Molecular Dynamics Simulation of the Crystallizable Fragment of IgG1—Insights for the Design of Fcabs


Abstract
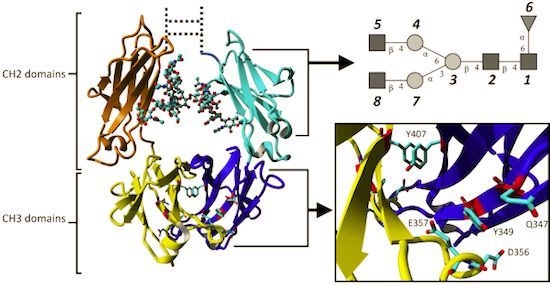
:
1. Introduction
2. Results and Discussion
2.1. Results
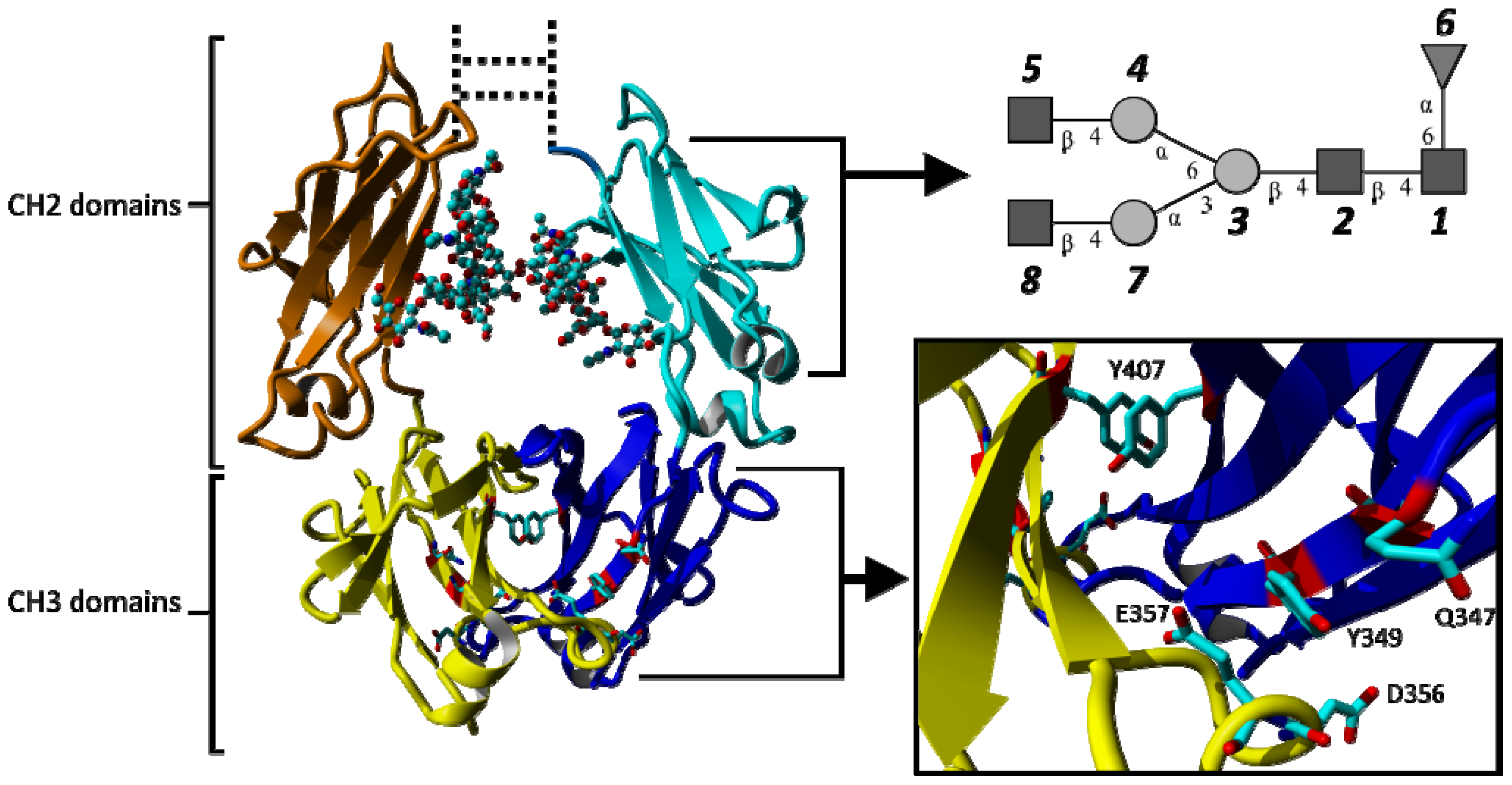
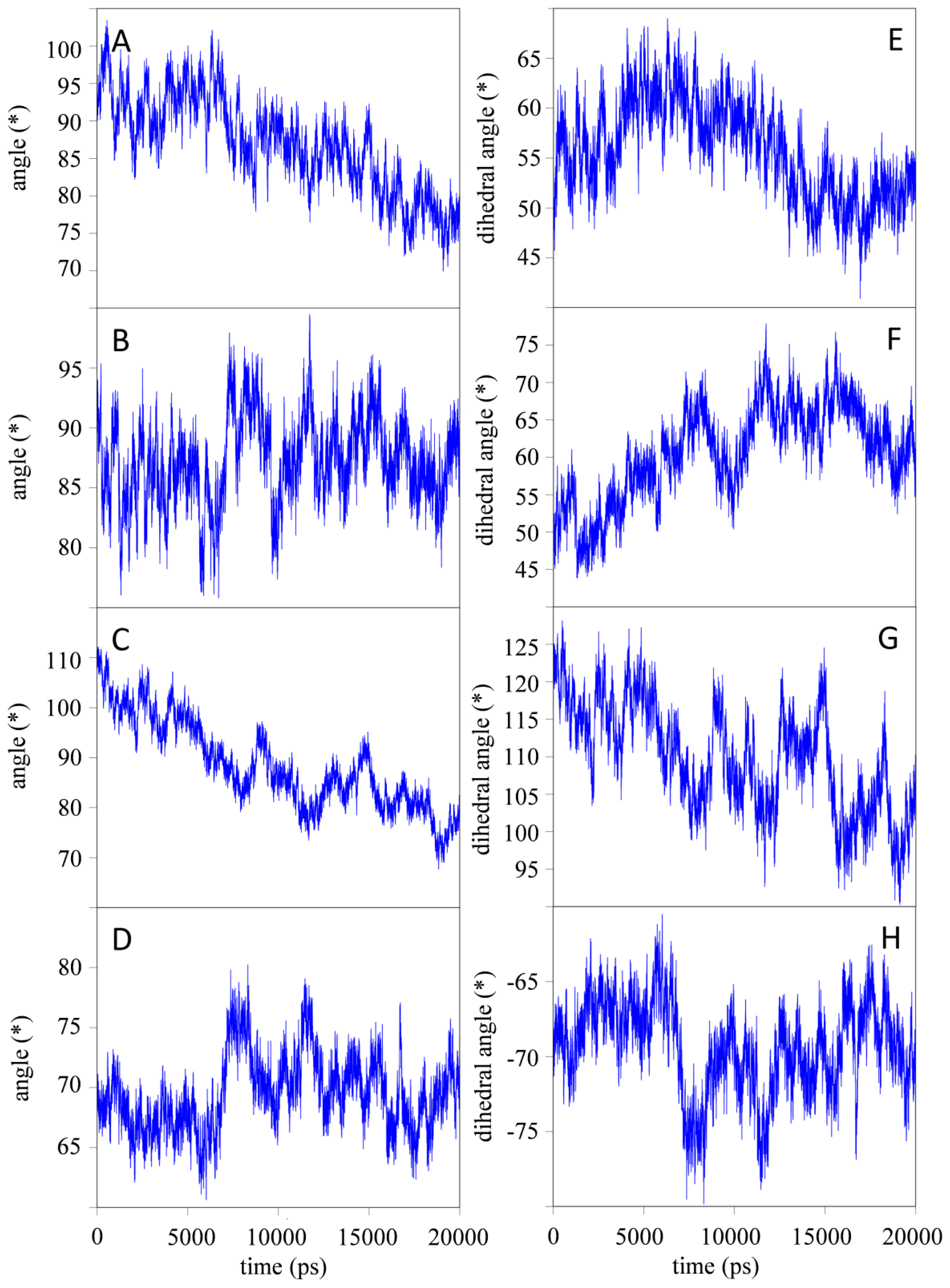
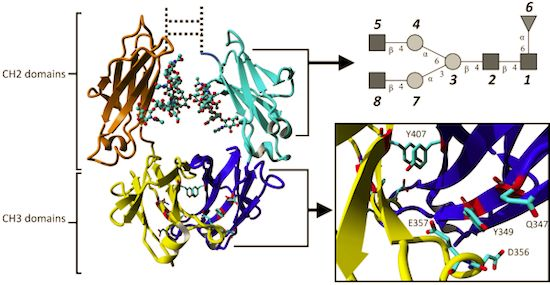
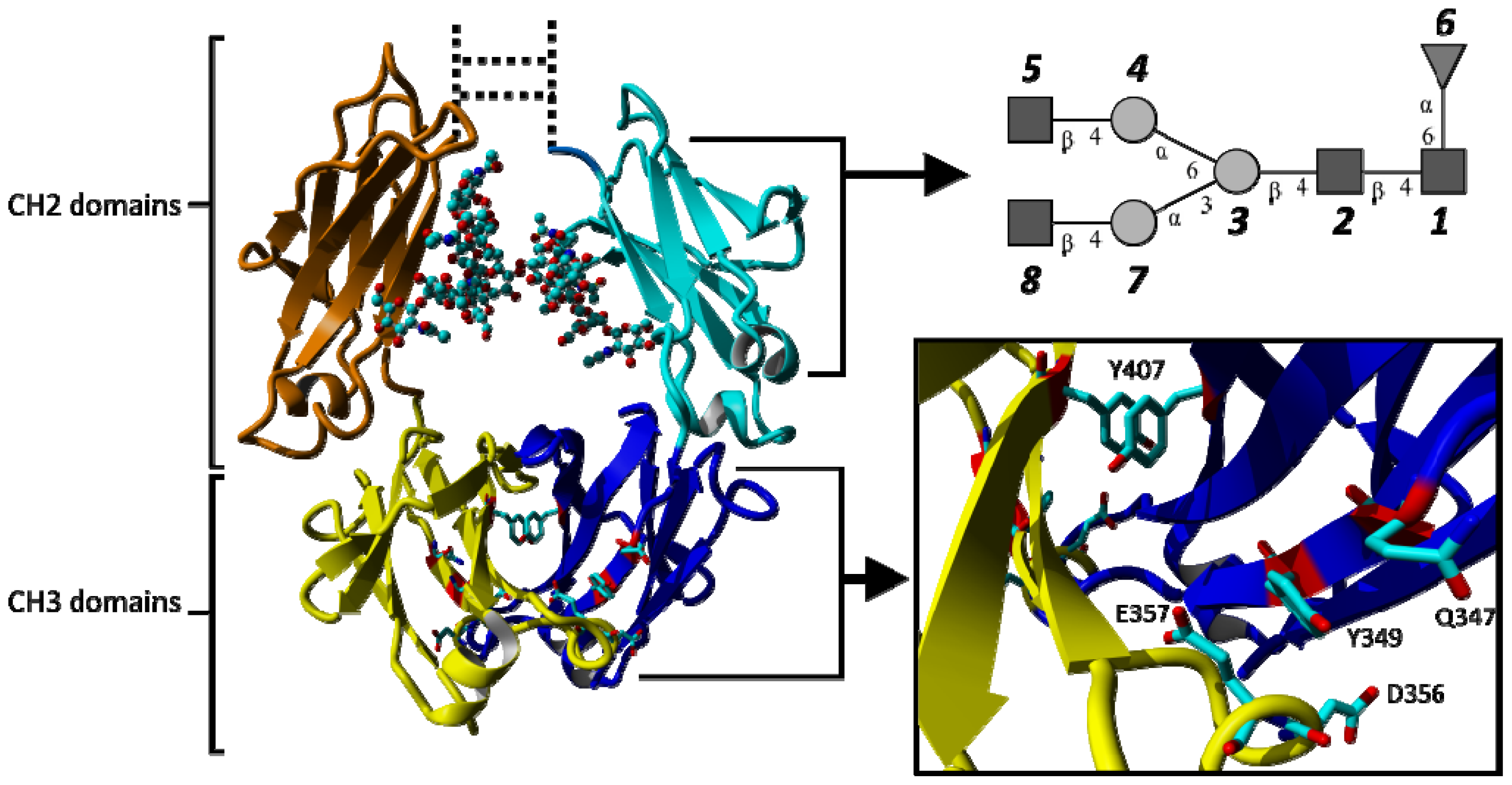
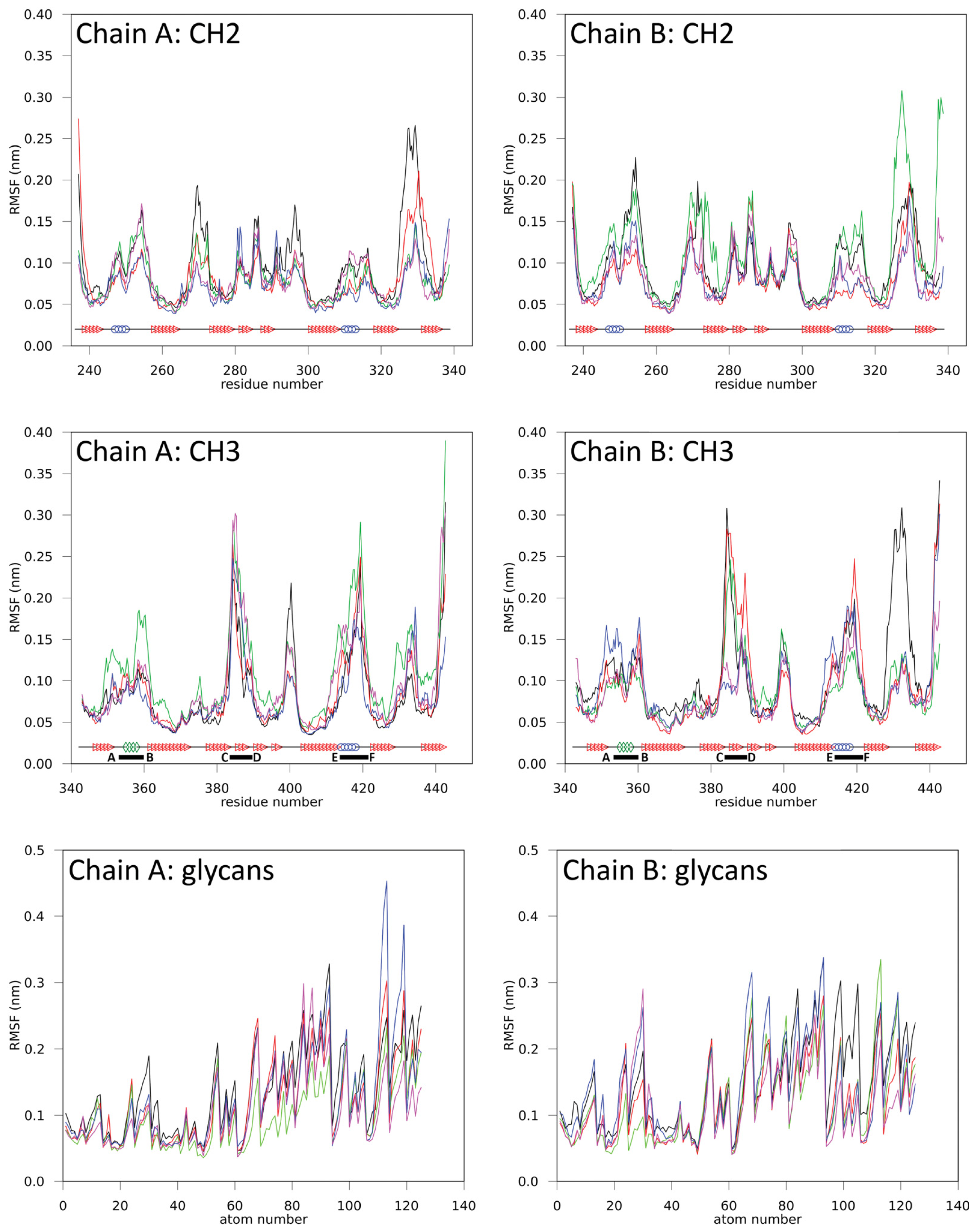
2.1.1. Structural Stability and Domain Dynamics
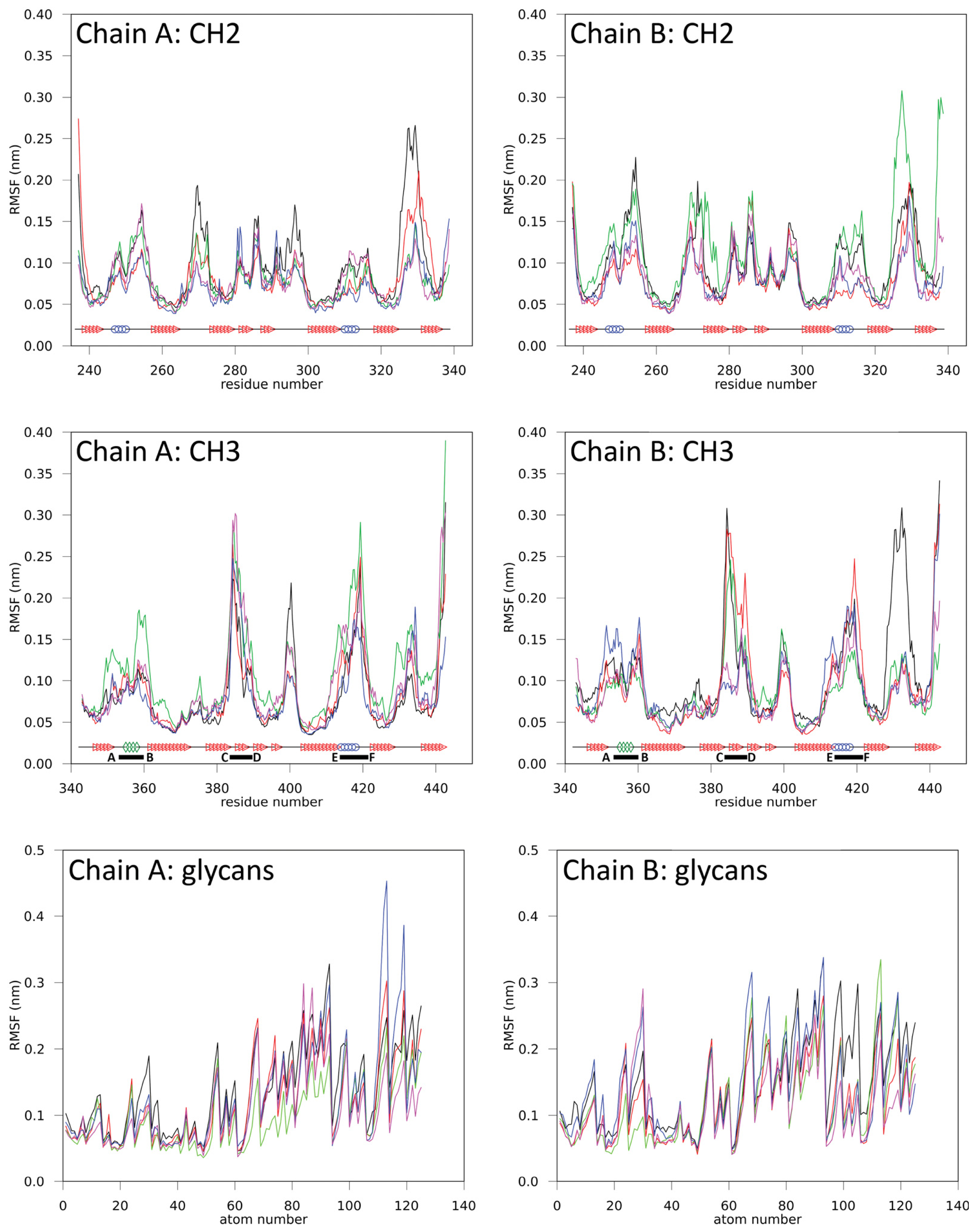
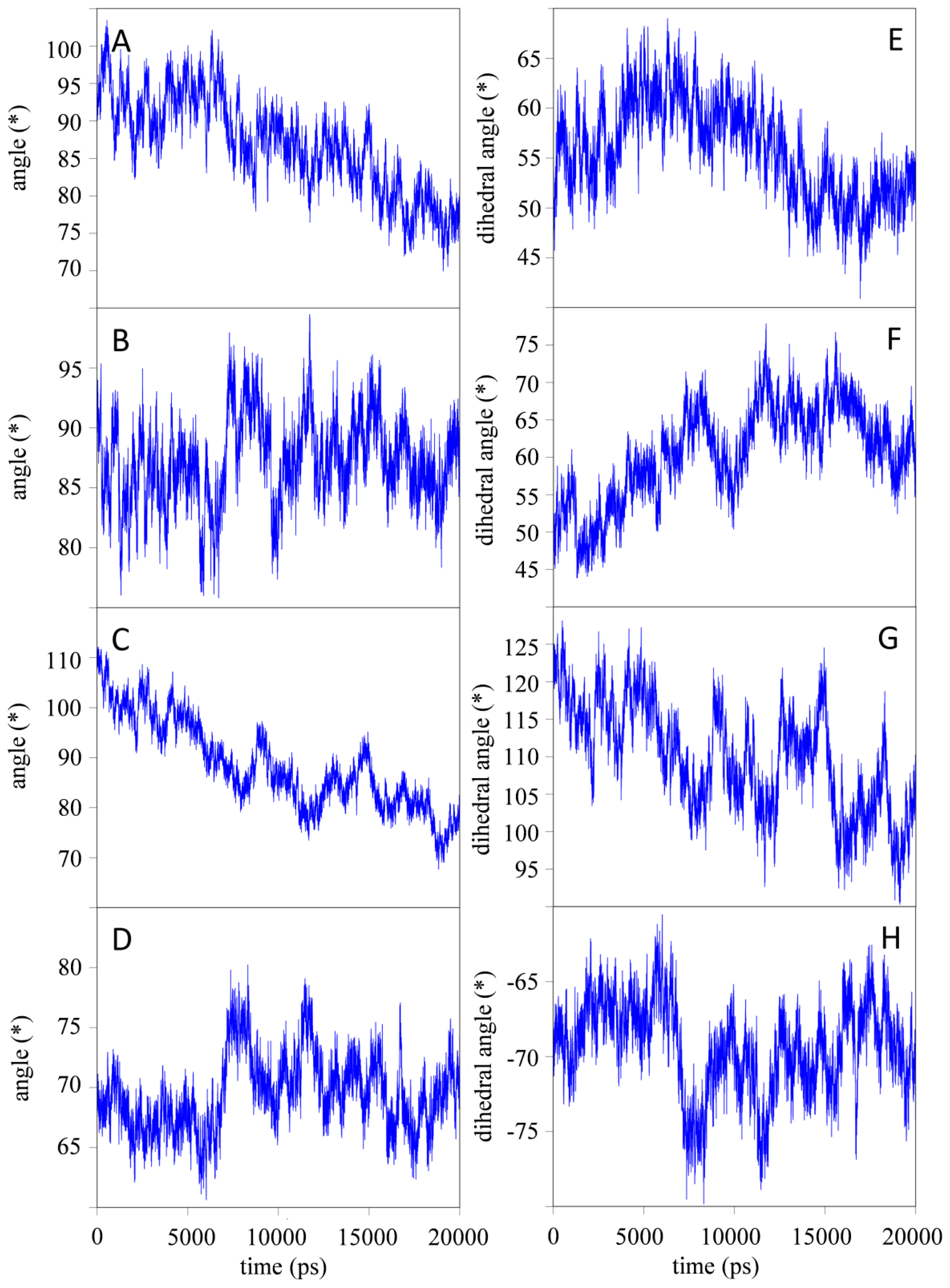
2.1.2. Impact of the Hinge Region and Dynamics of the Glycan Structure
2.1.3. Stabilizing Interactions
2.2. Discussion
3. Experimental Section
3.1. Simulation Setup
3.2. Data Analysis
4. Conclusions
Supplementary Information
ijms-15-00438-s001.pdfAcknowledgments
Conflicts of Interest
References
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar]
- Ehrlich, P. Über Den Jetzigen Stand Der Karzinomforschung (in Germany). In Beiträge zur Experimentellen Pathologie und Chemotherapie; Akademische Verlagsgesellschaft: Leipzig, Germany, 1909; pp. 117–164. [Google Scholar]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar]
- Teillaud, J. From Whole Monoclonal Antibodies to Single Domain Antibodies: Think Small. In Single Domain Antibodies; Saerens, D., Muyldermans, S., Eds.; Humana Press: Totowa, NJ, USA, 2012; Volume 911, pp. 3–13. [Google Scholar]
- Chames, P.; van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol 2009, 157, 220–233. [Google Scholar]
- Ruuls, S.R.; Lammerts van Bueren, J.J.; van de Winkel, J.G.; Parren, P.W. Novel human antibody therapeutics: The age of the Umabs. Biotechnol. J 2008, 3, 1157–1171. [Google Scholar]
- Jakobovits, A.; Amado, R.G.; Yang, X.; Roskos, L.; Schwab, G. From XenoMouse technology to Panitumumab, the first fully human antibody product from transgenic mice. Nat. Biotechnol 2007, 25, 1134–1143. [Google Scholar]
- Feldhaus, M.J.; Siegel, R.W. Yeast display of antibody fragments: A discovery and characterization platform. J. Immunol. Methods 2004, 290, 69–80. [Google Scholar]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol 2006, 6, 343–357. [Google Scholar]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. the impact of glycosylation on the biological function and structure of human immunoglobulins. Ann. Rev. Immunol 2007, 25, 21–50. [Google Scholar]
- Bradbury, A.R.; Sidhu, S.; Dubel, S.; McCafferty, J. Beyond natural antibodies: The power of in vitro display technologies. Nat. Biotechnol 2011, 29, 245–254. [Google Scholar]
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov 2010, 9, 767–774. [Google Scholar]
- Moore, P.L.; Gray, E.S.; Wibmer, C.K.; Bhiman, J.N.; Nonyane, M.; Sheward, D.J.; Hermanus, T.; Bajimaya, S.; Tumba, N.L.; Abrahams, M.R.; et al. Evolution of an HIV glycan-dependent broadly neutralizing antibody epitope through immune escape. Nat. Med 2012, 18, 1688–1692. [Google Scholar]
- Garber, K. Genentech’s alzheimer’s antibody trial to study disease prevention. Nat. Biotechnol 2012, 30, 731–732. [Google Scholar]
- Hino, T.; Arakawa, T.; Iwanari, H.; Yurugi-Kobayashi, T.; Ikeda-Suno, C.; Nakada-Nakura, Y.; Kusano-Arai, O.; Weyand, S.; Shimamura, T.; Nomura, N.; et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 2012, 482, 237–240. [Google Scholar]
- Vincent, K.J.; Zurini, M. Current strategies in antibody engineering: Fc engineering and pH-dependent antigen binding, bispecific antibodies and antibody drug conjugates. Biotechnol. J 2012, 7, 1444–1450. [Google Scholar]
- Hudson, P.J. Recombinant antibody fragments. Curr. Opin. Biotechnol 1998, 9, 395–402. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol 2005, 23, 1126–1136. [Google Scholar]
- De Marco, A. Biotechnological applications of recombinant single-domain antibody fragments. Microb. Cell Fact 2011. [Google Scholar] [CrossRef]
- Zahnd, C.; Pecorari, F.; Straumann, N.; Wyler, E.; Pluckthun, A. Selection and characterization of Her2 binding-designed ankyrin repeat proteins. J. Biol. Chem 2006, 281, 35167–35175. [Google Scholar]
- Orlova, A.; Magnusson, M.; Eriksson, T.L.; Nilsson, M.; Larsson, B.; Hoiden-Guthenberg, I.; Widstrom, C.; Carlsson, J.; Tolmachev, V.; Ståhl, S.; et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res 2006, 66, 4339–4348. [Google Scholar]
- Dimitrov, D.S. Therapeutic antibodies, vaccines and antibodyomes. MAbs 2010, 2, 347–356. [Google Scholar]
- Boersma, Y.L.; Pluckthun, A. DARPins and other repeat protein scaffolds: Advances in engineering and applications. Curr. Opin. Biotechnol 2011, 22, 849–857. [Google Scholar]
- Wozniak-Knopp, G.; Bartl, S.; Bauer, A.; Mostageer, M.; Woisetschlager, M.; Antes, B.; Ettl, K.; Kainer, M.; Weberhofer, G.; Wiederkum, S.; et al. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng. Des. Sel 2010, 23, 289–297. [Google Scholar]
- Ying, T.; Chen, W.; Gong, R.; Feng, Y.; Dimitrov, D.S. Soluble monomeric IgG1 Fc. J. Biol. Chem 2012, 287, 19399–19408. [Google Scholar]
- Ying, T.; Chen, W.; Feng, Y.; Wang, Y.; Gong, R.; Dimitrov, D.S. Engineered soluble monomeric IgG1 CH3 domain: Generation, mechanism of function, and implications for design of biological therapeutics. J. Biol. Chem 2013, 288, 25154–25164. [Google Scholar]
- Kabat, E.A.; Wu, T.T.; Gottesman, K.S.; Foeller, C. Sequences of Proteins of Immunological Interest; Diane Publishing Company: Collingdale, PA, USA, 1992. [Google Scholar]
- Kontermann, R. Dual targeting strategies with bispecific antibodies. MAbs 2012, 4, 182–197. [Google Scholar]
- Traxlmayr, M.W.; Wozniak-Knopp, G.; Antes, B.; Stadlmayr, G.; Ruker, F.; Obinger, C. Integrin binding human antibody constant domains—Probing the C-terminal structural loops for grafting the RGD motif. J. Biotechnol 2011, 155, 193–202. [Google Scholar]
- Traxlmayr, M.W.; Faissner, M.; Stadlmayr, G.; Hasenhindl, C.; Antes, B.; Ruker, F.; Obinger, C. Directed evolution of stabilized IgG1-Fc scaffolds by application of strong heat shock to libraries displayed on yeast. Biochim. Biophys. Acta 2012, 1824, 542–549. [Google Scholar]
- Hu, G.; Zhang, Q.; Chen, L.Y. Insights into scFv: Drug binding using the molecular dynamics simulation and free energy calculation. J. Mol. Model 2011, 17, 1919–1926. [Google Scholar]
- De Ruiter, A.; Mader, A.; Kunert, R.; Oostenbrink, C. Molecular simulations to rationalize humanized Ab2/3H6 activity. Aust. J. Chem 2011, 64, 900–909. [Google Scholar]
- Wang, X.; Kumar, S.; Buck, P.M.; Singh, S.K. Impact of deglycosylation and thermal stress on conformational stability of a full length murine IgG2a monoclonal antibody: Observations from molecular dynamics simulations. Proteins 2013, 81, 443–460. [Google Scholar]
- Kortkhonjia, E.; Brandman, R.; Zhou, J.Z.; Voelz, V.A.; Chorny, I.; Kabakoff, B.; Patapoff, T.W.; Dill, K.A.; Swartz, T.E. Probing antibody internal dynamics with fluorescence anisotropy and molecular dynamics simulations. MAbs 2013, 5, 306–322. [Google Scholar]
- Traxlmayr, M.W.; Hasenhindl, C.; Hackl, M.; Stadlmayr, G.; Rybka, J.D.; Borth, N.; Grillari, J.; Rüker, F.; Obinger, C. Construction of a stability landscape of the CH3 domain of human IgG1 by combining directed evolution with high throughput sequencing. J. Mol. Biol 2012, 423, 397–412. [Google Scholar]
- Hasenhindl, C.; Traxlmayr, M.W.; Wozniak-Knopp, G.; Jones, P.C.; Stadlmayr, G.; Rüker, F.; Obinger, C. Stability assessment on a library scale: A rapid method for the evaluation of the commutability and insertion of residues in C-terminal loops of the CH3 domains of IgG1-Fc. Protein Eng. Des. Sel 2013, 26, 675–682. [Google Scholar]
- Meier, S.; Duus, J. Carbohydrate dynamics: Antibody glycans wiggle and jiggle. Nat. Chem. Biol 2011, 7, 131–132. [Google Scholar]
- Schmid, N.; Christ, C.D.; Christen, M.; Eichenberger, A.P.; van Gunsteren, W.F. Architecture, implementation and parallelisation of the GROMOS software for biomolecular simulation. Comput. Phys. Commun 2012, 183, 890–903. [Google Scholar]
- Eichenberger, A.P.; Allison, J.R.; Dolenc, J.; Geerke, D.P.; Horta, B.A.C.; Meier, K.; Oostenbrink, C.; Schmid, N.; Steiner, D.; Wang, D.; et al. GROMOS++ Software for the analysis of biomolecular simulation trajectories. J. Chem. Theory Comput 2011, 7, 3379–3390. [Google Scholar]
- Saphire, E.O.; Parren, P.W.; Pantophlet, R.; Zwick, M.B.; Morris, G.M.; Rudd, P.M.; Dwek, R.A.; Stanfield, R.L.; Burton, D.R.; Wilson, I.A. Crystal structure of a neutralizing human IgG against HIV-1: A template for vaccine design. Science 2001, 293, 1155–1159. [Google Scholar]
- Schmid, N.; Eichenberger, A.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.; van Gunsteren, W. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J 2011, 40, 843–856. [Google Scholar]
- Hansen, H.S.; Hunenberger, P.H. A reoptimized GROMOS force field for hexopyranose-based carbohydrates accounting for the relative free energies of ring conformers, anomers, epimers, hydroxymethyl rotamers, and glycosidic linkage conformers. J. Comput. Chem 2011, 32, 998–1032. [Google Scholar]
- Berendsen, H.; Postma, J.; van Gunsteren, W.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces; Pullman, B., Ed.; D. Reidel Publishing Company: Boston, MA, USA, 1981; pp. 331–342. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar]
- Hockney, R.W. The potential calculation and some applications. Methods Comput. Phys 1970, 9, 136–211. [Google Scholar]
- Ryckaert, J.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of N-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- Heinz, T.N.; Hünenberger, P.H. A fast pairlist-construction algorithm for molecular simulations under periodic boundary conditions. J. Comput. Chem 2004, 25, 1474–1486. [Google Scholar]
- Tironi, I.G.; Sperb, R.; Smith, P.E.; van Gunsteren, W.F. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys 1995, 102, 5451–5459. [Google Scholar]
- Heinz, T.N.; van Gunsteren, W.F.; Hünenberger, P.H. Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J. Chem. Phys 2001, 115, 1125–1136. [Google Scholar]
- Schmid, N.; Botschi, M.; van Gunsteren, W.F. A GPU solvent-solvent interaction calculation accelerator for biomolecular simulations using the GROMOS software. J. Comput. Chem 2010, 31, 1636–1643. [Google Scholar]
- Vetterling, W.; Flannery, B. Numerical Recipes in C++: The Art of Scientific Computing; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
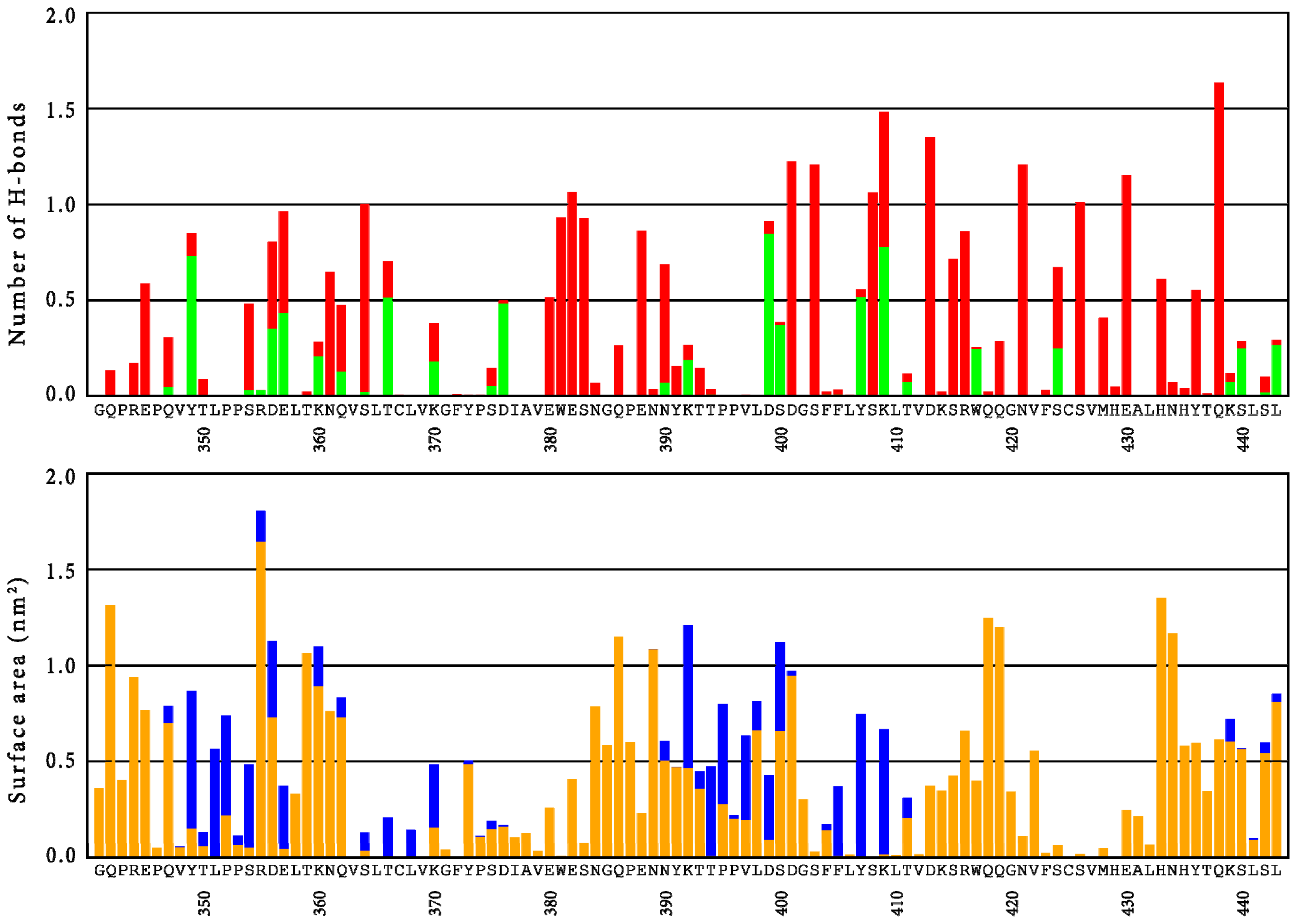{kind=link}
| Dihedrals | Atoms | Residue * | Chain | B7 | H7 | Q347E | B4 | H4 |
|---|---|---|---|---|---|---|---|---|
| 3 | O5-C5-C6-O6 | 1–6 | A | 86 | 99 | 23 | 1 | 6 |
| B | 60 | 115 | 11 | 91 | 23 | |||
| 9 | C4-O4-C1-O5 | 2–3 | A | 12 | 0 | 128 | 2346 | 748 |
| B | 2 | 1435 | 50 | 0 | 12 | |||
| 10 | C2-O2-C1-O5 | 4–5 | A | 70 | 320 | 18 | 44 | 0 |
| B | 892 | 418 | 140 | 236 | 28 | |||
| 18 | C2-O2-C1-O5 | 7–8 | A | 98 | 417 | 4 | 2 | 40 |
| B | 4 | 617 | 2 | 54 | 229 | |||
| From a | To a | B7 (%) | H7 (%) | Q347E (%) | B4 (%) | H4 (%) |
|---|---|---|---|---|---|---|
| Q/E347A | Y349A | 21 | 11 | 89 | 23 | 21 |
| Y349A | D356B | 20 | 43 | 22 | 14 | 12 |
| E357B | 6 | 18 | 1 | 0 | 0 | |
| Q/E347A | solvent | 0 | 9 | 7 | 1 | 0 |
| D356A | chain B | 77 | 20 | 16 | 16 | 6 |
| E357A | chain B | 4 | 63 | 1 | 0 | 3 |
| Q/E347B | Y349B | 16 | 14 | 95 | 18 | 5 |
| Y349B | D356A | 53 | 10 | 7 | 10 | 3 |
| E357A | 1 | 36 | 0 | 0 | 2 | |
| Q/E347B | solvent | 29 | 19 | 14 | 5 | 4 |
| D356B | chain A | 31 | 49 | 24 | 45 | 20 |
| E357B | chain A | 15 | 22 | 4 | 0 | 1 |
| chain A | chain B | 611 | 640 | 391 | 456 | 405 |
| chain A | glycans | 471 | 417 | 529 | 438 | 475 |
| chain B | glycans | 482 | 370 | 680 | 396 | 448 |
| glycans | glycans | 661 | 622 | 497 | 636 | 642 |
| chain A | solvent | 4220 | 9889 | 9630 | 1318 | 6998 |
| chain B | solvent | 10083 | 7714 | 8944 | 7498 | 5964 |
| glycans | solvent | 2178 | 2606 | 2771 | 1091 | 2235 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lai, B.; Hasenhindl, C.; Obinger, C.; Oostenbrink, C. Molecular Dynamics Simulation of the Crystallizable Fragment of IgG1—Insights for the Design of Fcabs. Int. J. Mol. Sci. 2014, 15, 438-455. https://doi.org/10.3390/ijms15010438
Lai B, Hasenhindl C, Obinger C, Oostenbrink C. Molecular Dynamics Simulation of the Crystallizable Fragment of IgG1—Insights for the Design of Fcabs. International Journal of Molecular Sciences. 2014; 15(1):438-455. https://doi.org/10.3390/ijms15010438
Chicago/Turabian StyleLai, Balder, Christoph Hasenhindl, Christian Obinger, and Chris Oostenbrink. 2014. "Molecular Dynamics Simulation of the Crystallizable Fragment of IgG1—Insights for the Design of Fcabs" International Journal of Molecular Sciences 15, no. 1: 438-455. https://doi.org/10.3390/ijms15010438