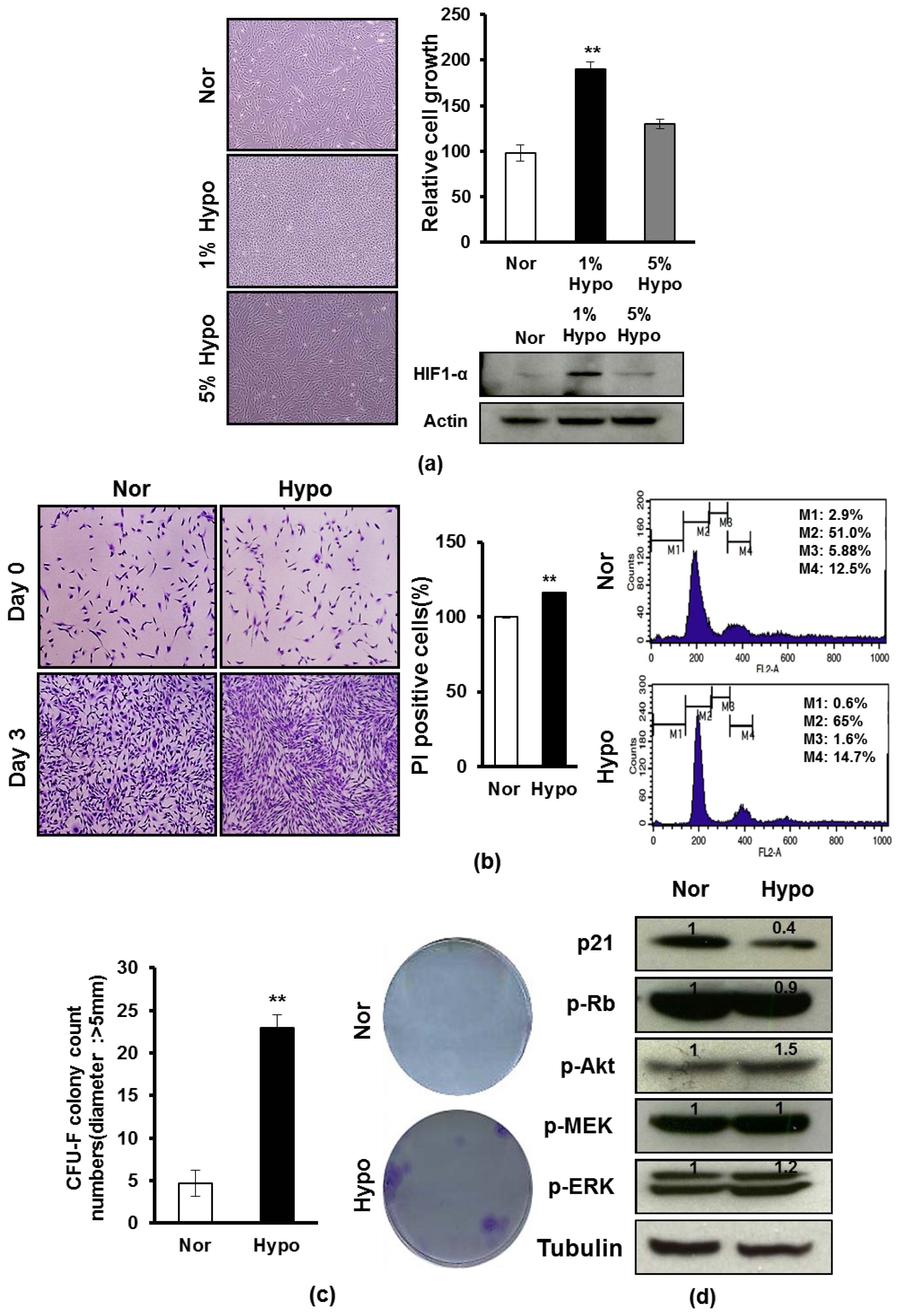
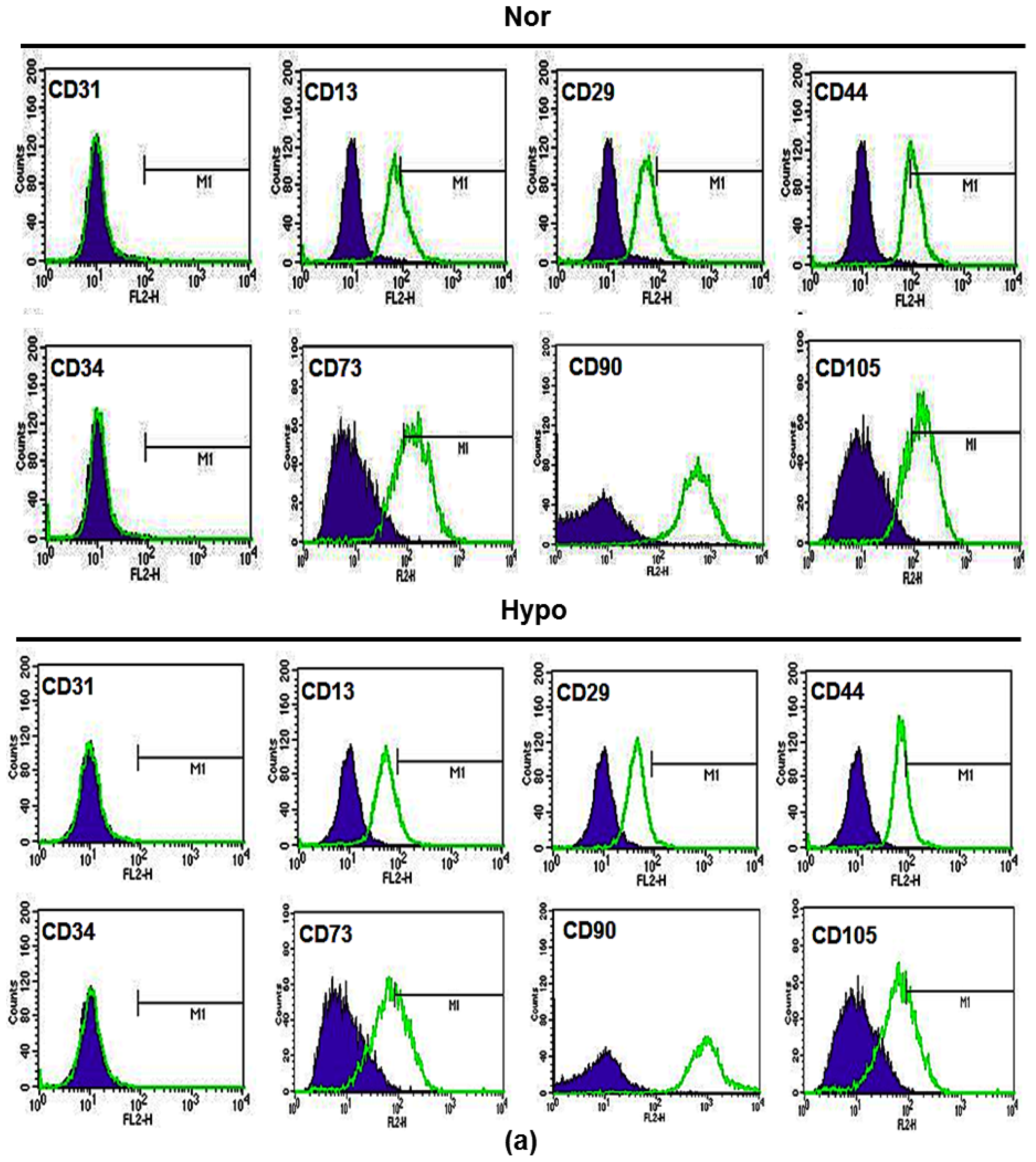
2.1. Hypoxia Promotes Proliferation and Survival of AF-MSCs
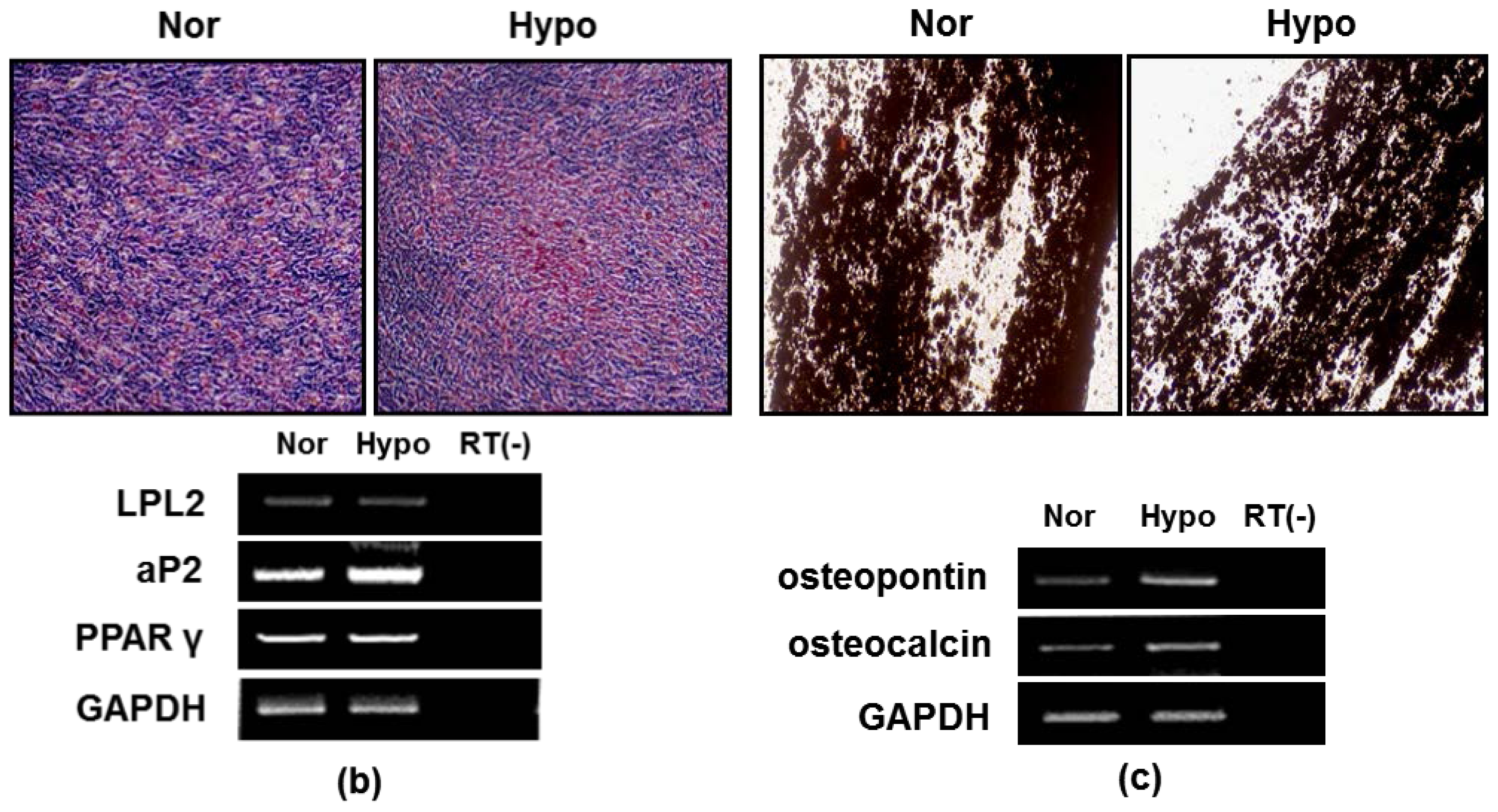
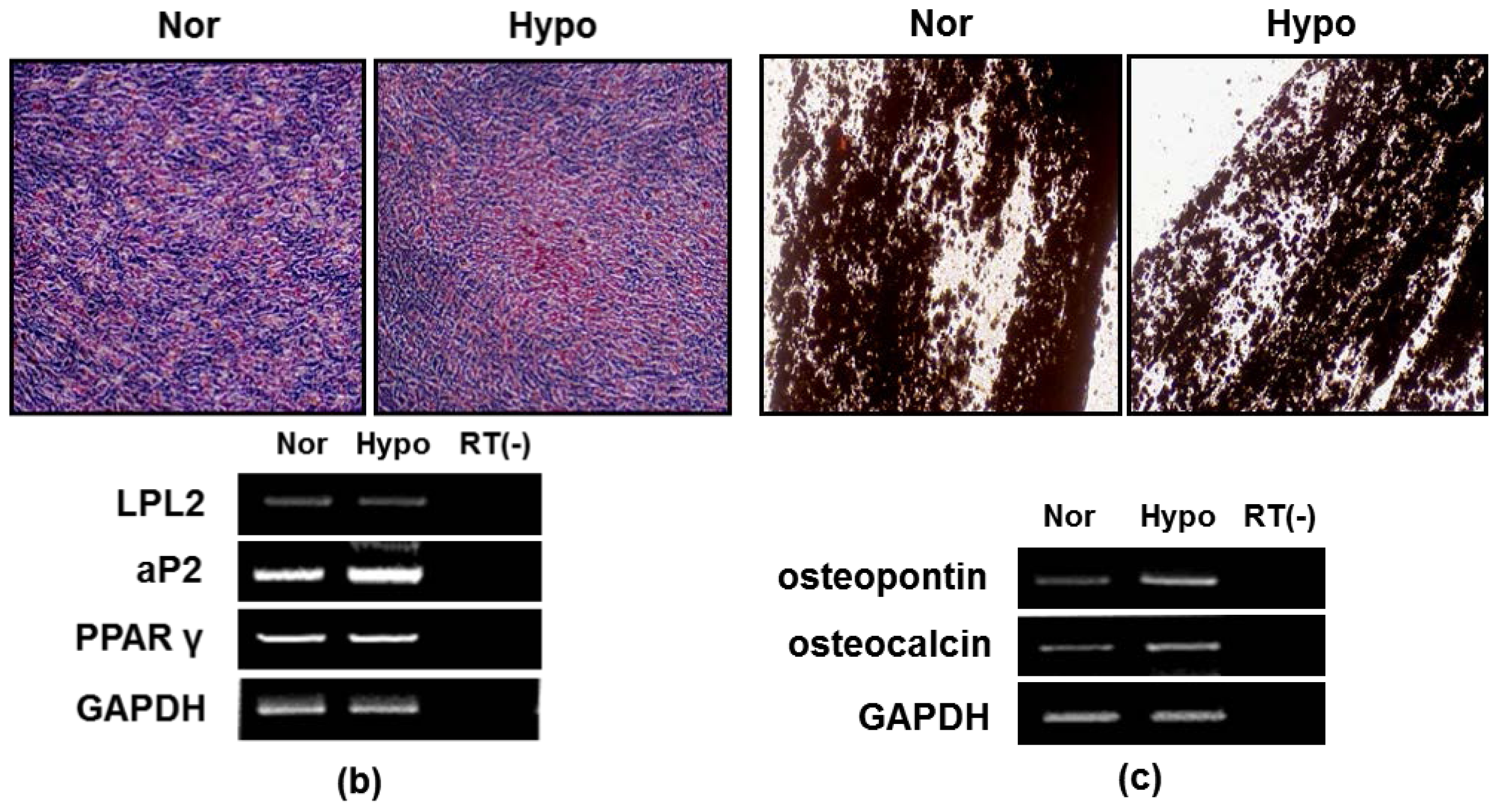
2.2. Hypoxia Maintenances Mesenchymal Differentiation Potentials
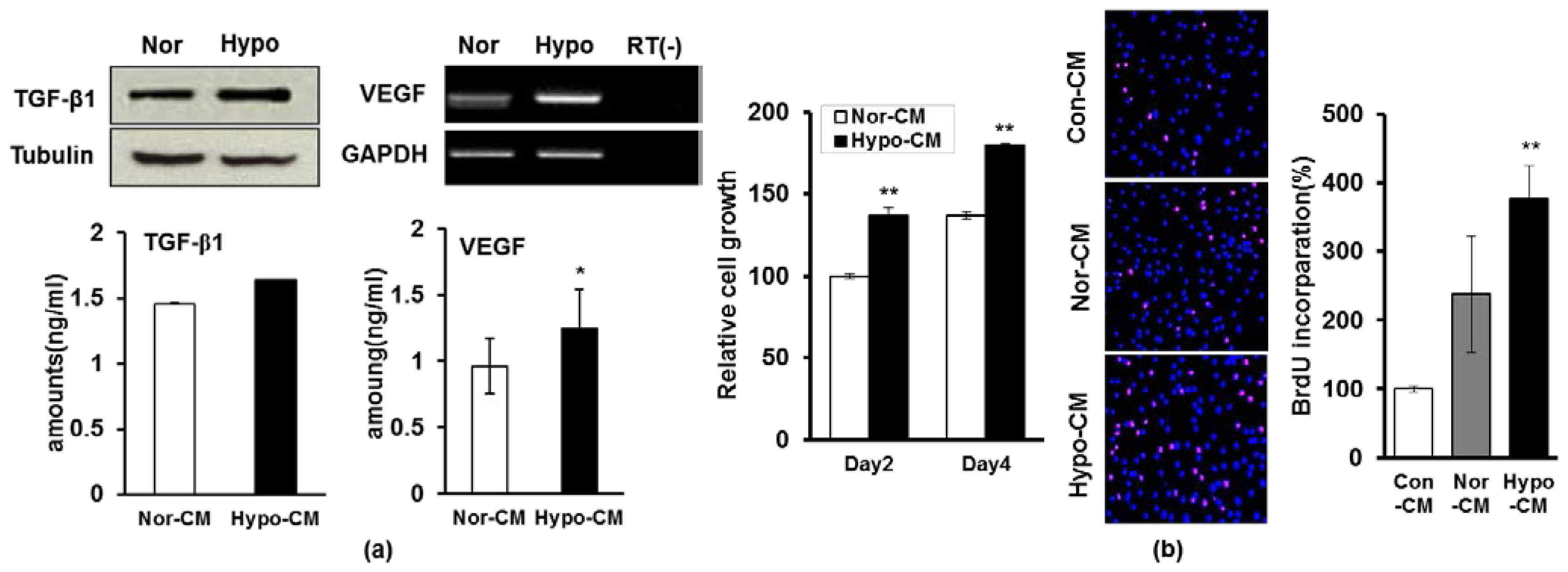
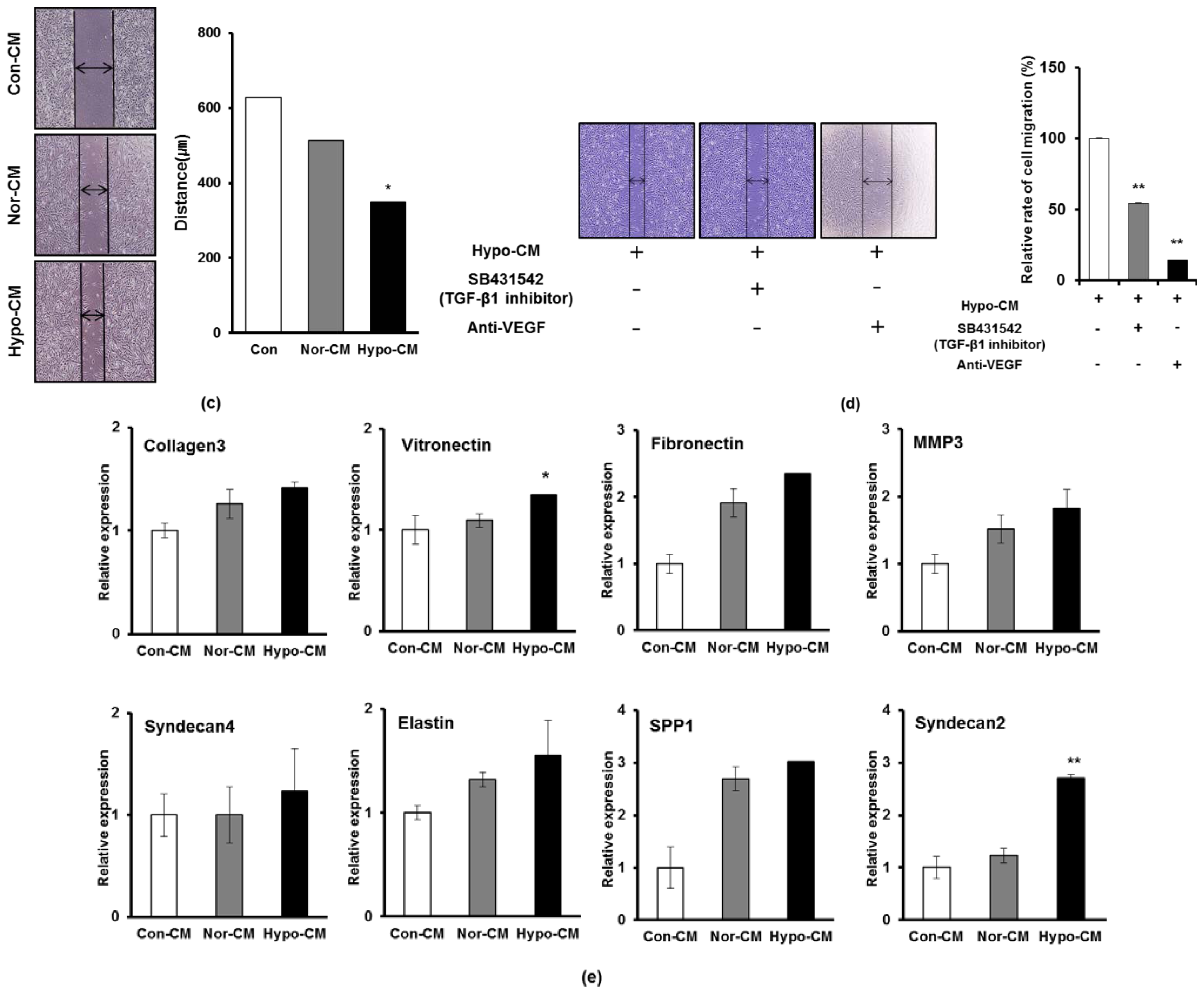
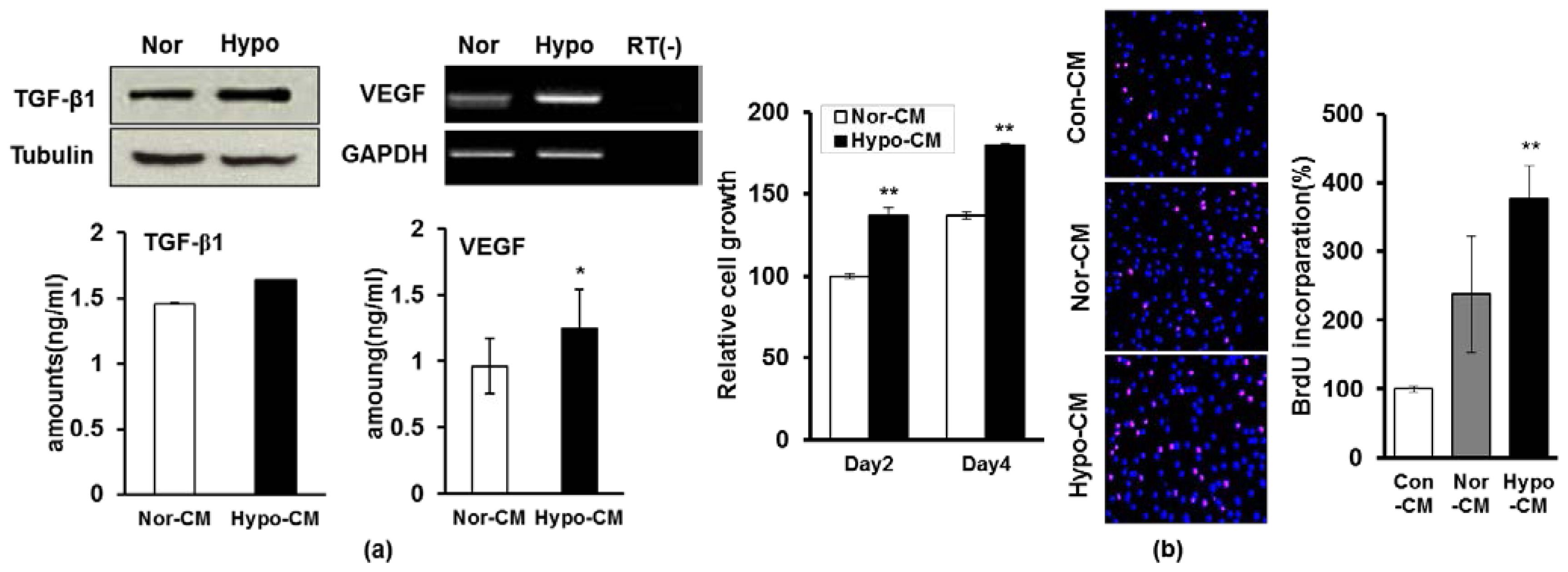
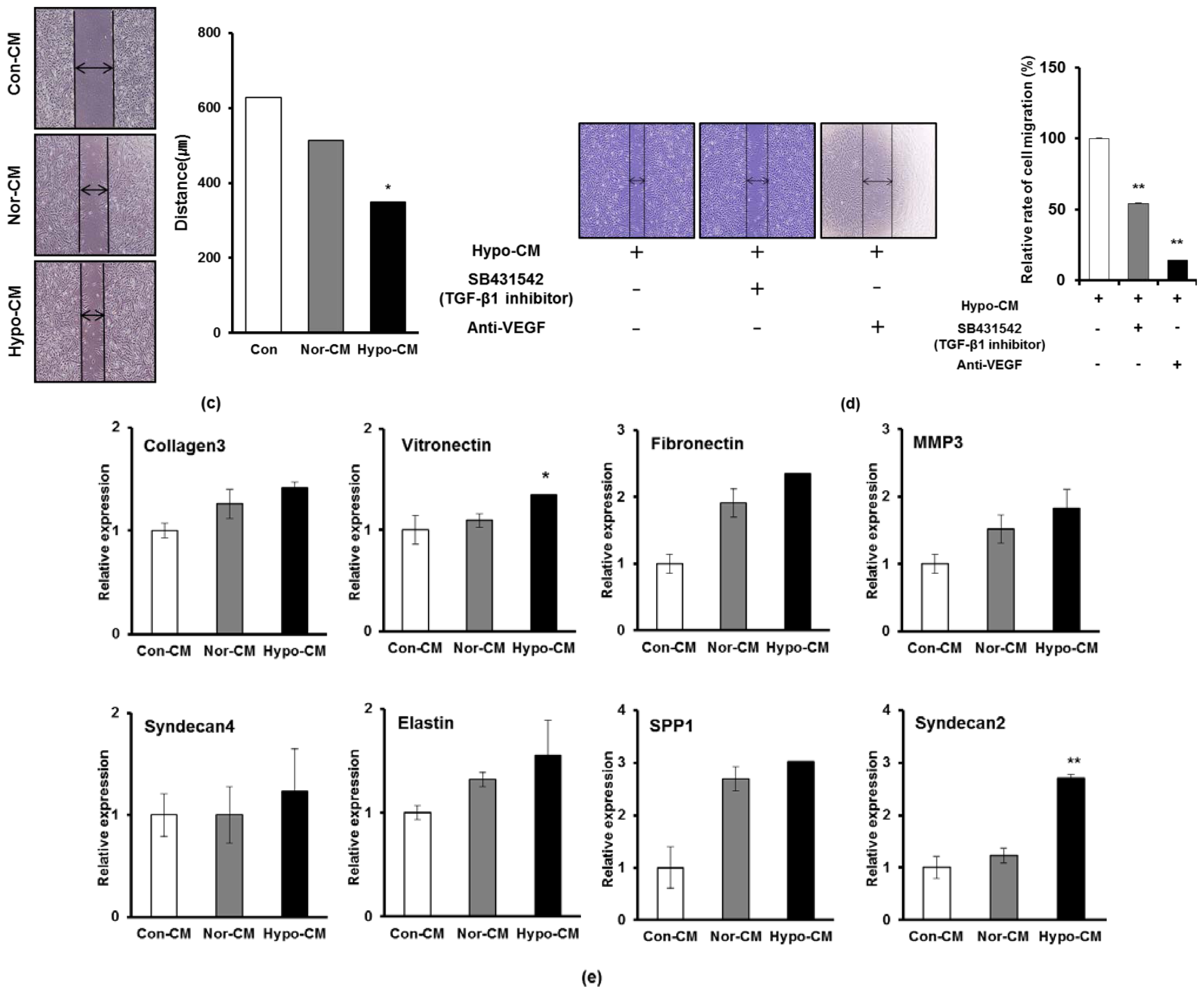
2.3. Hypoxia Facilitates the Secretion of Paracrine Factors of AF-MSCs and Hypoxic Conditioned Medium Accelerates the Proliferation and Migration of Human Dermal Fibroblasts
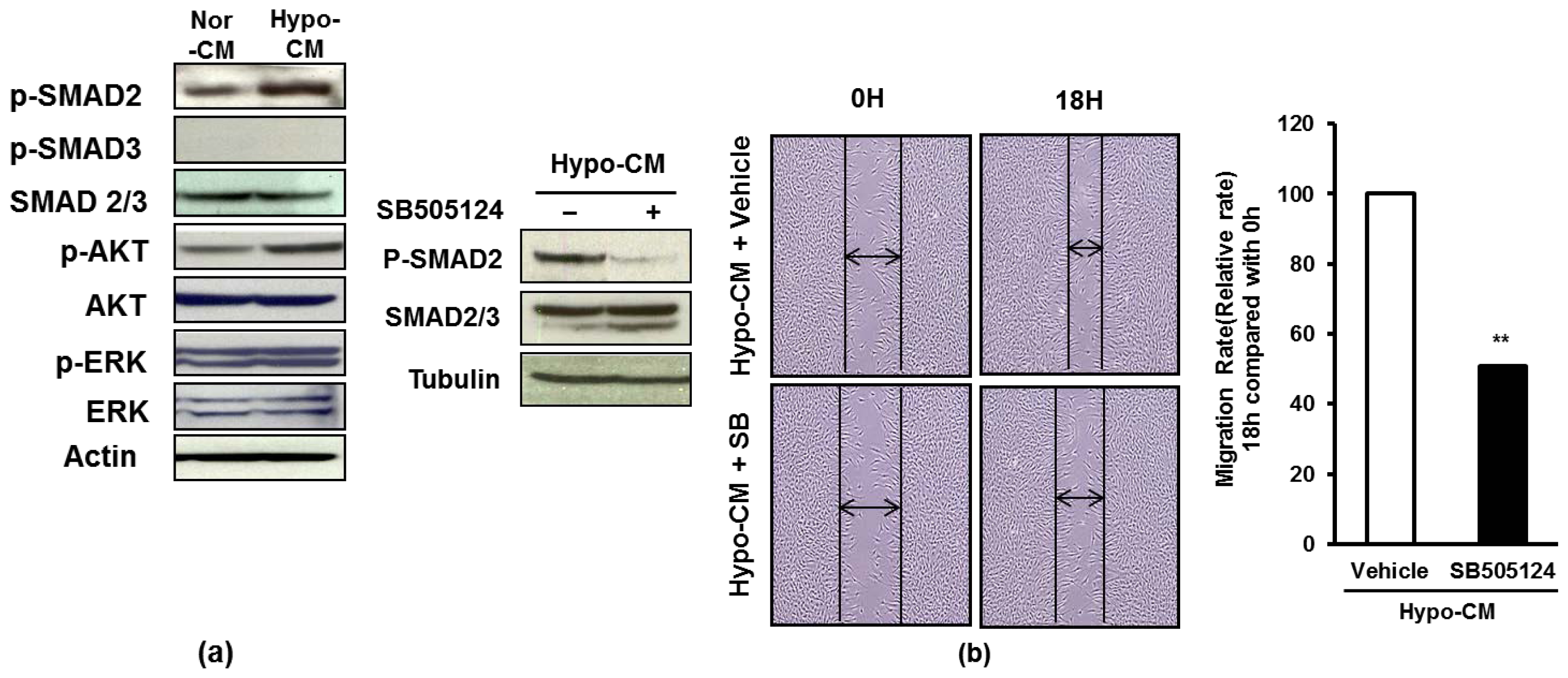
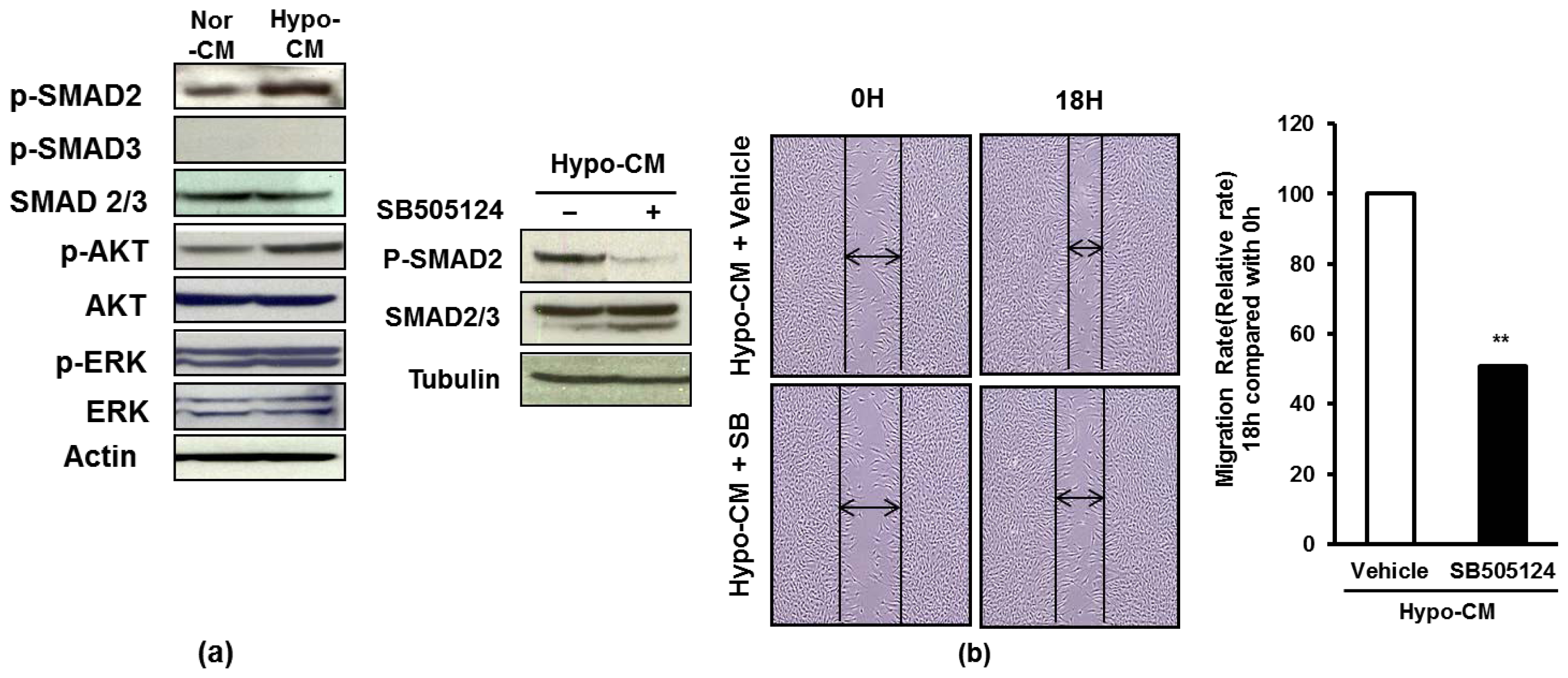
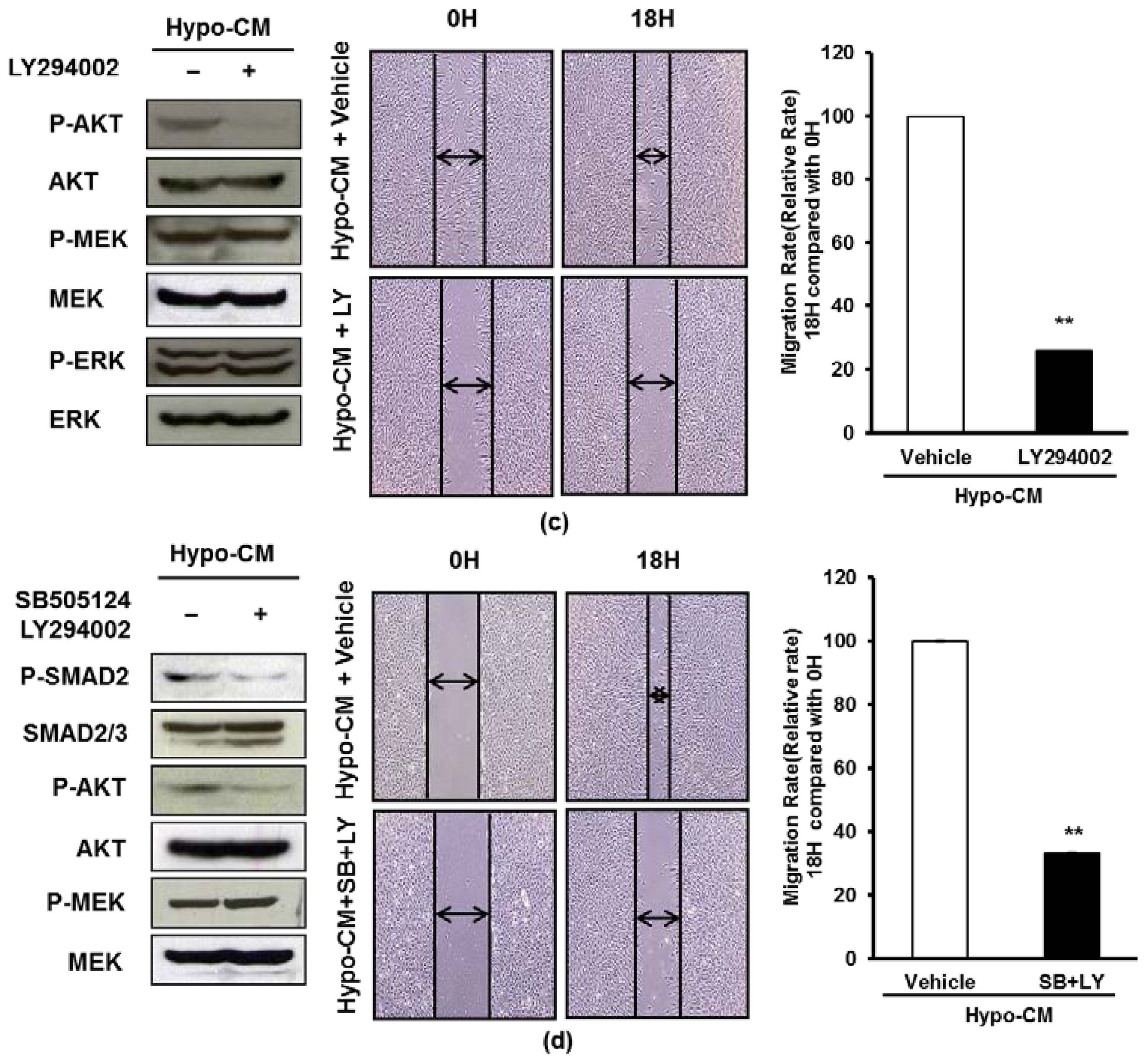
2.4. AF-MSCs Hypoxic Conditioned Medium Regulates TGF-β/SMAD2 and PI3K/AKT Pathway in Human Dermal Fibroblast
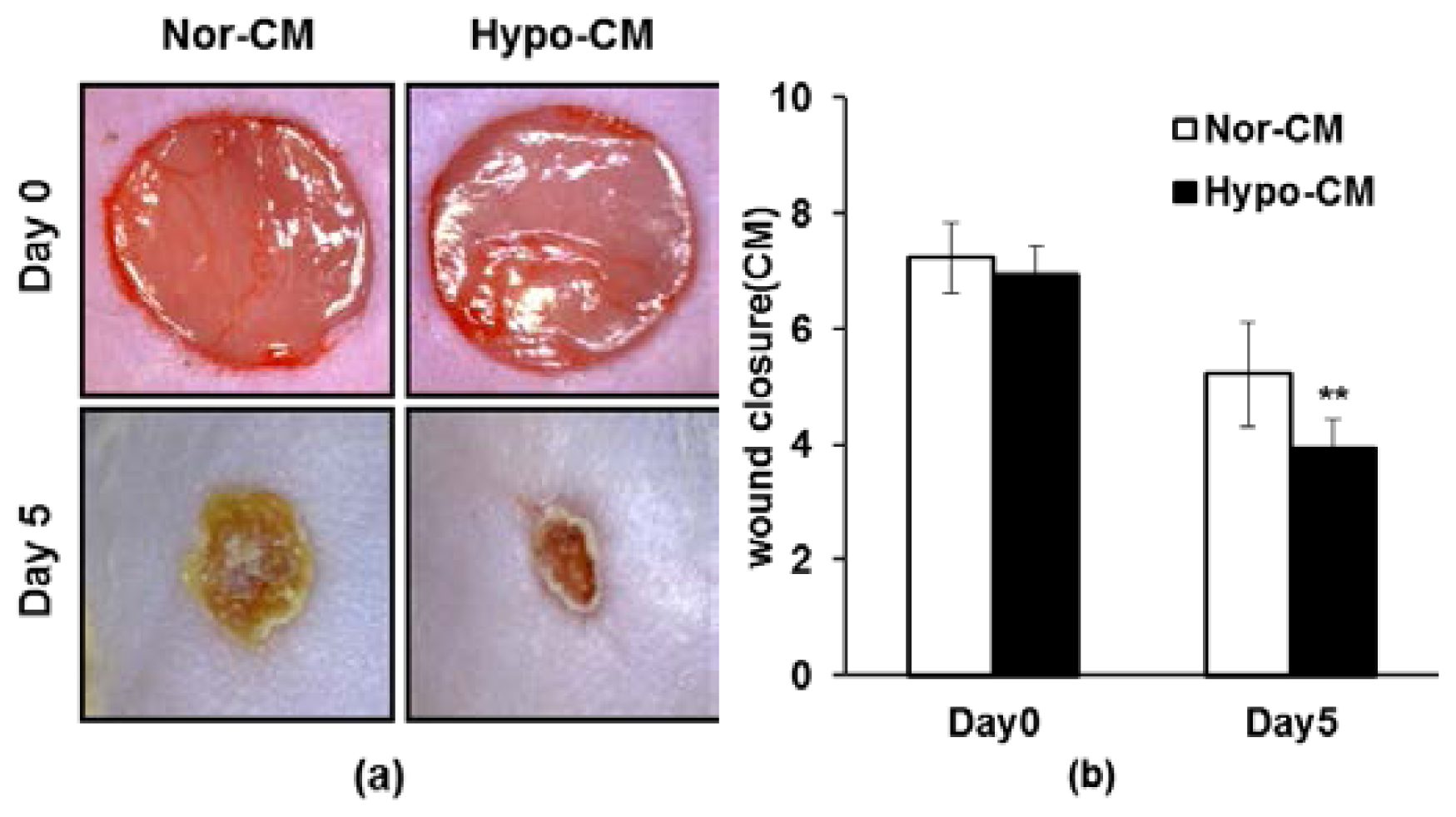
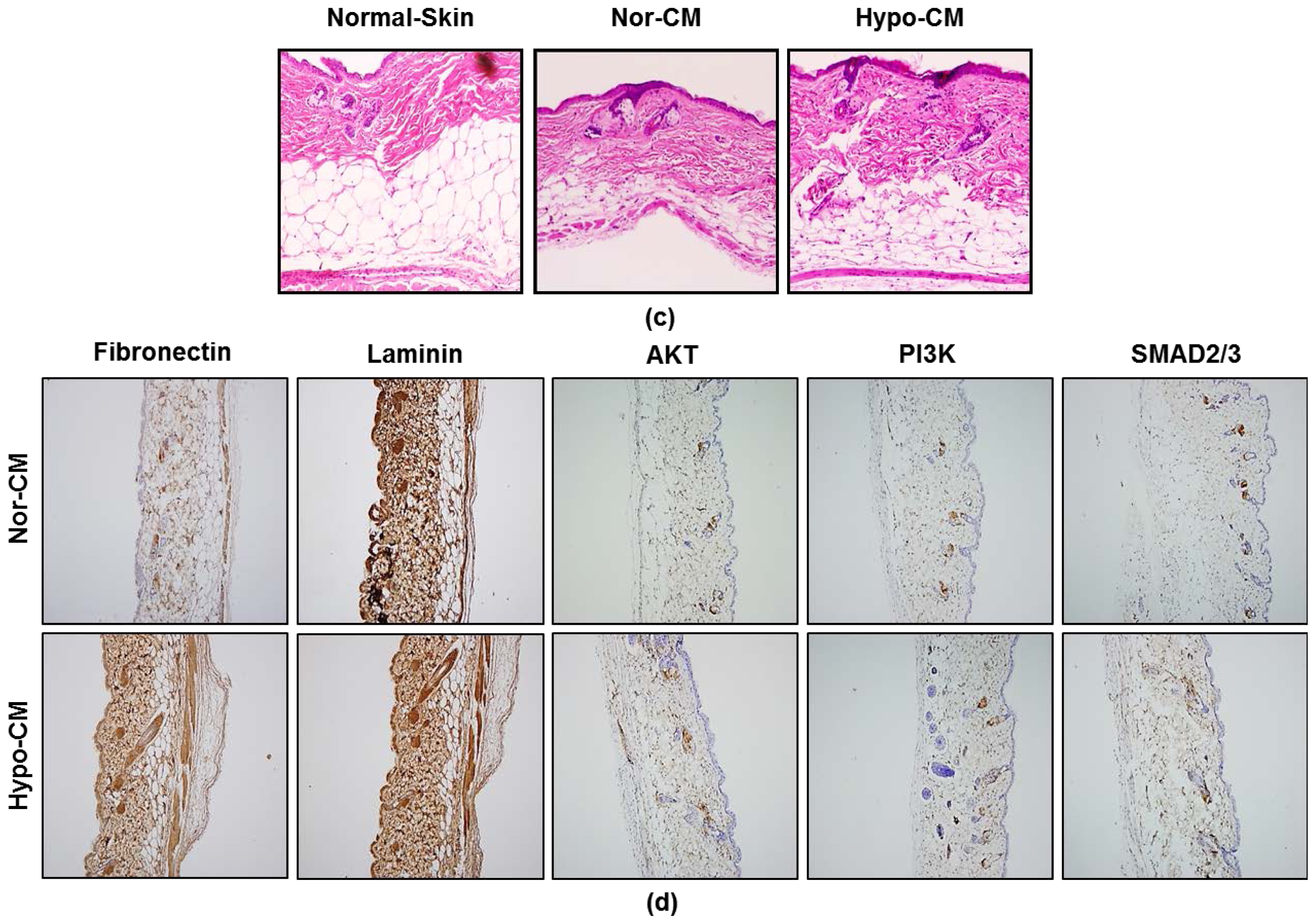
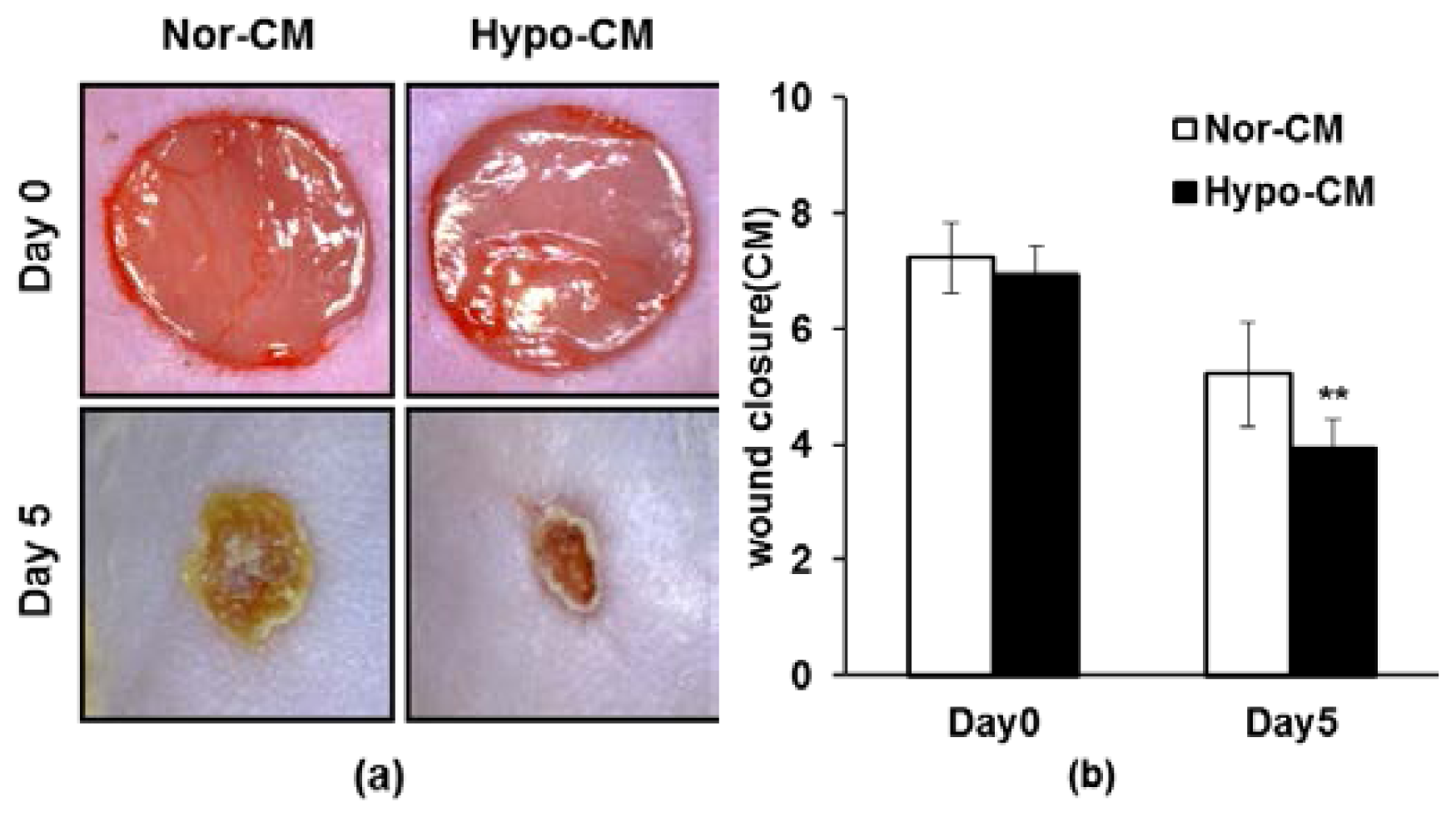
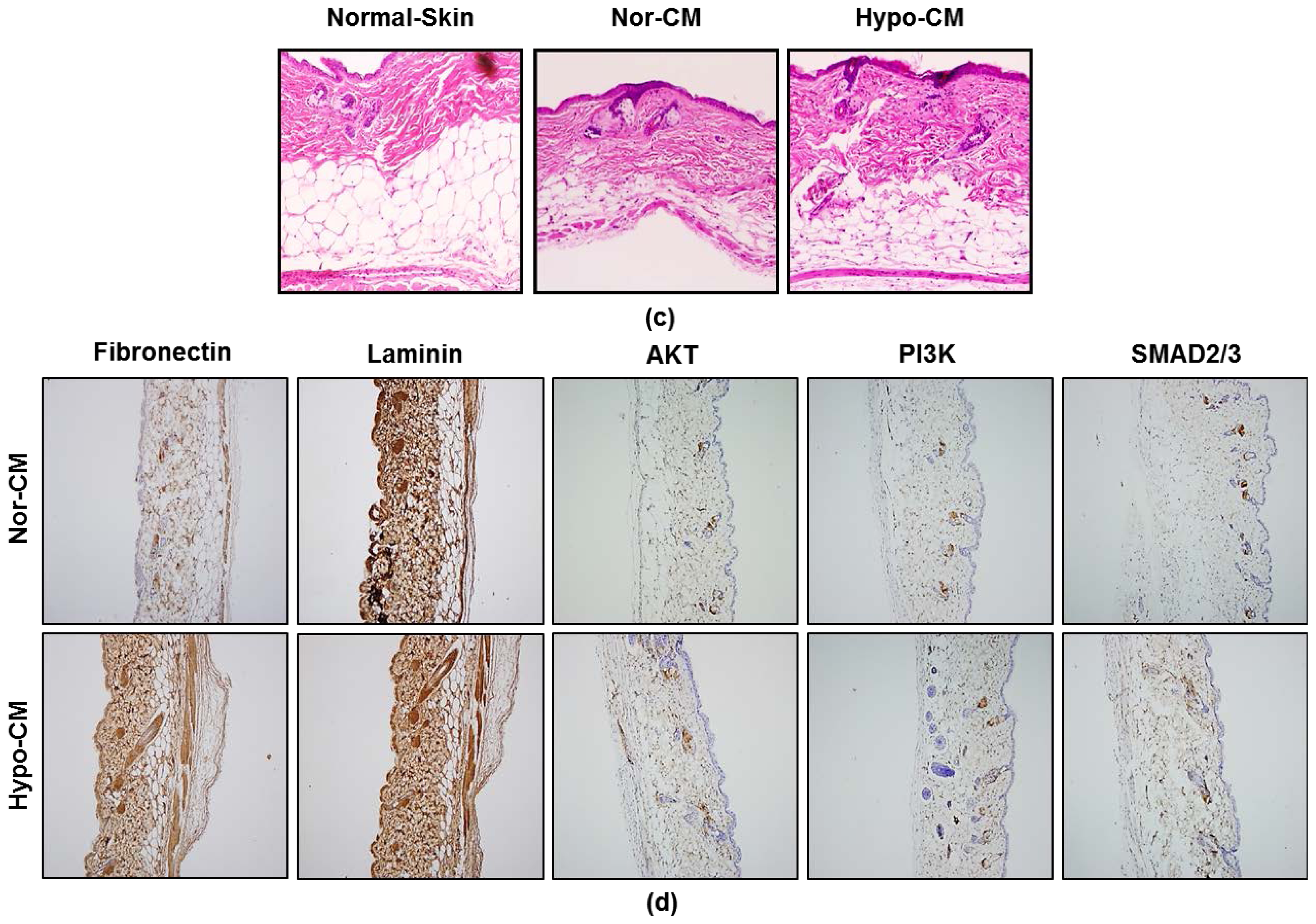
2.5. AF-MSCs Hypoxic Conditioned Medium Accelerates Wound Healing in Vivo
3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. Survival and Proliferation of AF-MSCs
4.3. Colony-Forming Unit Fibroblast (CFU-F) Assay
4.4. Preparation of Conditioned Media from Normoxia (AF-MSC-norCM) or Hypoxia-Treated AF-MSCs (AF-MSC-hypoCM)
4.5. Adipogenic and Osteogenic Differentiation
4.6. BrdU Assay
4.7. RT-PCR and Quantitative Real Time Polymerase Chain Reaction
4.8. Western Blot
4.9. ELISA
4.10. Immunofluorescence (IF)
4.11. In Vitro Wound Healing Assay
4.12. In Vivo Wound Healing Assay
4.13. Microarray Analysis
4.14. Data Acquisition and Analysis
4.15. Statistical Analysis
5. Conclusions
Supplementary Information
ijms-15-00605-s001.pdfAcknowledgments
Conflicts of Interest
References
- Martin, P. Wound healing-aiming for perfect skin regeneration. Science 1997, 276, 75–81. [Google Scholar]
- Falanga, V. Wound healing and its impairment in the diabetic foot. Lancet 2005, 366, 1736–1743. [Google Scholar]
- Singer, A.J.; Clark, R.A.F. Cutaneous wound healing. N. Engl. J. Med 1999, 341, 738–746. [Google Scholar]
- Pillouer-Prost, A.L. Fibroblasts: What’s new in cellular biology? J. Cosmet. Laser Ther 2003, 5, 232–238. [Google Scholar]
- Kim, W.-S.; Park, B.-S.; Sung, J.-H.; Yang, J.-M.; Park, S.-B.; Kwak, S.-J.; Park, J.-S. Wound healing effect of adipose-derived stem cells: A critical role of secretory factors on human dermal fibroblasts. J. Dermatol. Sci 2007, 48, 15–24. [Google Scholar]
- Tsai, M.S.; Lee, J.L.; Chang, Y.J.; Hwang, S.M. Isolation of human multipotent mesenchymal stem cells from second-trimester amniotic fluid using a novel two-stage culture protocol. Hum. Reprod 2004, 19, 1450–1456. [Google Scholar]
- Prusa, A.-R.; Hengstschlager, M. Amniotic fluid cells and human stem cell research—A new connection. Med. Sci. Monit 2002, 8, RA253–RA257. [Google Scholar]
- Tsai, M.-S.; Hwang, S.-M.; Tsai, Y.-L.; Cheng, F.-C.; Lee, J.-L.; Chang, Y.-J. Clonal amniotic fluid-derived stem cells express characteristics of both mesenchymal and neural stem cells. Biol. Reprod 2006, 74, 545–551. [Google Scholar]
- De Coppi, P.; Bartsch, G.; Siddiqui, M.M.; Xu, T.; Santos, C.C.; Perin, L.; Mostoslavsky, G.; Serre, A.C.; Snyder, E.Y.; Yoo, J.J. Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol 2007, 25, 100–106. [Google Scholar]
- Scherjon, S.A.; Kleijburg-van der Keur, C.; Noort, W.A.; Claas, F.H.; Willemze, R.; Fibbe, W.E.; Kanhai, H.H. Amniotic fluid as a novel source of mesenchymal stem cells for therapeutic transplantation. Blood 2003, 102, 1548–1549. [Google Scholar]
- Horwitz, E.M.; Gordon, P.L.; Koo, W.K.; Marx, J.C.; Neel, M.D.; McNall, R.Y.; Muul, L.; Hofmann, T. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proc. Natl. Acad. Sci. USA 2002, 99, 8932–8937. [Google Scholar]
- Bajada, S.; Harrison, P.; Ashton, B.; Cassar-Pullicino, V.; Ashammakhi, N.; Richardson, J. Successful treatment of refractory tibial nonunion using calcium sulphate and bone marrow stromal cell implantation. J. Bone Joint Surg. Br 2007, 89, 1382–1386. [Google Scholar]
- Chen, S.-L.; Fang, W.-W.; Ye, F.; Liu, Y.-H.; Qian, J.; Shan, S.-J.; Zhang, J.-J.; Chunhua, R.Z.; Liao, L.-M.; Lin, S. Effect on left ventricular function of intracoronary transplantation of autologous bone marrow mesenchymal stem cell in patients with acute myocardial infarction. Am. J. Cardiol 2004, 94, 92–95. [Google Scholar]
- Katritsis, D.G.; Sotiropoulou, P.A.; Karvouni, E.; Karabinos, I.; Korovesis, S.; Perez, S.A.; Voridis, E.M.; Papamichail, M. Transcoronary transplantation of autologous mesenchymal stem cells and endothelial progenitors into infarcted human myocardium. Catheter. Cardiovasc. Interv 2005, 65, 321–329. [Google Scholar]
- Chernykh, E.; Shevela, E.Y.; Leplina, O.Y.; Tikhonova, M.; Ostanin, A.; Kulagin, A.; Pronkina, N.; Muradov, Z.M.; Stupak, V.; Kozlov, V. Characteristics of bone marrow cells under conditions of impaired innervation in patients with spinal trauma. Bull. Exp. Biol. Med 2006, 141, 117–120. [Google Scholar]
- Fei, X.; Jiang, S.; Zhang, S.; Li, Y.; Ge, J.; He, B.; Goldstein, S.; Ruiz, G. Isolation, culture, and identification of amniotic fluid-derived mesenchymal stem cells. Cell Biochem. Biophys 2013, 67, 689–694. [Google Scholar]
- Chen, L.; Tredget, E.E.; Wu, P.Y.; Wu, Y. Paracrine factors of mesenchymal stem cells recruit macrophages and endothelial lineage cells and enhance wound healing. PLoS One 2008, 3, e1886. [Google Scholar]
- Walter, M.; Wright, K.T.; Fuller, H.; MacNeil, S.; Johnson, W.E.B. Mesenchymal stem cell-conditioned medium accelerates skin wound healing: An in vitro study of fibroblast and keratinocyte scratch assays. Exp. Cell Res 2010, 316, 1271–1281. [Google Scholar]
- Yoon, B.S.; Moon, J.-H.; Jun, E.K.; Kim, J.; Maeng, I.; Kim, J.S.; Lee, J.H.; Baik, C.S.; Kim, A.; Cho, K.S. Secretory profiles and wound healing effects of human amniotic fluid-derived mesenchymal stem cells. Stem Cells Dev 2009, 19, 887–902. [Google Scholar]
- Lee, E.Y.; Xia, Y.; Kim, W.S.; Kim, M.H.; Kim, T.H.; Kim, K.J.; Park, B.S.; Sung, J.H. Hypoxia-enhanced wound-healing function of adipose-derived stem cells: Increase in stem cell proliferation and up-regulation of VEGF and bFGF. Wound Repair Regen 2009, 17, 540–547. [Google Scholar]
- Packer, L.; Fuehr, K. Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature 1977, 267, 423–425. [Google Scholar]
- Lash, G.E.; Fitzpatrick, T.E.; Graham, C.H. Effect of hypoxia on cellular adhesion to vitronectin and fibronectin. Biochem. Biophys. Res. Commun 2001, 287, 622–629. [Google Scholar]
- Carmeliet, P.; Dor, Y.; Herbert, J.-M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar]
- Loike, J.; Cao, L.; Brett, J.; Ogawa, S.; Silverstein, S.; Stern, D. Hypoxia induces glucose transporter expression in endothelial cells. Am. J. Physiol. Cell Physiol 1992, 263, C326–C333. [Google Scholar]
- Horino, Y.; Takahashi, S.; Miura, T.; Takahashi, Y. Prolonged hypoxia accelerates the posttranscriptional process of collagen synthesis in cultured fibroblasts. Life Sci 2002, 71, 3031–3045. [Google Scholar]
- Minchenko, A.; Salceda, S.; Bauer, T.; Caro, J. Hypoxia regulatory elements of the human vascular endothelial growth factor gene. Cell. Mol. Biol. Res 1994, 40, 35–39. [Google Scholar]
- Lennon, D.P.; Edmison, J.M.; Caplan, A.I. Cultivation of rat marrow-derived mesenchymal stem cells in reduced oxygen tension: Effects on in vitro and in vivo osteochondrogenesis. J. Cell. Physiol 2001, 187, 345–355. [Google Scholar]
- Crisostomo, P.R.; Wang, Y.; Markel, T.A.; Wang, M.; Lahm, T.; Meldrum, D.R. Human mesenchymal stem cells stimulated by TNF-α, LPS, or hypoxia produce growth factors by an NFκB- but not JNK-dependent mechanism. Am. J. Physiol. Cell Physiol 2008, 294, C675–C682. [Google Scholar]
- Moon, J.-H.; Kwak, S.S.; Park, G.; Jung, H.-Y.; Yoon, B.S.; Park, J.; Ryu, K.S.; Choi, S.-C.; Maeng, I.; Kim, B. Isolation and characterization of multipotent human keloid-derived mesenchymal-like stem cells. Stem Cells Dev 2008, 17, 713–724. [Google Scholar]
- Colter, D.C.; Sekiya, I.; Prockop, D.J. Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc. Natl. Acad. Sci. USA 2001, 98, 7841–7845. [Google Scholar]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar]
- Young, H.E.; Steele, T.A.; Bray, R.A.; Hudson, J.; Floyd, J.A.; Hawkins, K.; Thomas, K.; Austin, T.; Edwards, C.; Cuzzourt, J. Human reserve pluripotent mesenchymal stem cells are present in the connective tissues of skeletal muscle and dermis derived from fetal, adult, and geriatric donors. Anat. Rec 2001, 264, 51–62. [Google Scholar]
- Kern, S.; Eichler, H.; Stoeve, J.; Klüter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar]
- Park, G.; Yoon, B.S.; Moon, J.-H.; Kim, B.; Jun, E.K.; Oh, S.; Kim, H.; Song, H.J.; Noh, J.Y.; Oh, C. Green tea polyphenol epigallocatechin-3-gallate suppresses collagen production and proliferation in keloid fibroblasts via inhibition of the STAT3-signaling pathway. J. Investig. Dermatol 2008, 128, 2429–2441. [Google Scholar]
- Rosch, R.; Junge, K.; Binnebösel, M.; Mirgartz, N.; Klinge, U.; Schumpelick, V. Improved abdominal wall wound healing by helium pneumoperitoneum. Surg. Endosc 2006, 20, 1892–1896. [Google Scholar]
- Jung, S.-A.; Lee, H.K.; Yoon, J.S.; Kim, S.-J.; Kim, C.Y.; Song, H.; Hwang, K.-C.; Lee, J.B.; Lee, J.H. Upregulation of TGF-β-induced tissue transglutaminase expression by PI3K-Akt pathway activation in human subconjunctival fibroblasts. Investig. Ophthalmol. Vis. Sci 2007, 48, 1952–1958. [Google Scholar]
- Galiano, R.D.; Michaels, V.; Dobryansky, M.; Levine, J.P.; Gurtner, G.C. Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen 2004, 12, 485–492. [Google Scholar]
- Yang, J.D.; Choi, D.S.; Cho, Y.K.; Kim, T.K.; Lee, J.W.; Choi, K.Y.; Chung, H.Y.; Cho, B.C.; Byun, J.S. Effect of amniotic fluid stem cells and amniotic fluid cells on the wound healing process in a white rat model. Arch. Plast. Surg 2013, 40, 496–504. [Google Scholar]
- Lord-Dufour, S.; Copland, I.B.; Levros, L.C.; Post, M.; Das, A.; Khosla, C.; Galipeau, J.; Rassart, E.; Annabi, B. Evidence for transcriptional regulation of the glucose-6-phosphate transporter by HIF-1α: Targeting G6PT with mumbaistatin analogs in hypoxic mesenchymal stromal cells. Stem Cells 2009, 27, 489–497. [Google Scholar]
- Tsai, C.-C.; Chen, Y.-J.; Yew, T.-L.; Chen, L.-L.; Wang, J.-Y.; Chiu, C.-H.; Hung, S.-C. Hypoxia inhibits senescence and maintains mesenchymal stem cell properties through down-regulation of E2A-p21 by HIF-TWIST. Blood 2011, 117, 459–469. [Google Scholar]
- D’Ippolito, G.; Diabira, S.; Howard, G.A.; Roos, B.A.; Schiller, P.C. Low oxygen tension inhibits osteogenic differentiation and enhances stemness of human MIAMI cells. Bone 2006, 39, 513–522. [Google Scholar]
- Rosova, I.; Dao, M.; Capoccia, B.; Link, D.; Nolta, J.A. Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells 2008, 26, 2173–2182. [Google Scholar]
- Kwon, D.S.; Kwon, C.H.; Kim, J.H.; Woo, J.S.; Jung, J.S.; Kim, Y.K. Signal transduction of MEK/ERK and PI3K/Akt activation by hypoxia/reoxygenation in renal epithelial cells. Eur. J. Cell Biol 2006, 85, 1189–1199. [Google Scholar]
- Liu, H.; Xue, W.; Ge, G.; Luo, X.; Li, Y.; Xiang, H.; Ding, X.; Tian, P.; Tian, X. Hypoxic preconditioning advances CXCR4 and CXCR7 expression by activating HIF-1α in MSCs. Biochem. Biophys. Res. Commun 2010, 401, 509–515. [Google Scholar]
- Chen, J.; Crawford, R.; Chen, C.; Xiao, Y. The key regulatory roles of the PI3K/Akt signalling pathway in the functionalities of mesenchymal stem cells and applications in tissue regeneration. Tissue Eng 2013, 19, 516–528. [Google Scholar]
- Kinnaird, T.; Stabile, E.; Burnett, M.; Lee, C.; Barr, S.; Fuchs, S.; Epstein, S. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ. Res 2004, 94, 678–685. [Google Scholar]
- Lu, Y.; Azad, N.; Wang, L.; Iyer, A.K.; Castranova, V.; Jiang, B.-H.; Rojanasakul, Y. Phosphatidylinositol-3-kinase/Akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am. J. Respir. Cell Mol. Biol 2010, 42, 432–441. [Google Scholar]
- Meckmongkol, T.T.; Harmon, R.; McKeown-Longo, P.; van de Water, L. The fibronectin synergy site modulates TGF-β-dependent fibroblast contraction. Biochem. Biophys. Res. Commun 2007, 360, 709–714. [Google Scholar]
- Hamada, K.; Sasaki, T.; Koni, P.A.; Natsui, M.; Kishimoto, H.; Sasaki, J.; Yajima, N.; Horie, Y.; Hasegawa, G.; Naito, M. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev 2005, 19, 2054–2065. [Google Scholar]
- Shih, N.R.; Jo, O.D.; Yanagawa, N. Effects of PHEX antisense in human osteoblast cells. J. Am. Soc. Nephrol 2002, 13, 394–399. [Google Scholar]
- Furstoss, O.; Dorey, K.; Simon, V.; Barilà, D.; Superti-Furga, G.; Roche, S. c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis. EMBO J 2002, 21, 514–524. [Google Scholar]
- Mairet-Coello, G.; Tury, A.; DiCicco-Bloom, E. Insulin-like growth factor-1 promotes G1/S cell cycle progression through bidirectional regulation of cyclins and cyclin-dependent kinase inhibitors via the phosphatidylinositol 3-kinase/Akt pathway in developing rat cerebral cortex. J. Neurosci 2009, 29, 775–788. [Google Scholar]
- Kho, Y.; Kim, S.; Yoon, B.S.; Moon, J.-H.; Kim, B.; Kwak, S.; Woo, J.; Oh, S.; Hong, K.; Kim, S. Induction of serum amyloid A genes is associated with growth and apoptosis of HC11 mammary epithelial cells. Biosci. Biotechnol. Biochem 2008, 72, 70–81. [Google Scholar]
- Miller, G.E.; Chen, E. Life stress and diminished expression of genes encoding glucocorticoid receptor and beta2-adrenergic receptor in children with asthma. Proc. Natl. Acad. Sci. USA 2006, 103, 5496–5501. [Google Scholar]
- Neuhoff, S.; Moers, J.; Rieks, M.; Grunwald, T.; Jensen, A.; Dermietzel, R.; Meier, C. Proliferation, differentiation, and cytokine secretion of human umbilical cord blood-derived mononuclear cells in vitro. Exp. Hematol. 2007, 35, 1119–1131. [Google Scholar]
- Yoon, B.S.; Yoo, S.J.; Lee, J.E.; You, S.; Lee, H.T.; Yoon, H.S. Enhanced differentiation of human embryonic stem cells into cardiomyocytes by combining hanging drop culture and 5-azacytidine treatment. Differentiation 2006, 74, 149–159. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological process | Gene Symbol |
|---|---|
| Glycolysis | PFKP, ALDOC, ENO2, TPI1P2, PGM1 |
| Response to hypoxia | VLDLR, PLOD1, TFRC, PLOD2, ALDOC, BNIP3, EGLN1 |
| Regulation of cellular component movement | MAP2KQ1, HMOX1, INSR |
| Response to nutrient levels | VLDLR, AQP3, LIPG, HMOX1, INSR, SUOX, STC2 |
| Regulation of cell migration | INSR, MAP2K1, HMOX1 |
| Transcription regulator activity | SAP30, RFX2, BTG1, ID3, NKIL3, SCAI, KLF7 |
| Cytokines | VEGFR, FLT1, IL4, IL6, IL15, IL17A, IL32, IL33, IL2RG, IL3RA, FGF20, FGFR3, FGFR2, FGF1 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jun, E.K.; Zhang, Q.; Yoon, B.S.; Moon, J.-H.; Lee, G.; Park, G.; Kang, P.J.; Lee, J.H.; Kim, A.; You, S. Hypoxic Conditioned Medium from Human Amniotic Fluid-Derived Mesenchymal Stem Cells Accelerates Skin Wound Healing through TGF-β/SMAD2 and PI3K/Akt Pathways. Int. J. Mol. Sci. 2014, 15, 605-628. https://doi.org/10.3390/ijms15010605
Jun EK, Zhang Q, Yoon BS, Moon J-H, Lee G, Park G, Kang PJ, Lee JH, Kim A, You S. Hypoxic Conditioned Medium from Human Amniotic Fluid-Derived Mesenchymal Stem Cells Accelerates Skin Wound Healing through TGF-β/SMAD2 and PI3K/Akt Pathways. International Journal of Molecular Sciences. 2014; 15(1):605-628. https://doi.org/10.3390/ijms15010605
Chicago/Turabian StyleJun, Eun Kyoung, Qiankun Zhang, Byung Sun Yoon, Jai-Hee Moon, Gilju Lee, Gyuman Park, Phil Jun Kang, Jung Han Lee, Areee Kim, and Seungkwon You. 2014. "Hypoxic Conditioned Medium from Human Amniotic Fluid-Derived Mesenchymal Stem Cells Accelerates Skin Wound Healing through TGF-β/SMAD2 and PI3K/Akt Pathways" International Journal of Molecular Sciences 15, no. 1: 605-628. https://doi.org/10.3390/ijms15010605
APA StyleJun, E. K., Zhang, Q., Yoon, B. S., Moon, J.-H., Lee, G., Park, G., Kang, P. J., Lee, J. H., Kim, A., & You, S. (2014). Hypoxic Conditioned Medium from Human Amniotic Fluid-Derived Mesenchymal Stem Cells Accelerates Skin Wound Healing through TGF-β/SMAD2 and PI3K/Akt Pathways. International Journal of Molecular Sciences, 15(1), 605-628. https://doi.org/10.3390/ijms15010605