Resveratrol Partially Prevents Rotenone-Induced Neurotoxicity in Dopaminergic SH-SY5Y Cells through Induction of Heme Oxygenase-1 Dependent Autophagy


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
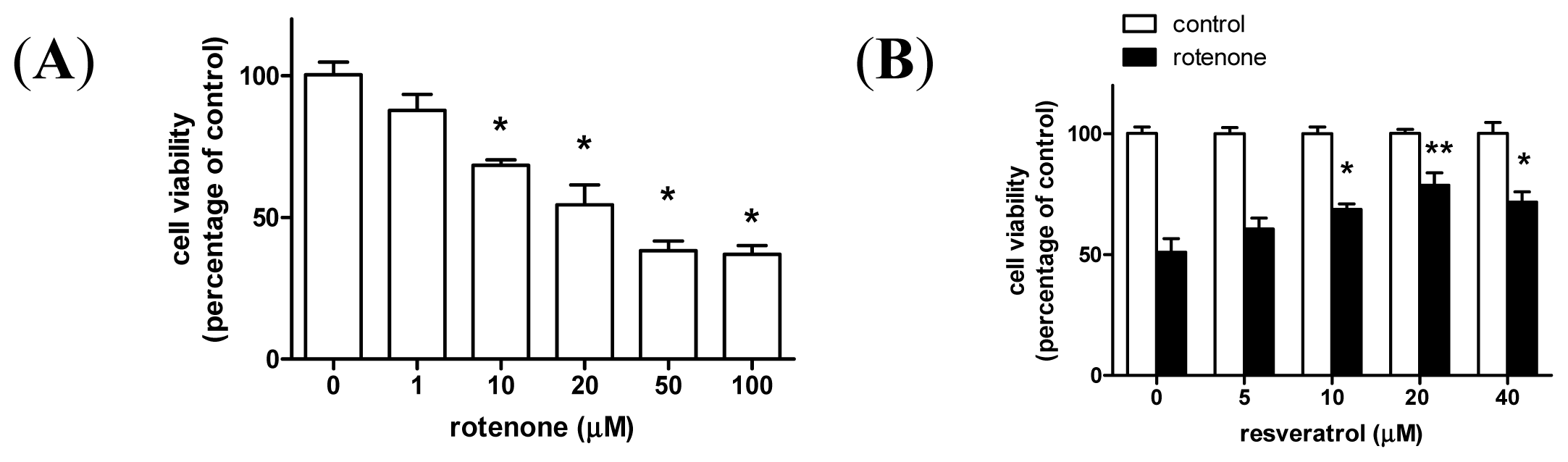
2. Results and Discussion
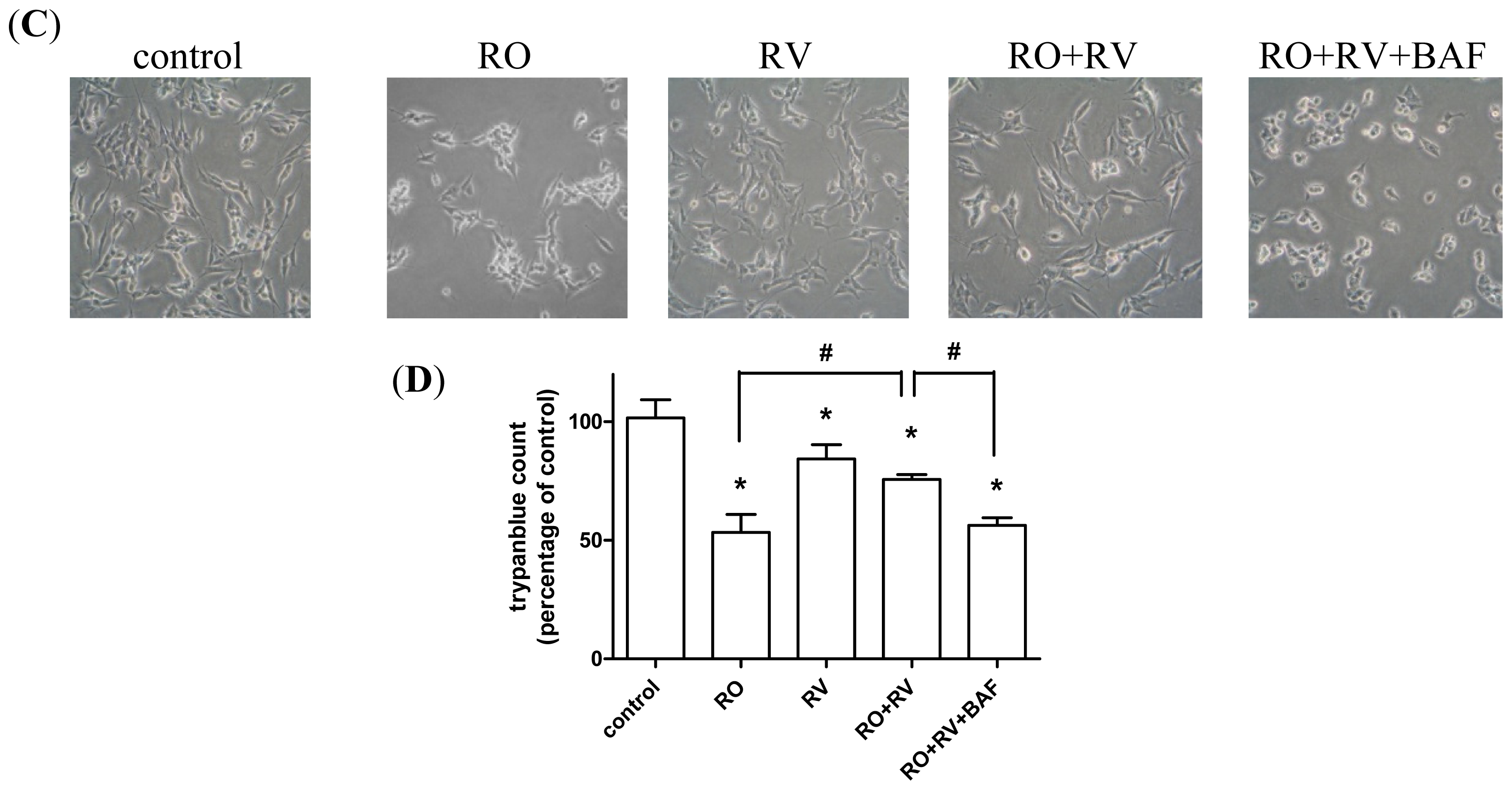
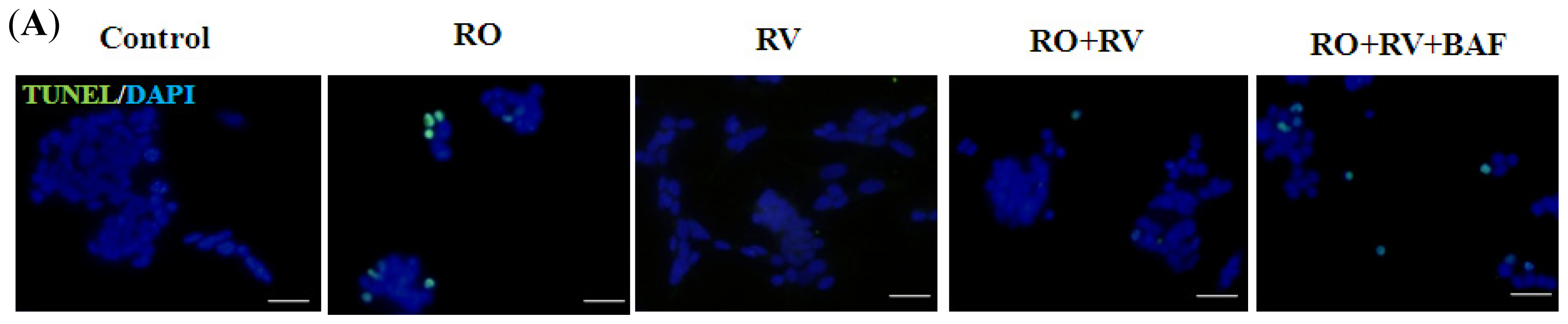
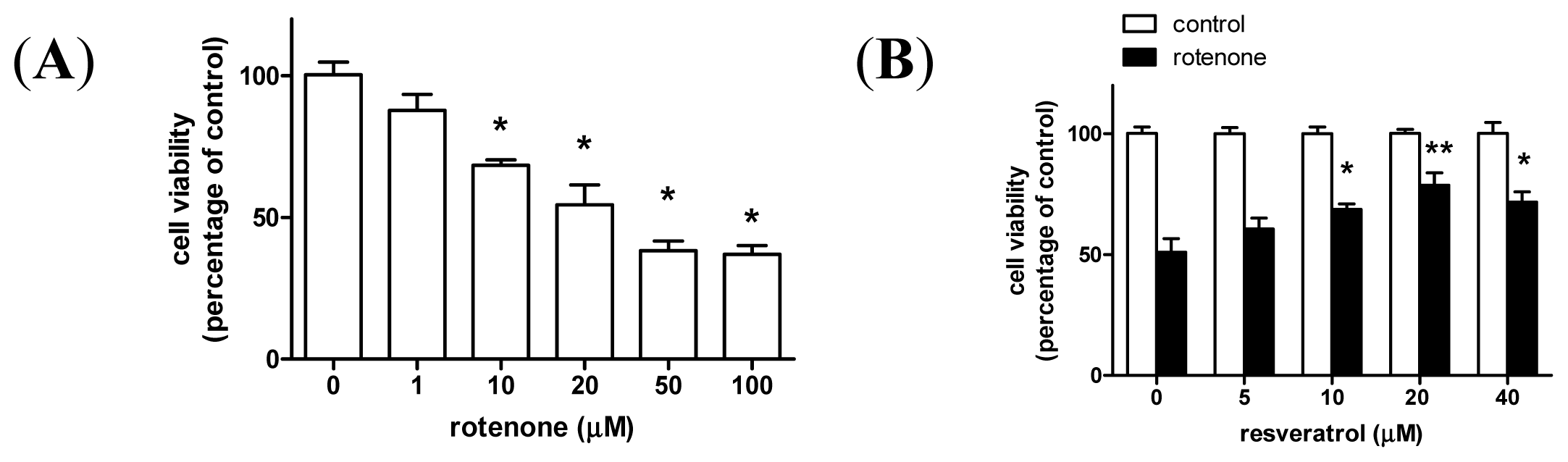
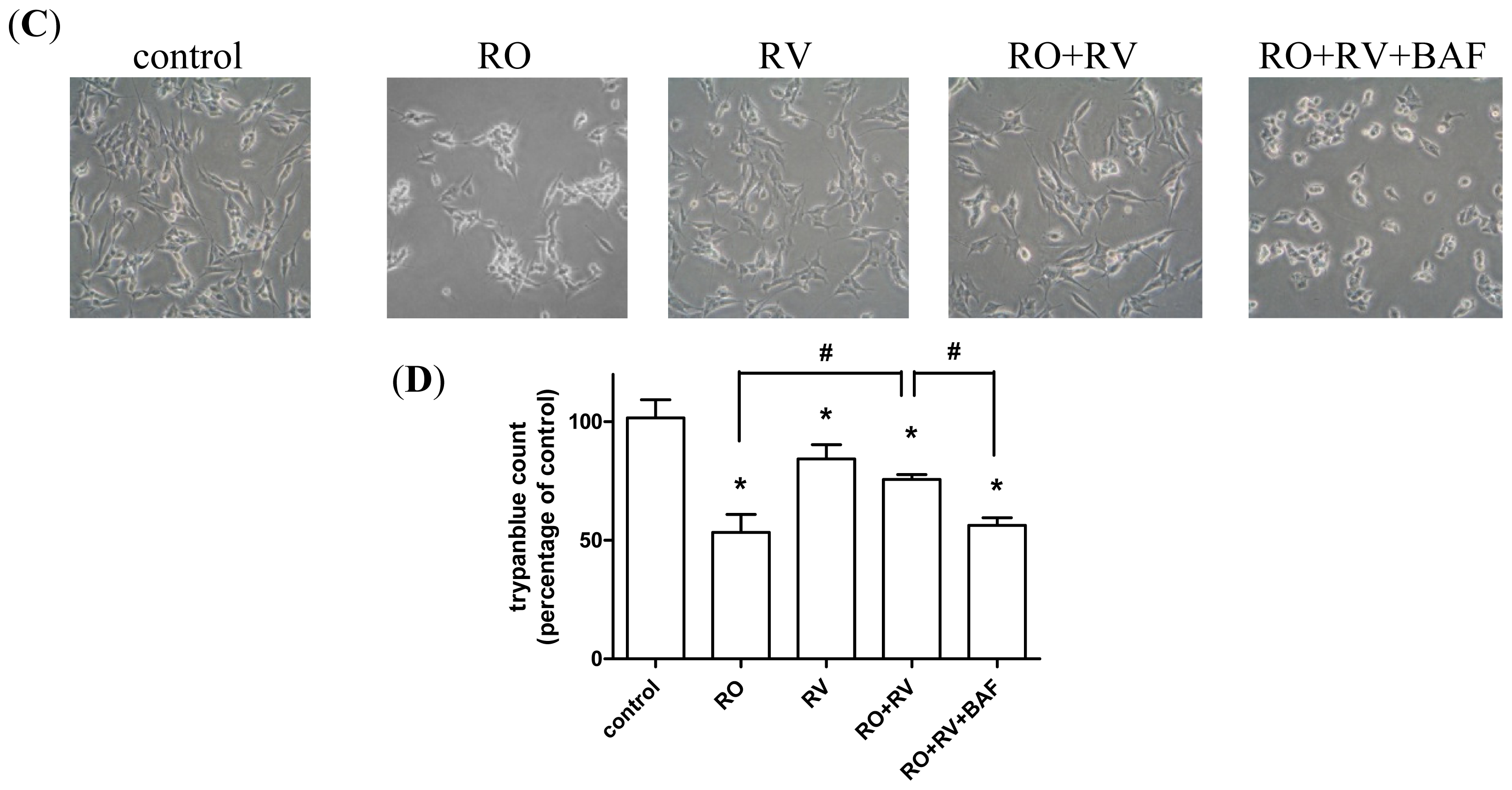
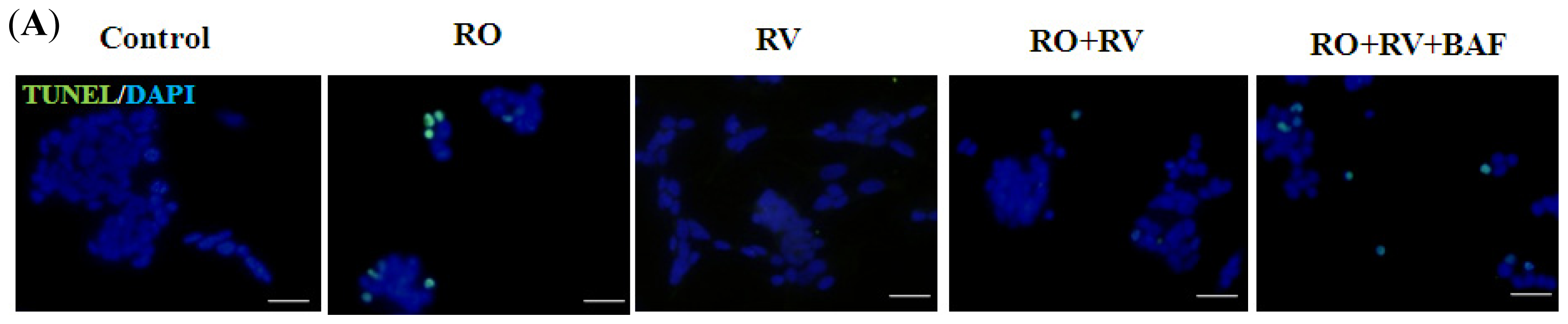
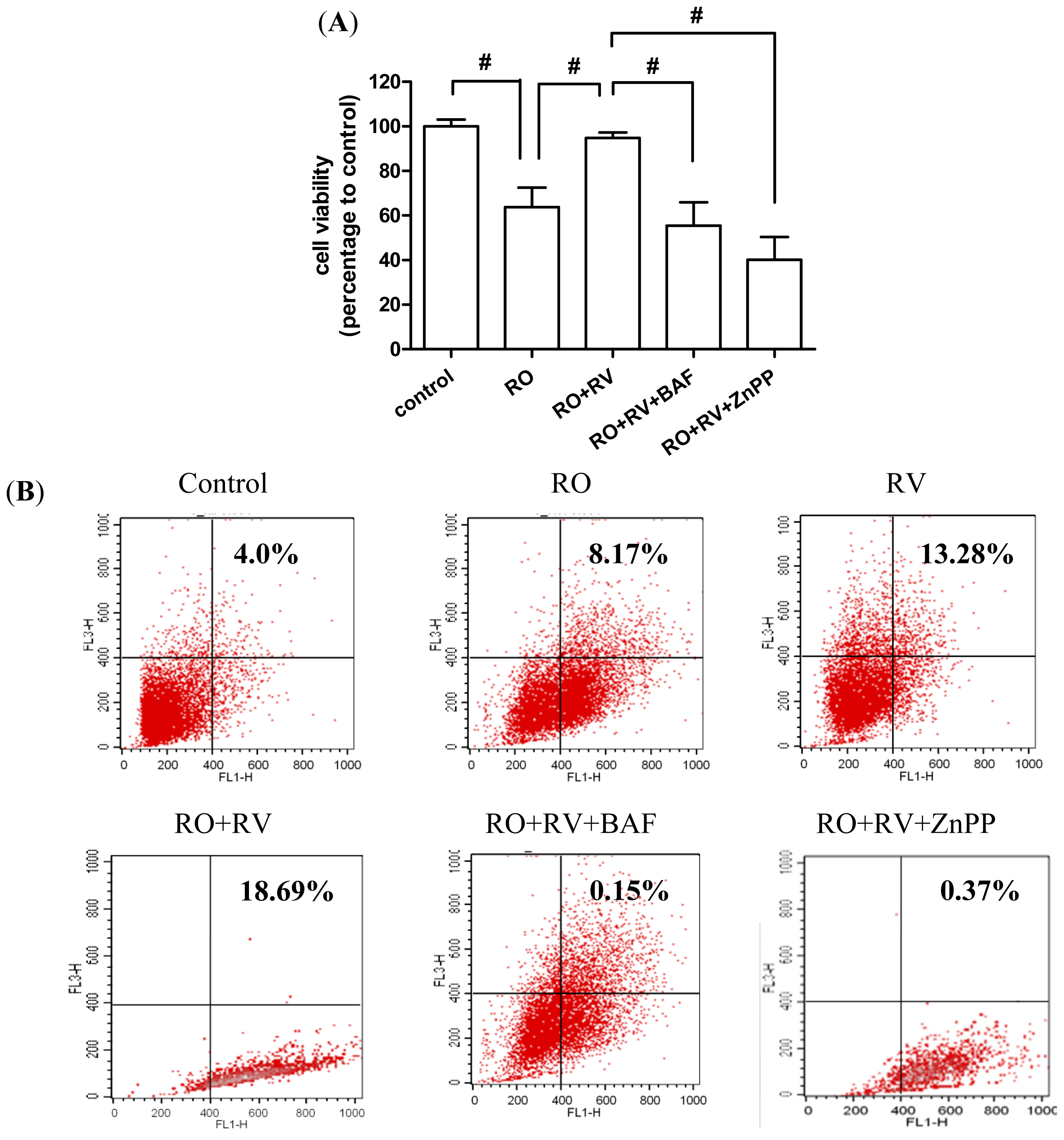
2.1. Resveratrol Prevents Rotenone-Induced Apoptosis in an Autophagy-Dependent Manner
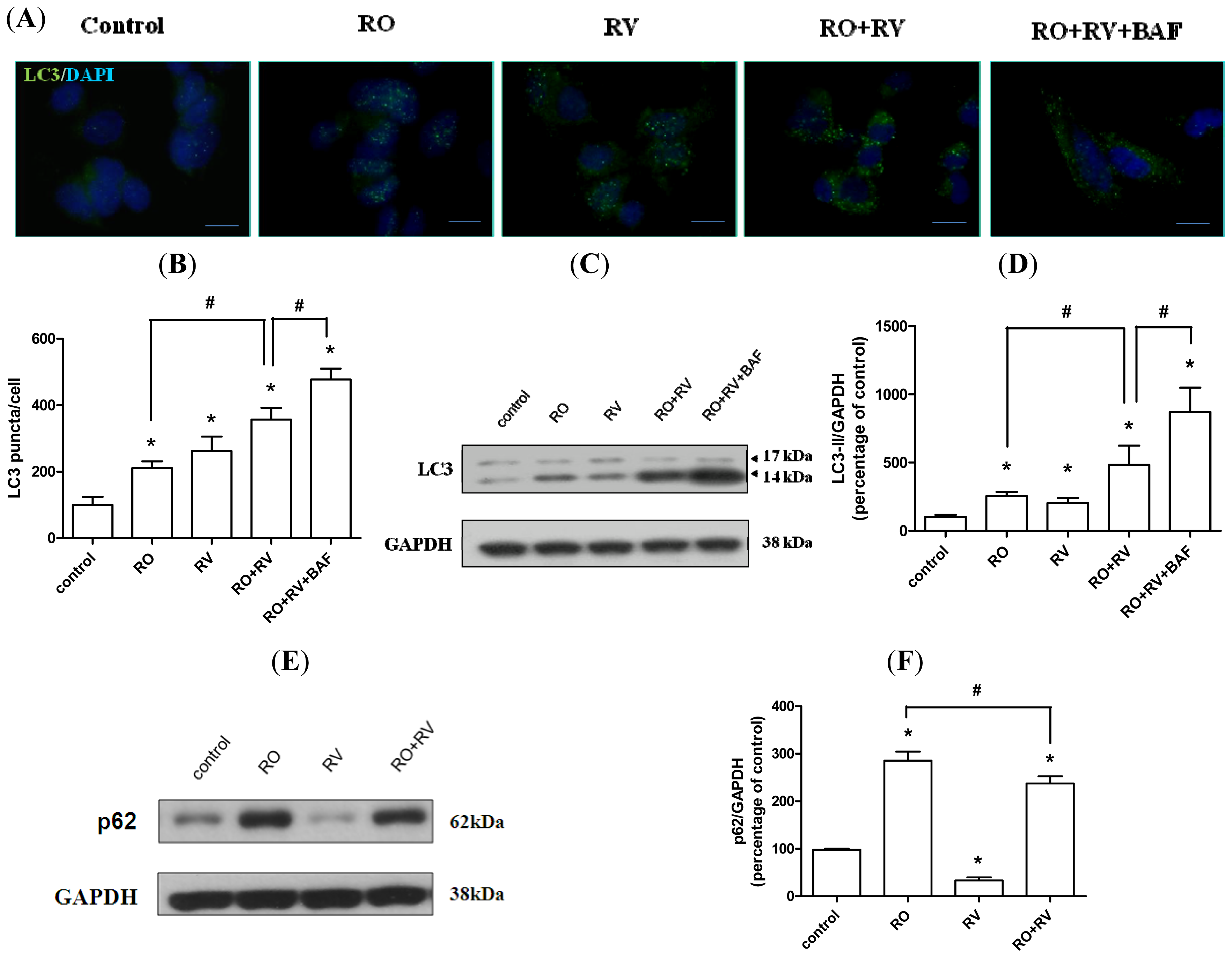
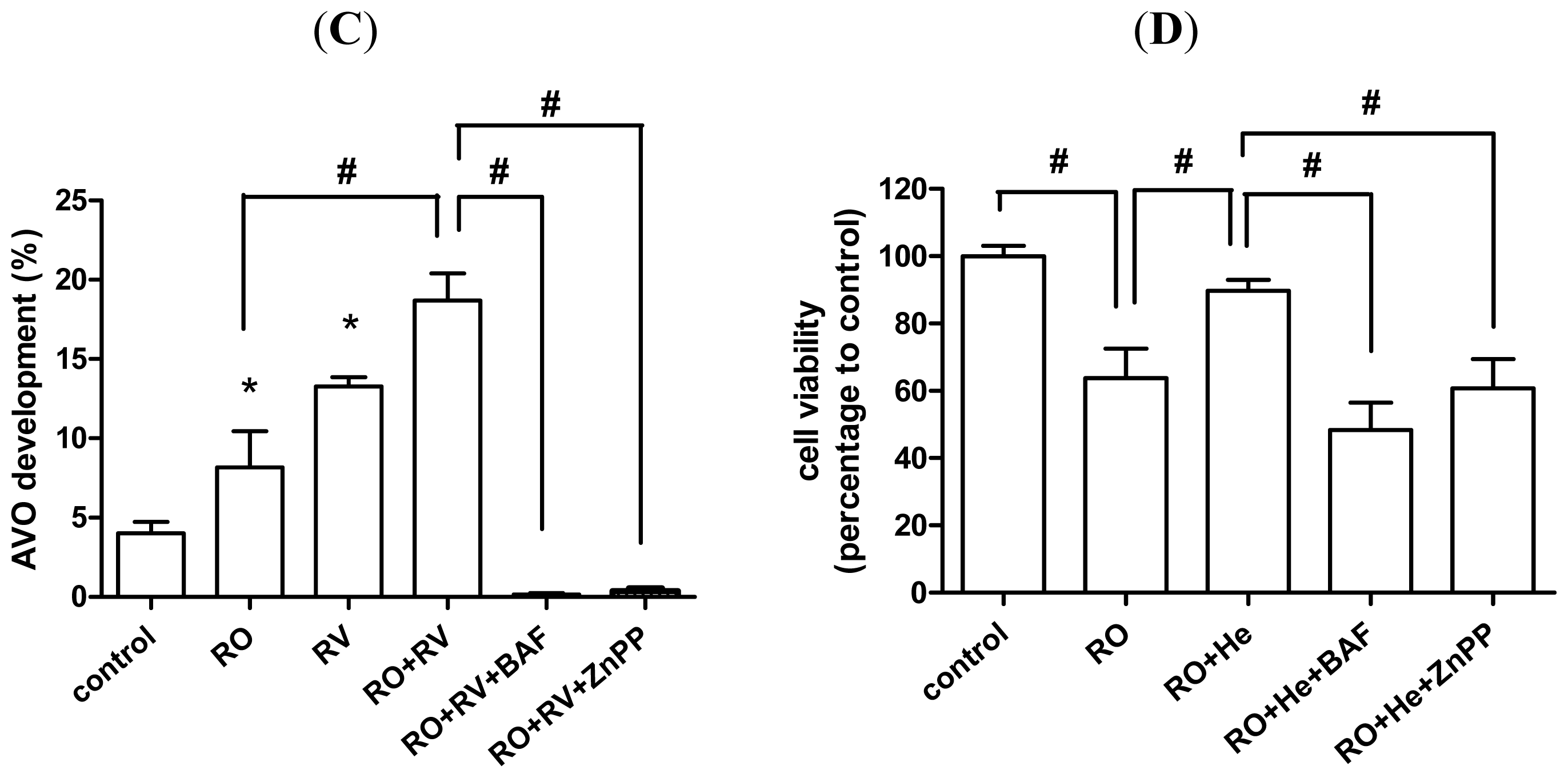
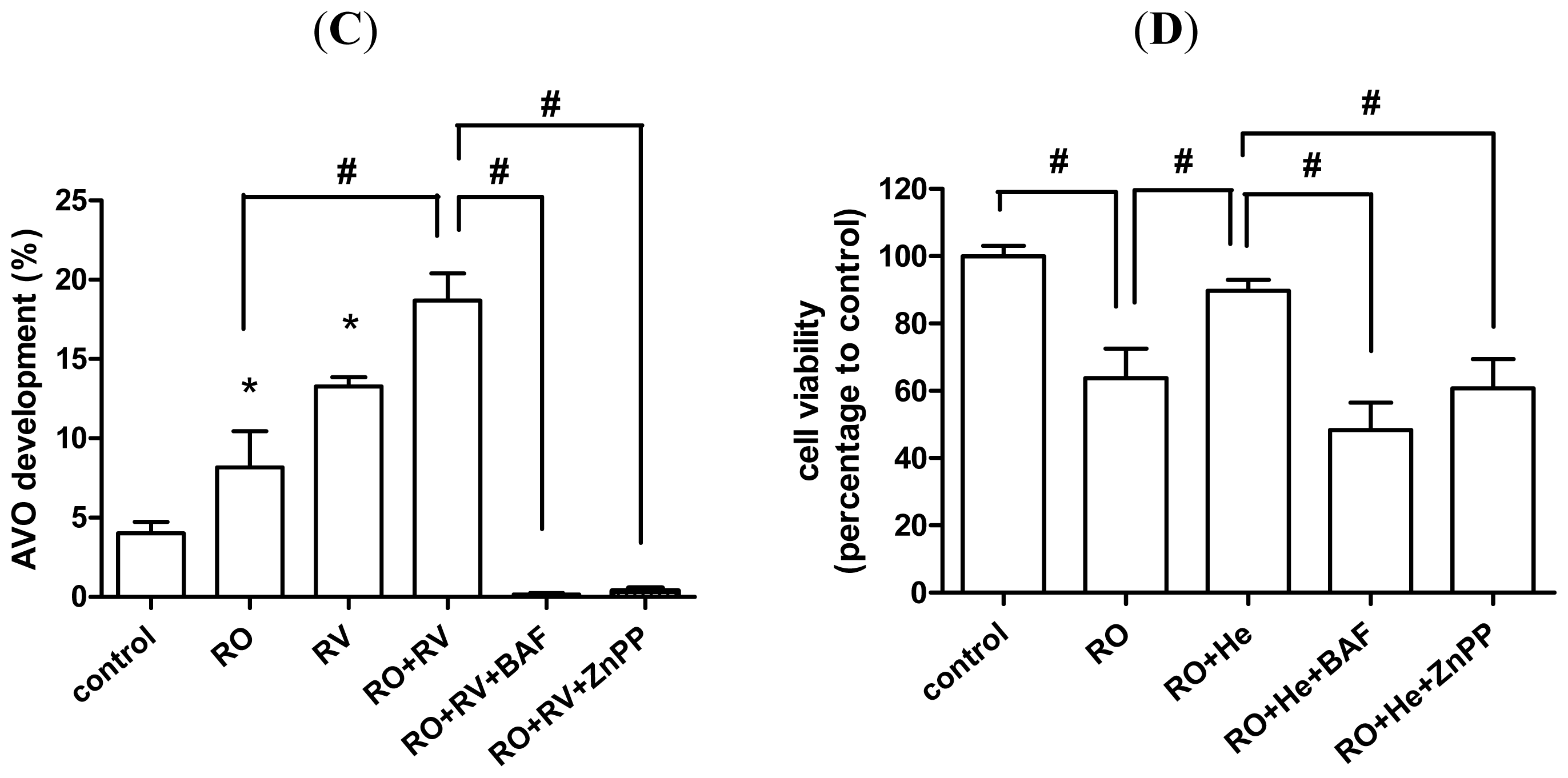
2.2. Resveratrol Increases Rotenone-Induced Autophagosome Formation
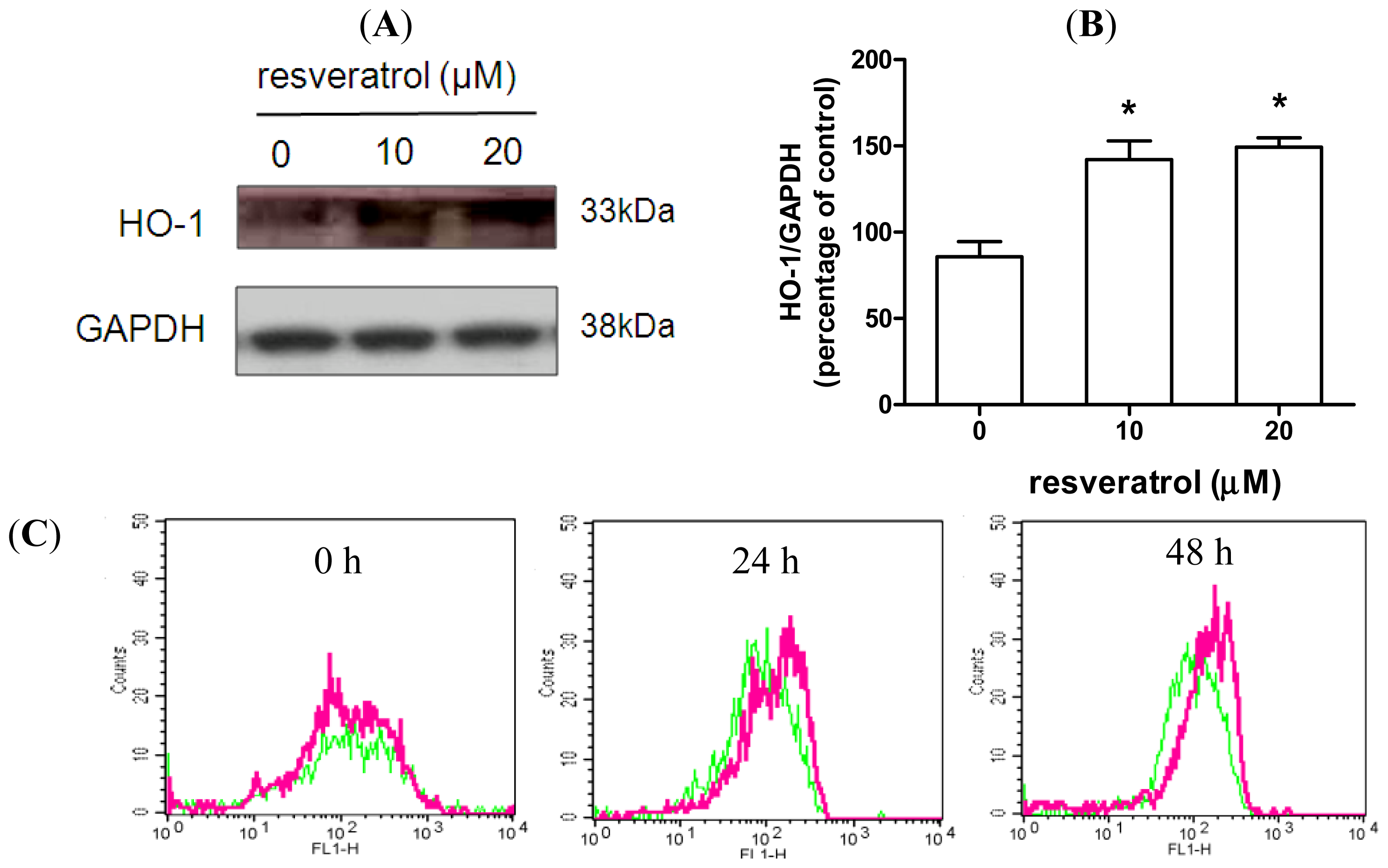
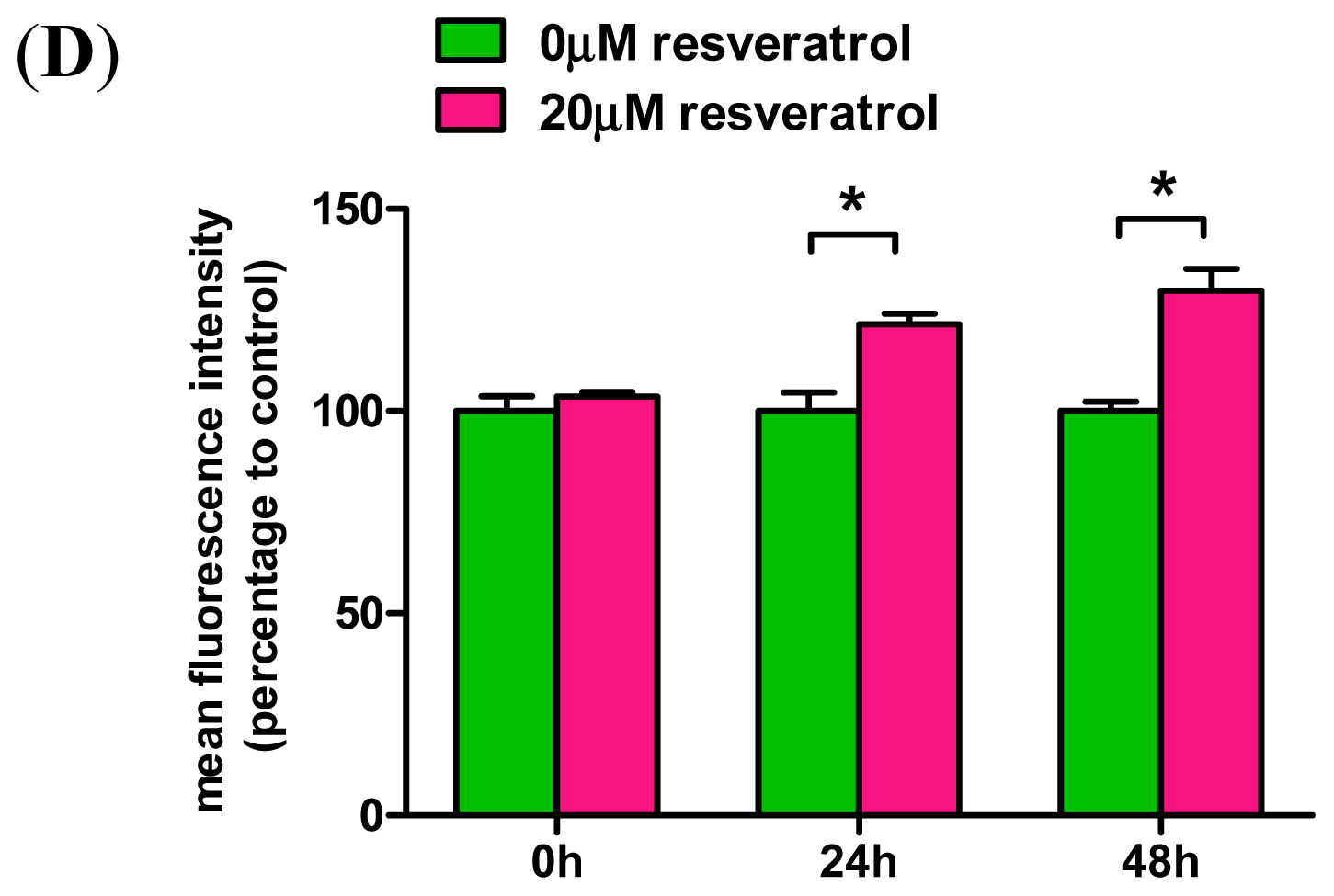
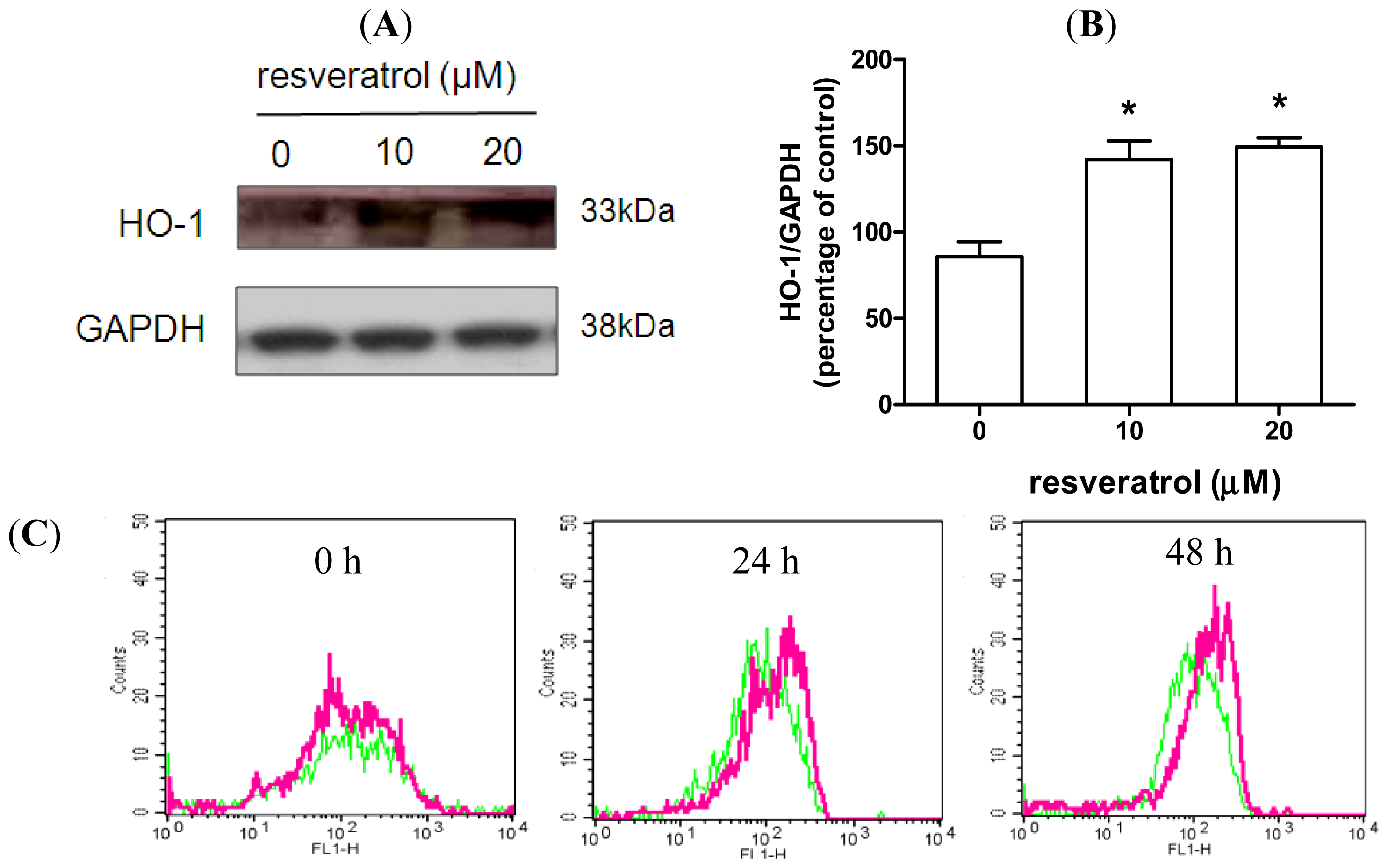
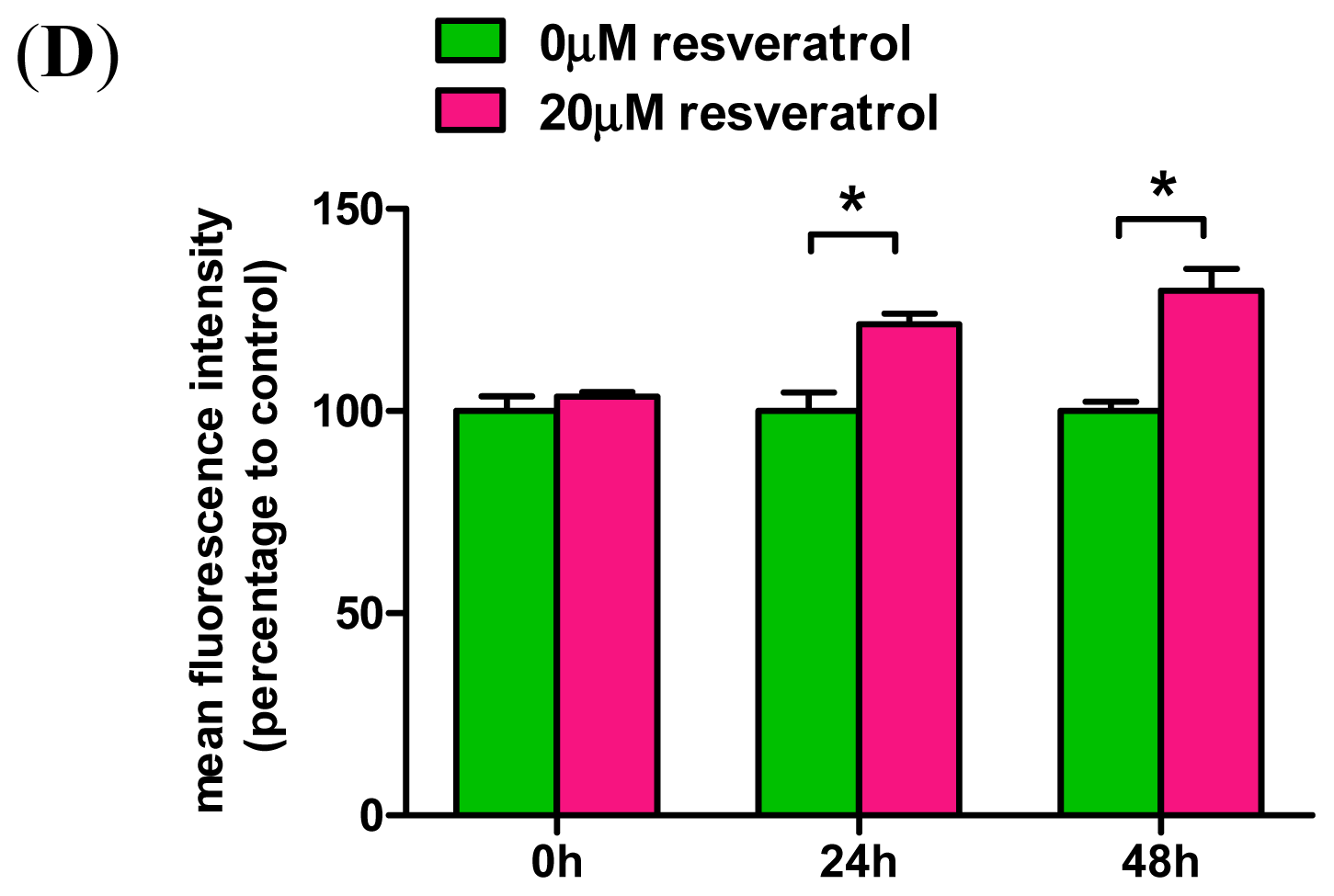
2.3. Resveratrol Prevents Rotenone-Mediated Inhibition of HO-1 Expression
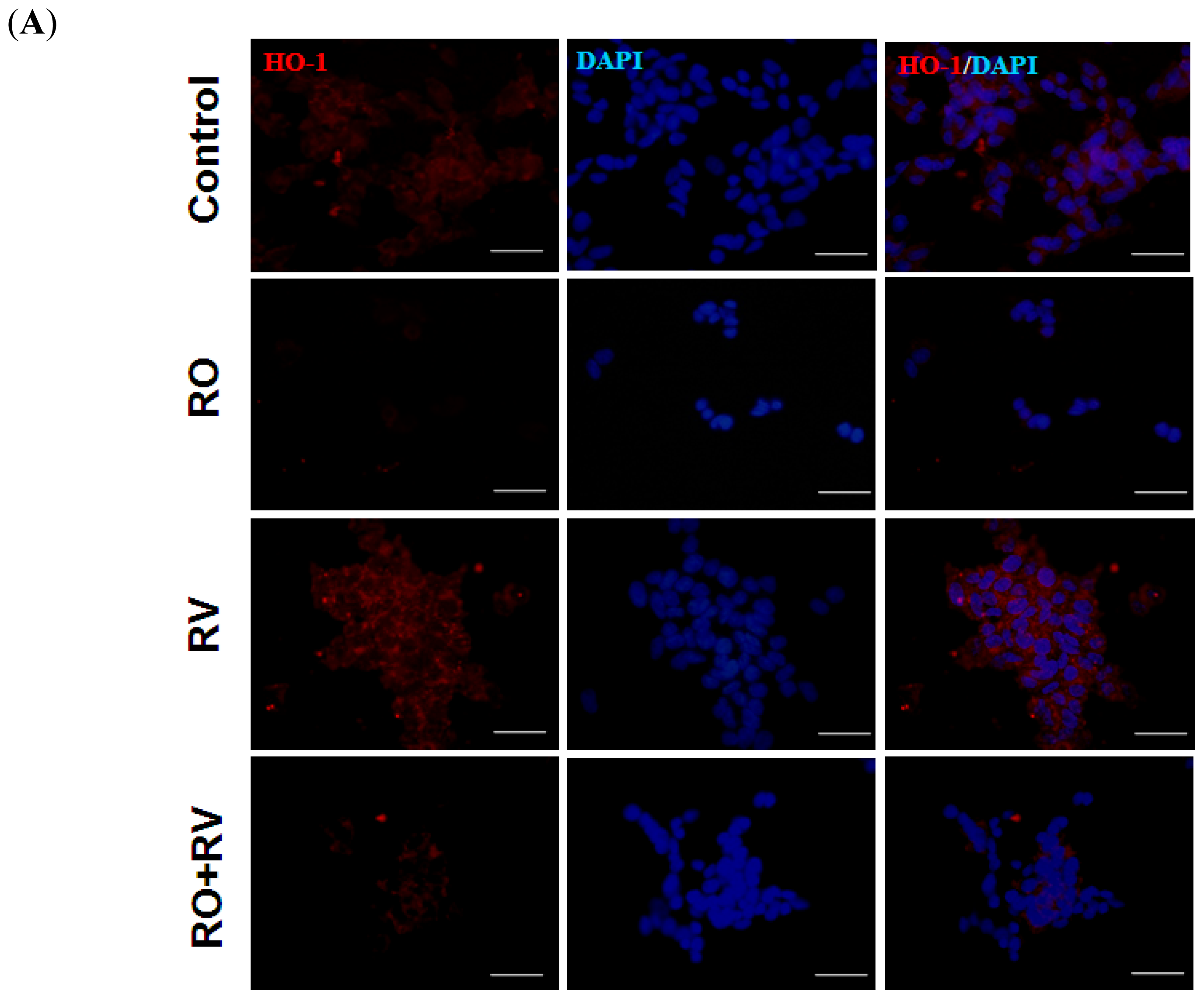
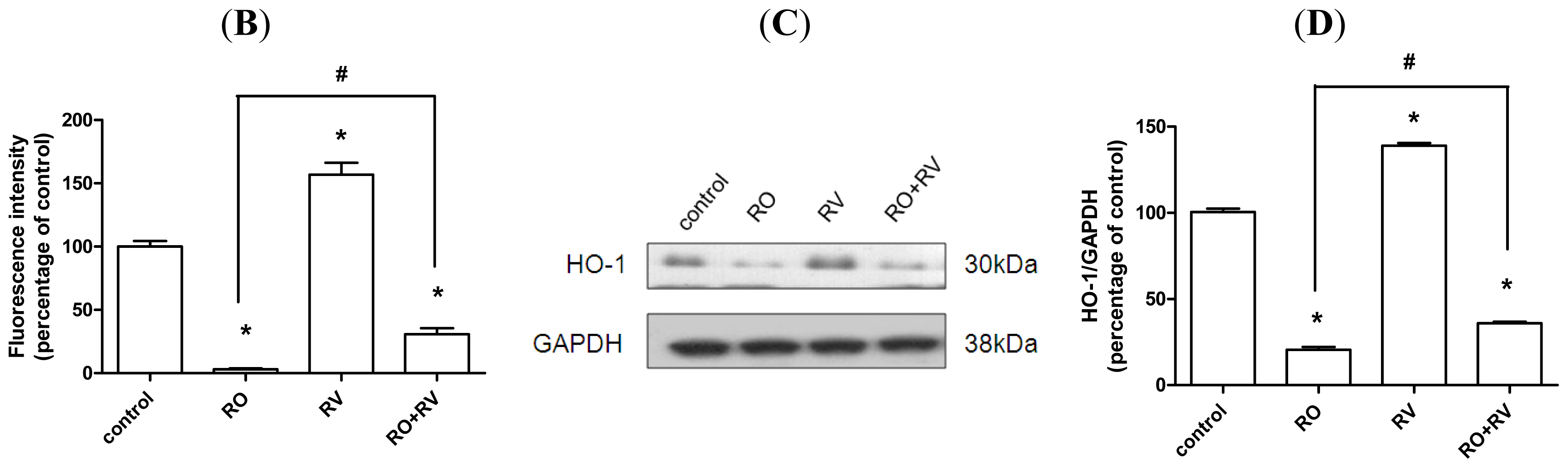
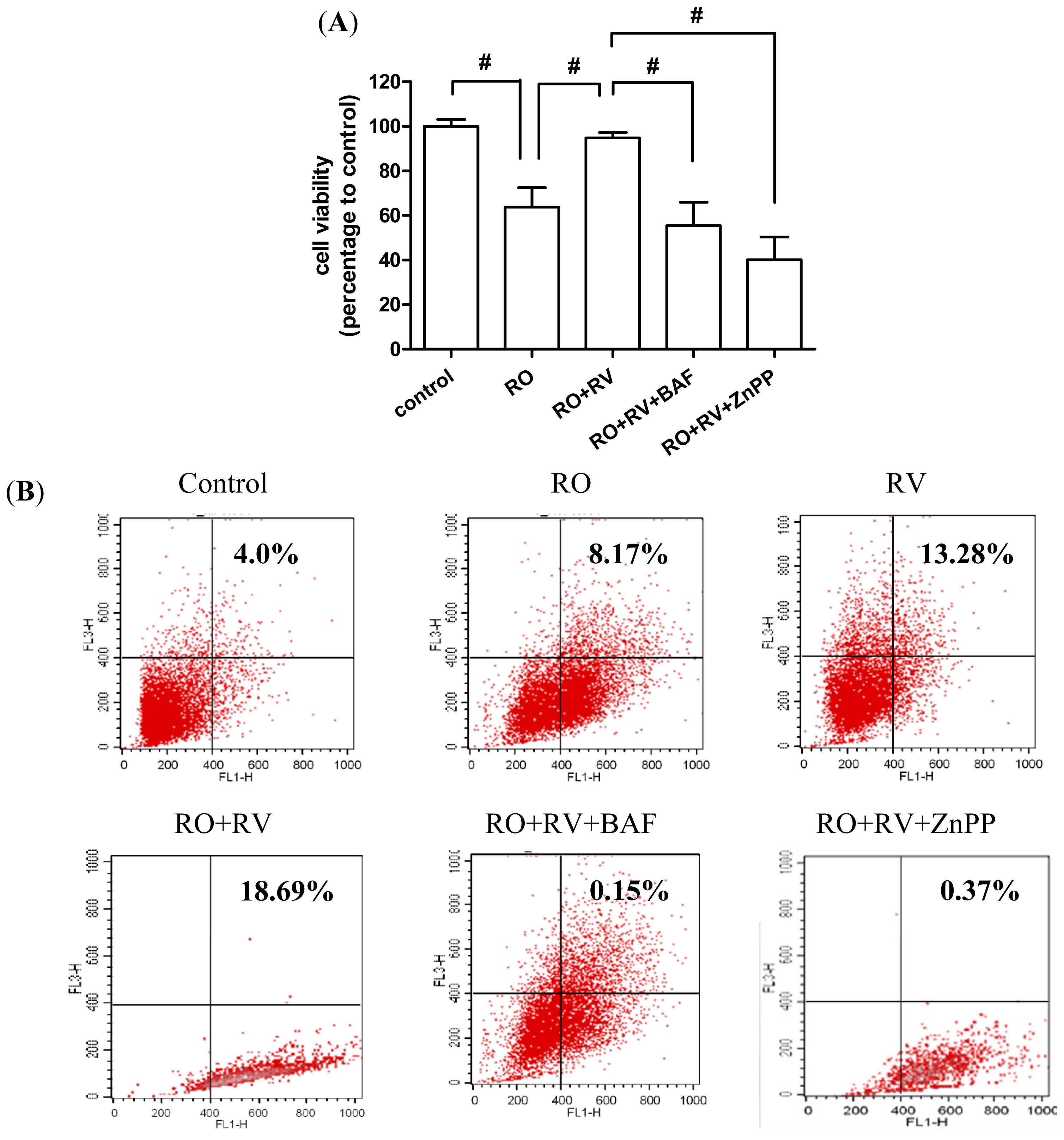
2.4. Resveratrol Prevents Rotenone-Induced Neuronal Death through HO-1 Dependent Autophagy
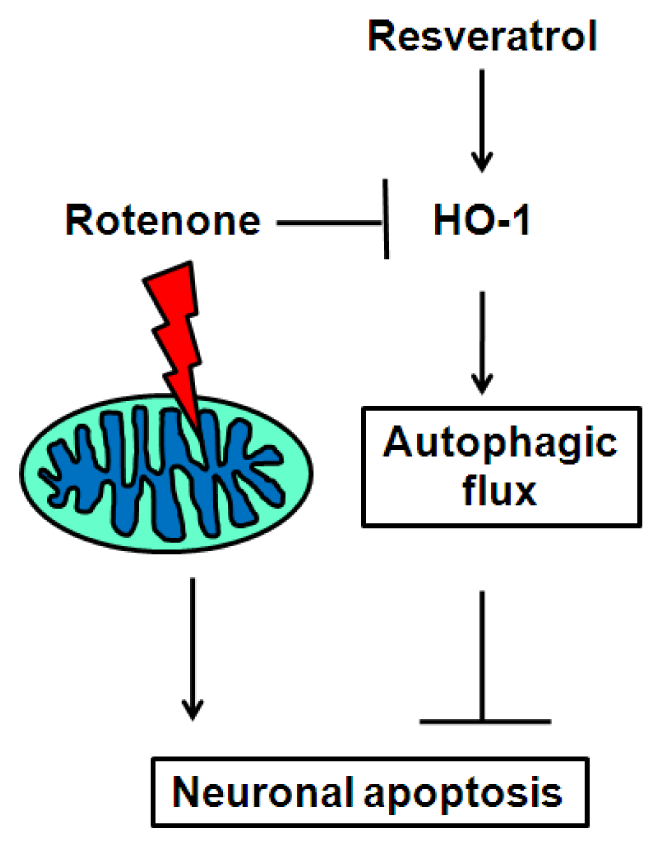
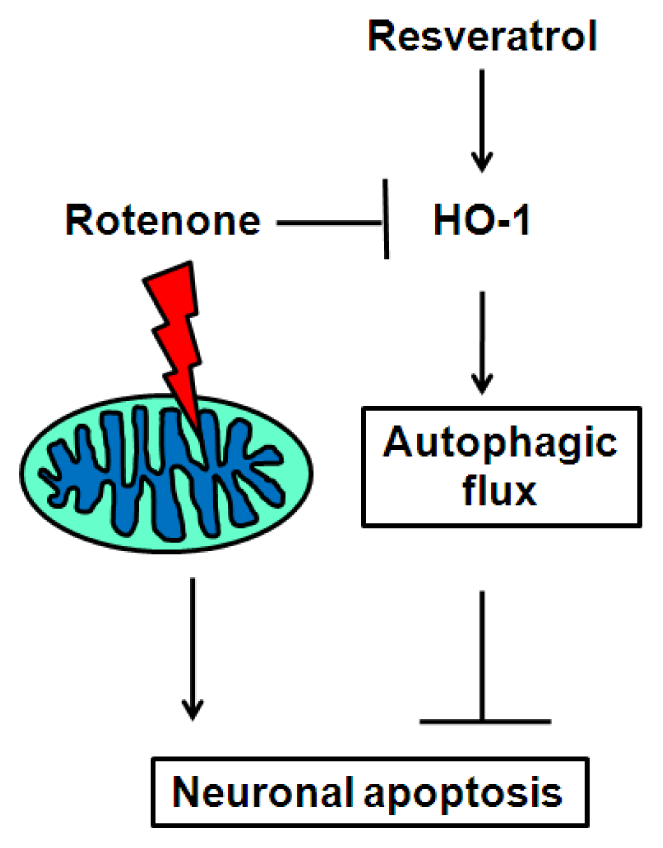
2.5. Discussion
3. Experimental section
3.1. Animals and Experimental Model of Parkinson’s Disease
3.2. Cell Culture
3.3. Isolation of Striatal Mitochondria and Detection of Oxidized Proteins
3.4. Immunohistochemical Staining
3.5. Electron Microscopy
3.6. Cell-Survival Assay
3.7. Immunoblotting
3.8. Immunocytochemistry
3.9. Flow Cytometry
4. Conclusions
Supplementary Information
ijms-15-01625-s001.pdfAcknowledgments
Conflicts of Interest
References
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic Biol. Med 2013, 62, 132–144. [Google Scholar]
- Sherer, T.B.; Betarbet, R.; Greenamyre, J.T. Pathogenesis of Parkinson’s disease. Curr. Opin. Investig.Drugs 2001, 2, 657–662. [Google Scholar]
- Jellinger, K.A. Neuropathology of sporadic Parkinson’s disease: Evaluation and changes of concepts. Mov. Disord 2012, 27, 8–30. [Google Scholar]
- Minami, Y.; Yamamoto, R.; Nishikouri, M.; Fukao, A.; Hisamichi, S. Mortality and cancer incidence in patients with Parkinson’s disease. J. Neurol 2000, 247, 429–434. [Google Scholar]
- Posada, I.J.; Benito-Leon, J.; Louis, E.D.; Trincado, R.; Villarejo, A.; Medrano, M.J.; Bermejo-Pareja, F. Mortality from Parkinson’s disease: A population-based prospective study (NEDICES). Mov. Disord 2011, 26, 2522–2529. [Google Scholar]
- Greenamyre, J.T.; Sherer, T.B.; Betarbet, R.; Panov, A.V. Complex I and Parkinson’s disease. IUBMB Life 2001, 52, 135–141. [Google Scholar]
- Lin, T.K.; Liou, C.W.; Chen, S.D.; Chuang, Y.C.; Tiao, M.M.; Wang, P.W.; Chen, J.B.; Chuang, J.H. Mitochondrial dysfunction and biogenesis in the pathogenesis of Parkinson’s disease. Chang Gung Med. J 2009, 32, 589–599. [Google Scholar]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar]
- Beal, M.F. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann. N. Y. Acad. Sci 2003, 991, 120–131. [Google Scholar]
- Choi, W.S.; Palmiter, R.D.; Xia, Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol 2011, 192, 873–882. [Google Scholar]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar]
- Bueler, H. Mitochondrial dynamics, cell death and the pathogenesis of Parkinson’s disease. Apoptosis 2010, 15, 1336–1353. [Google Scholar]
- Perier, C.; Bove, J.; Vila, M. Mitochondria and programmed cell death in Parkinson’s disease: Apoptosis and beyond. Antioxid. Redox Signal 2012, 16, 883–895. [Google Scholar]
- Yao, Z.; Wood, N.W. Cell death pathways in Parkinson’s disease: Role of mitochondria. Antioxid. Redox Signal 2009, 11, 2135–2149. [Google Scholar]
- Miller, R.L.; James-Kracke, M.; Sun, G.Y.; Sun, A.Y. Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem. Res 2009, 34, 55–65. [Google Scholar]
- Radad, K.; Rausch, W.D.; Gille, G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem. Int 2006, 49, 379–386. [Google Scholar]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol 2008, 7, 97–109. [Google Scholar]
- Niranjan, R. The Role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson’s disease: Focus on astrocytes. Mol. Neurobiol 2013. [Epub ahead of print]. [Google Scholar]
- Emborg, M.E. Evaluation of animal models of Parkinson’s disease for neuroprotective strategies. J. Neurosci. Methods 2004, 139, 121–143. [Google Scholar]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol 1997, 12, 25–31. [Google Scholar]
- Celardo, I.; Martins, L.M.; Gandhi, S. Unravelling mitochondrial pathways to Parkinson’s disease. Br. J. Pharmacol 2013. [Google Scholar] [CrossRef]
- Janda, E.; Isidoro, C.; Carresi, C.; Mollace, V. Defective autophagy in Parkinson’s disease: Role of oxidative stress. Mol. Neurobiol 2012, 46, 639–661. [Google Scholar]
- Tassoni, A.; Fornale, S.; Franceschetti, M.; Musiani, F.; Michael, A.J.; Perry, B.; Bagni, N. Jasmonates and Na-orthovanadate promote resveratrol production in Vitis vinifera cv. Barbera cell cultures. New Phytol 2005, 166, 895–905. [Google Scholar]
- Harikumar, K.B.; Aggarwal, B.B. Resveratrol: A multitargeted agent for age-associated chronic diseases. Cell Cycle 2008, 7, 1020–1035. [Google Scholar]
- Soleas, G.J.; Diamandis, E.P.; Goldberg, D.M. Resveratrol: A molecule whose time has come? And gone? Clin. Biochem 1997, 30, 91–113. [Google Scholar]
- Lin, T.K.; Huang, L.T.; Huang, Y.H.; Tiao, M.M.; Tang, K.S.; Liou, C.W. The effect of the red wine polyphenol resveratrol on a rat model of biliary obstructed cholestasis: Involvement of anti-apoptotic signalling, mitochondrial biogenesis and the induction of autophagy. Apoptosis 2012, 17, 871–879. [Google Scholar]
- Hsu, K.F.; Wu, C.L.; Huang, S.C.; Wu, C.M.; Hsiao, J.R.; Yo, Y.T.; Chen, Y.H.; Shiau, A.L.; Chou, C.Y. Cathepsin L mediates resveratrol-induced autophagy and apoptotic cell death in cervical cancer cells. Autophagy 2009, 5, 451–460. [Google Scholar]
- Trincheri, N.F.; Follo, C.; Nicotra, G.; Peracchio, C.; Castino, R.; Isidoro, C. Resveratrol-induced apoptosis depends on the lipid kinase activity of Vps34 and on the formation of autophagolysosomes. Carcinogenesis 2008, 29, 381–389. [Google Scholar]
- Trincheri, N.F.; Nicotra, G.; Follo, C.; Castino, R.; Isidoro, C. Resveratrol induces cell death in colorectal cancer cells by a novel pathway involving lysosomal cathepsin D. Carcinogenesis 2007, 28, 922–931. [Google Scholar]
- Menzies, F.M.; Moreau, K.; Rubinsztein, D.C. Protein misfolding disorders and macroautophagy. Curr. Opin. Cell Biol 2011, 23, 190–197. [Google Scholar]
- Okamoto, K.; Kondo-Okamoto, N. Mitochondria and autophagy: Critical interplay between the two homeostats. Biochim. Biophys. Acta 2012, 1820, 595–600. [Google Scholar]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev 2006, 86, 583–650. [Google Scholar]
- Ryter, S.W.; Choi, A.M. Heme oxygenase-1: Molecular mechanisms of gene expression in oxygen-related stress. Antioxid Redox Signal 2002, 4, 625–632. [Google Scholar]
- Hu, C.M.; Chen, Y.H.; Chiang, M.T.; Chau, L.Y. Heme oxygenase-1 inhibits angiotensin II-induced cardiac hypertrophy in vitro and in vivo. Circulation 2004, 110, 309–316. [Google Scholar]
- Le, W.D.; Xie, W.J.; Appel, S.H. Protective role of heme oxygenase-1 in oxidative stress-induced neuronal injury. J. Neurosci. Res 1999, 56, 652–658. [Google Scholar]
- Mateo, I.; Infante, J.; Sanchez-Juan, P.; Garcia-Gorostiaga, I.; Rodriguez-Rodriguez, E.; Vazquez-Higuera, J.L.; Berciano, J.; Combarros, O. Serum heme oxygenase-1 levels are increased in Parkinson’s disease but not in Alzheimer’s disease. Acta Neurol. Scand 2010, 121, 136–138. [Google Scholar]
- Schipper, H.M. Heme oxygenase expression in human central nervous system disorders. Free Radic Biol. Med 2004, 37, 1995–2011. [Google Scholar]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem 2009, 110, 469–485. [Google Scholar]
- Kimura, S.; Fujita, N.; Noda, T.; Yoshimori, T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol 2009, 452, 1–12. [Google Scholar]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol 2005, 171, 603–614. [Google Scholar]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar]
- Espada, S.; Ortega, F.; Molina-Jijon, E.; Rojo, A.I.; Perez-Sen, R.; Pedraza-Chaverri, J.; Miras-Portugal, M.T.; Cuadrado, A. The purinergic P2Y(13) receptor activates the Nrf2/HO-1 axis and protects against oxidative stress-induced neuronal death. Free Radic Biol. Med 2010, 49, 416–426. [Google Scholar]
- Shi, Y.; Liang, X.C.; Zhang, H.; Wu, Q.L.; Qu, L.; Sun, Q. Quercetin protects rat dorsal root ganglion neurons against high glucose-induced injury in vitro through Nrf-2/HO-1 activation and NF-kappaB inhibition. Acta Pharmacol. Sin 2013, 34, 1140–1148. [Google Scholar]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res 2005, 65, 3336–3346. [Google Scholar]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. J. Neurosci 2012, 32, 7585–7593. [Google Scholar]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar]
- Kundu, M.; Thompson, C.B. Macroautophagy versus mitochondrial autophagy: A question of fate? Cell Death Differ 2005, 12 Suppl 2, 1484–1489. [Google Scholar]
- Thorburn, A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis 2008, 13, 1–9. [Google Scholar]
- Dadakhujaev, S.; Noh, H.S.; Jung, E.J.; Cha, J.Y.; Baek, S.M.; Ha, J.H.; Kim, D.R. Autophagy protects the rotenone-induced cell death in alpha-synuclein overexpressing SH-SY5Y cells. Neurosci. Lett 2010, 472, 47–52. [Google Scholar]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar]
- Wu, Y.; Li, X.; Xie, W.; Jankovic, J.; Le, W.; Pan, T. Neuroprotection of deferoxamine on rotenone-induced injury via accumulation of HIF-1 alpha and induction of autophagy in SH-SY5Y cells. Neurochem. Int 2010, 57, 198–205. [Google Scholar]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 2011, 19, 163–174. [Google Scholar]
- Filomeni, G.; Graziani, I.; de Zio, D.; Dini, L.; Centonze, D.; Rotilio, G.; Ciriolo, M.R. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging 2012, 33, 767–785. [Google Scholar]
- Ryu, E.J.; Harding, H.P.; Angelastro, J.M.; Vitolo, O.V.; Ron, D.; Greene, L.A. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J. Neurosci 2002, 22, 10690–10698. [Google Scholar]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci 2000, 3, 1301–1306. [Google Scholar]
- Lin, T.K.; Cheng, C.H.; Chen, S.D.; Liou, C.W.; Huang, C.R.; Chuang, Y.C. Mitochondrial dysfunction and oxidative stress promote apoptotic cell death in the striatum via cytochrome c/caspase-3 signaling cascade following chronic rotenone intoxication in rats. Int. J. Mol. Sci 2012, 13, 8722–8739. [Google Scholar]
- Mader, B.J.; Pivtoraiko, V.N.; Flippo, H.M.; Klocke, B.J.; Roth, K.A.; Mangieri, L.R.; Shacka, J.J. Rotenone inhibits autophagic flux prior to inducing cell death. ACS Chem. Neurosci 2012, 3, 1063–1072. [Google Scholar]
- Punnonen, E.L.; Autio, S.; Marjomaki, V.S.; Reunanen, H. Autophagy, cathepsin L transport, and acidification in cultured rat fibroblasts. J. Histochem. Cytochem 1992, 40, 1579–1587. [Google Scholar]
- Uchiyama, Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch. Histol. Cytol 2001, 64, 233–246. [Google Scholar]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar]
- Panahian, N.; Yoshiura, M.; Maines, M.D. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J. Neurochem 1999, 72, 1187–1203. [Google Scholar]
- Shah, Z.A.; Nada, S.E.; Dore, S. Heme oxygenase 1, beneficial role in permanent ischemic stroke and in Gingko biloba (EGb 761) neuroprotection. Neuroscience 2011, 180, 248–255. [Google Scholar]
- Hung, S.Y.; Liou, H.C.; Kang, K.H.; Wu, R.M.; Wen, C.C.; Fu, W.M. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol. Pharmacol 2008, 74, 1564–1575. [Google Scholar]
- Carchman, E.H.; Rao, J.; Loughran, P.A.; Rosengart, M.R.; Zuckerbraun, B.S. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology 2011, 53, 2053–2062. [Google Scholar]
- Waltz, P.; Carchman, E.H.; Young, A.C.; Rao, J.; Rosengart, M.R.; Kaczorowski, D.; Zuckerbraun, B.S. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy 2011, 7, 315–320. [Google Scholar]
- Bolisetty, S.; Traylor, A.M.; Kim, J.; Joseph, R.; Ricart, K.; Landar, A.; Agarwal, A. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J. Am. Soc. Nephrol 2010, 21, 1702–1712. [Google Scholar]
- Chen, S.D.; Lin, T.K.; Lin, J.W.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Activation of calcium/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res 2010, 88, 3144–3154. [Google Scholar]




© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lin, T.-K.; Chen, S.-D.; Chuang, Y.-C.; Lin, H.-Y.; Huang, C.-R.; Chuang, J.-H.; Wang, P.-W.; Huang, S.-T.; Tiao, M.-M.; Chen, J.-B.; et al. Resveratrol Partially Prevents Rotenone-Induced Neurotoxicity in Dopaminergic SH-SY5Y Cells through Induction of Heme Oxygenase-1 Dependent Autophagy. Int. J. Mol. Sci. 2014, 15, 1625-1646. https://doi.org/10.3390/ijms15011625
Lin T-K, Chen S-D, Chuang Y-C, Lin H-Y, Huang C-R, Chuang J-H, Wang P-W, Huang S-T, Tiao M-M, Chen J-B, et al. Resveratrol Partially Prevents Rotenone-Induced Neurotoxicity in Dopaminergic SH-SY5Y Cells through Induction of Heme Oxygenase-1 Dependent Autophagy. International Journal of Molecular Sciences. 2014; 15(1):1625-1646. https://doi.org/10.3390/ijms15011625
Chicago/Turabian StyleLin, Tsu-Kung, Shang-Der Chen, Yao-Chung Chuang, Hung-Yu Lin, Chi-Ren Huang, Jiin-Haur Chuang, Pei-Wen Wang, Sheng-Teng Huang, Mao-Meng Tiao, Jin-Bor Chen, and et al. 2014. "Resveratrol Partially Prevents Rotenone-Induced Neurotoxicity in Dopaminergic SH-SY5Y Cells through Induction of Heme Oxygenase-1 Dependent Autophagy" International Journal of Molecular Sciences 15, no. 1: 1625-1646. https://doi.org/10.3390/ijms15011625
APA StyleLin, T.-K., Chen, S.-D., Chuang, Y.-C., Lin, H.-Y., Huang, C.-R., Chuang, J.-H., Wang, P.-W., Huang, S.-T., Tiao, M.-M., Chen, J.-B., & Liou, C.-W. (2014). Resveratrol Partially Prevents Rotenone-Induced Neurotoxicity in Dopaminergic SH-SY5Y Cells through Induction of Heme Oxygenase-1 Dependent Autophagy. International Journal of Molecular Sciences, 15(1), 1625-1646. https://doi.org/10.3390/ijms15011625