Spectroscopic Studies of Model Photo-Receptors: Validation of a Nanosecond Time-Resolved Micro-Spectrophotometer Design Using Photoactive Yellow Protein and α-Phycoerythrocyanin

Abstract
:
1. Introduction
2. Results and Discussions
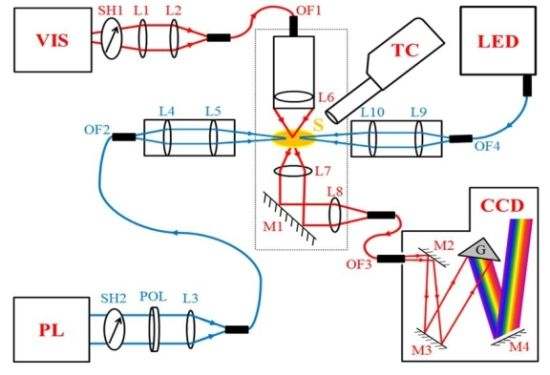
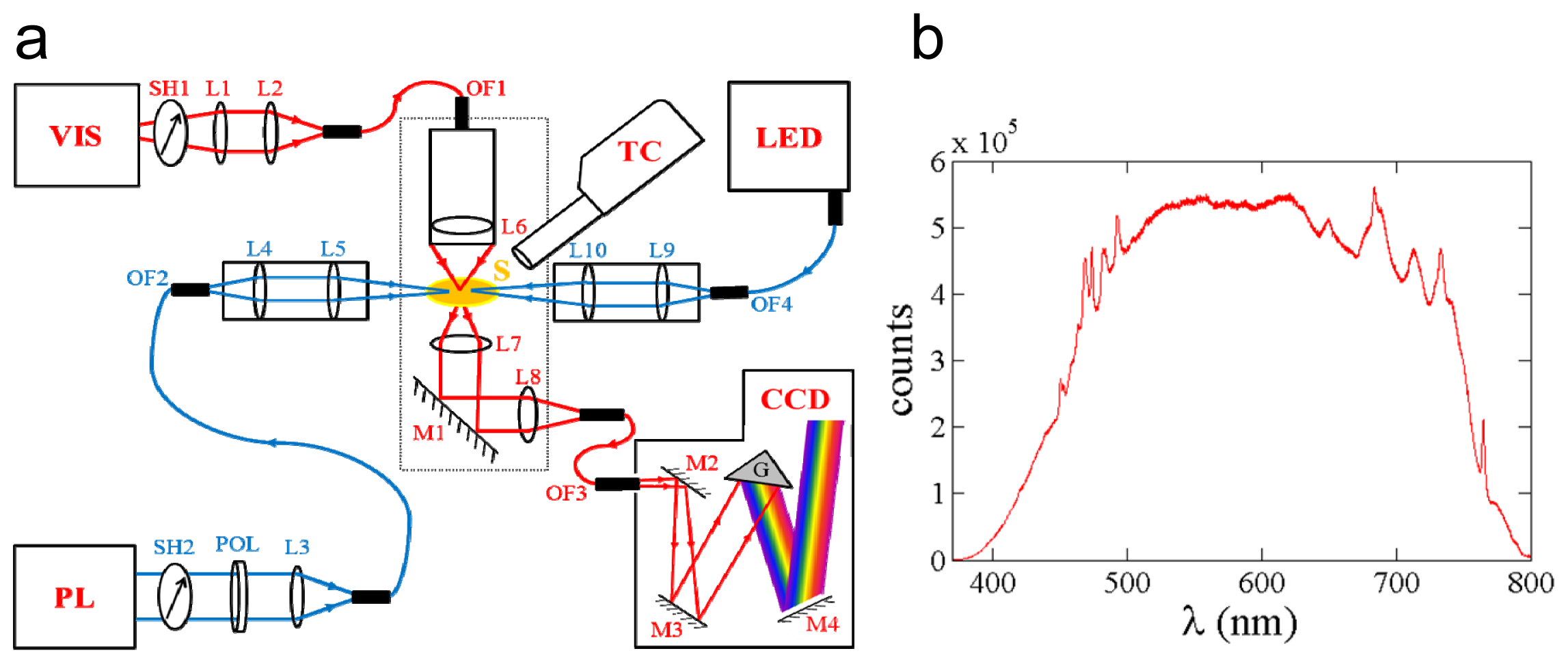
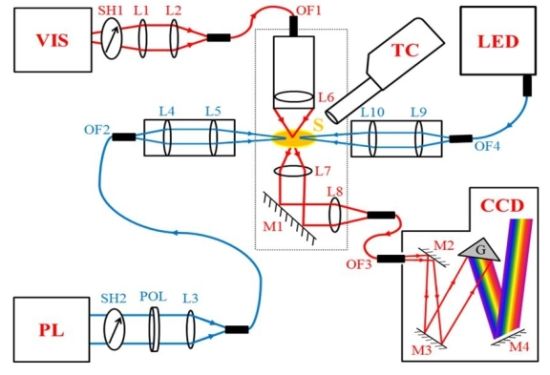
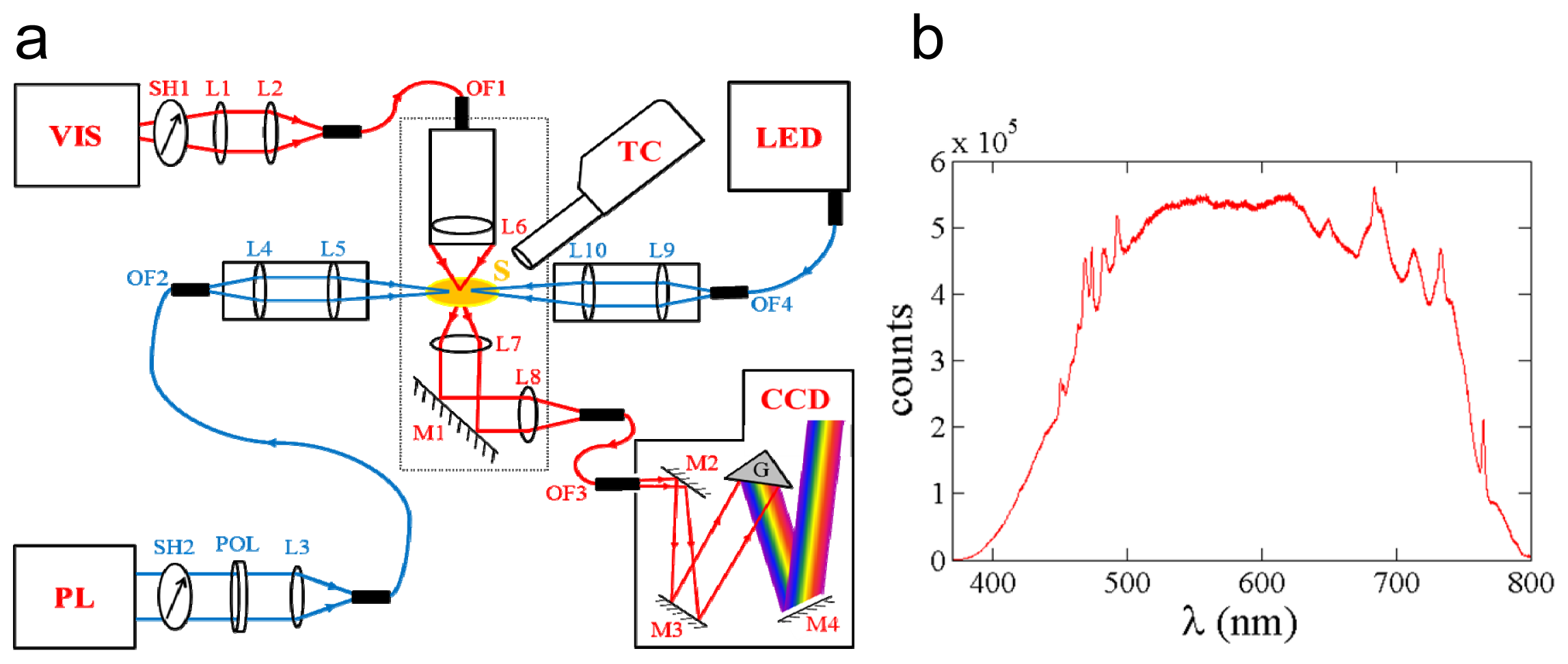
2.1. Design of the MSP
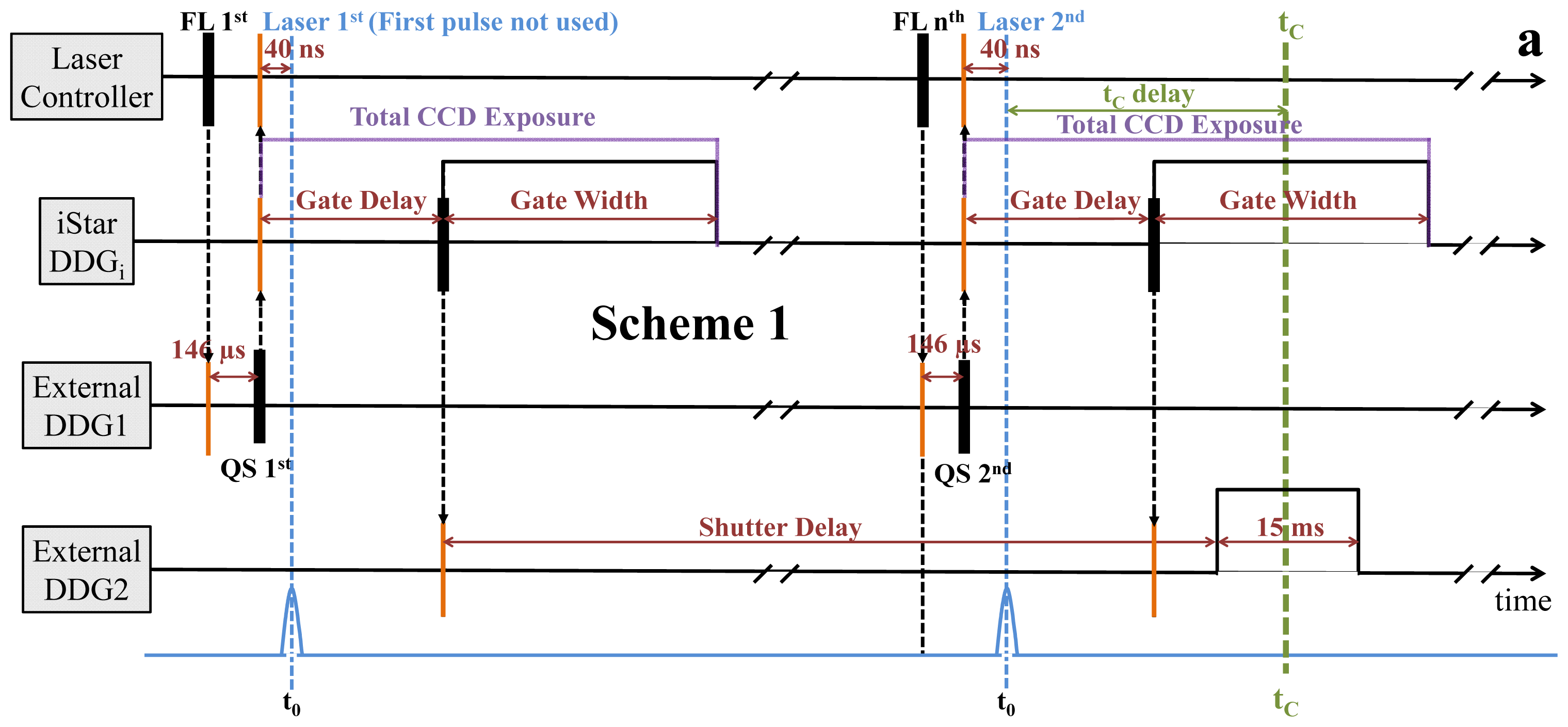
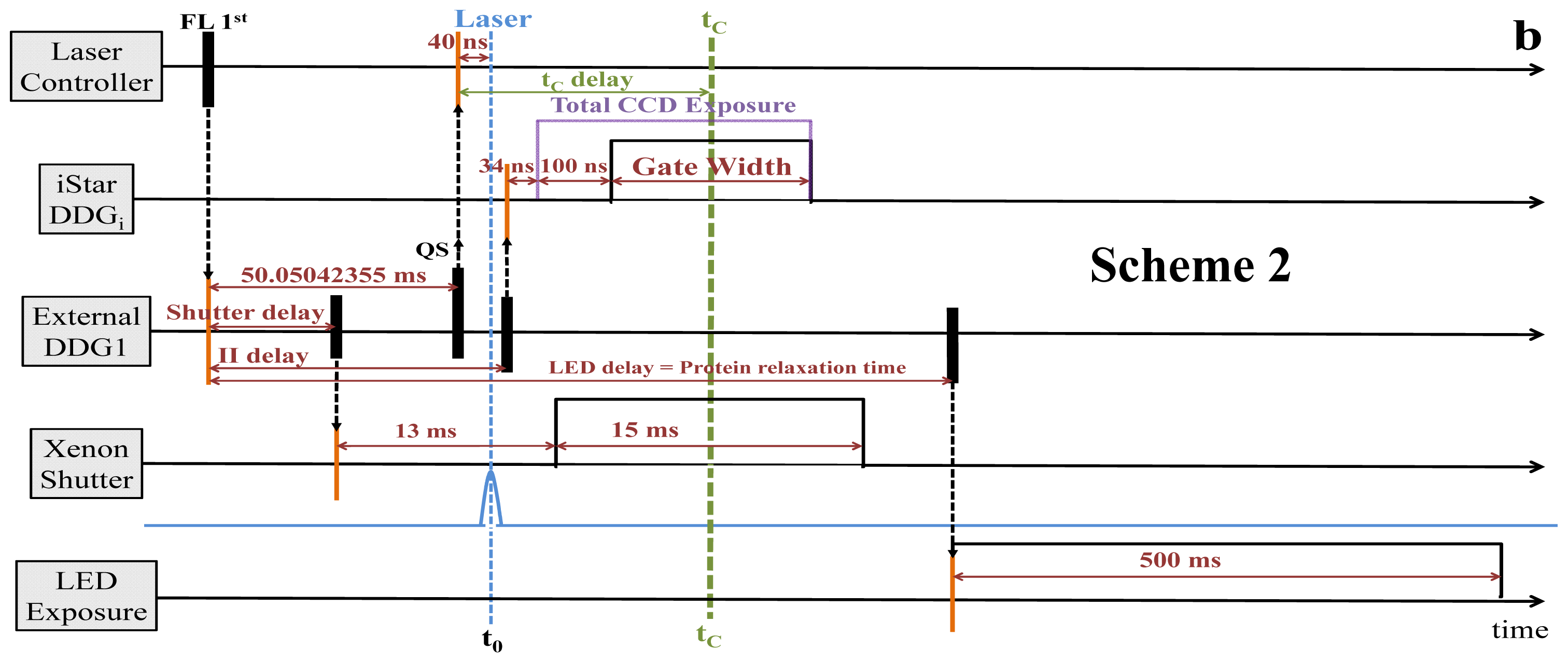
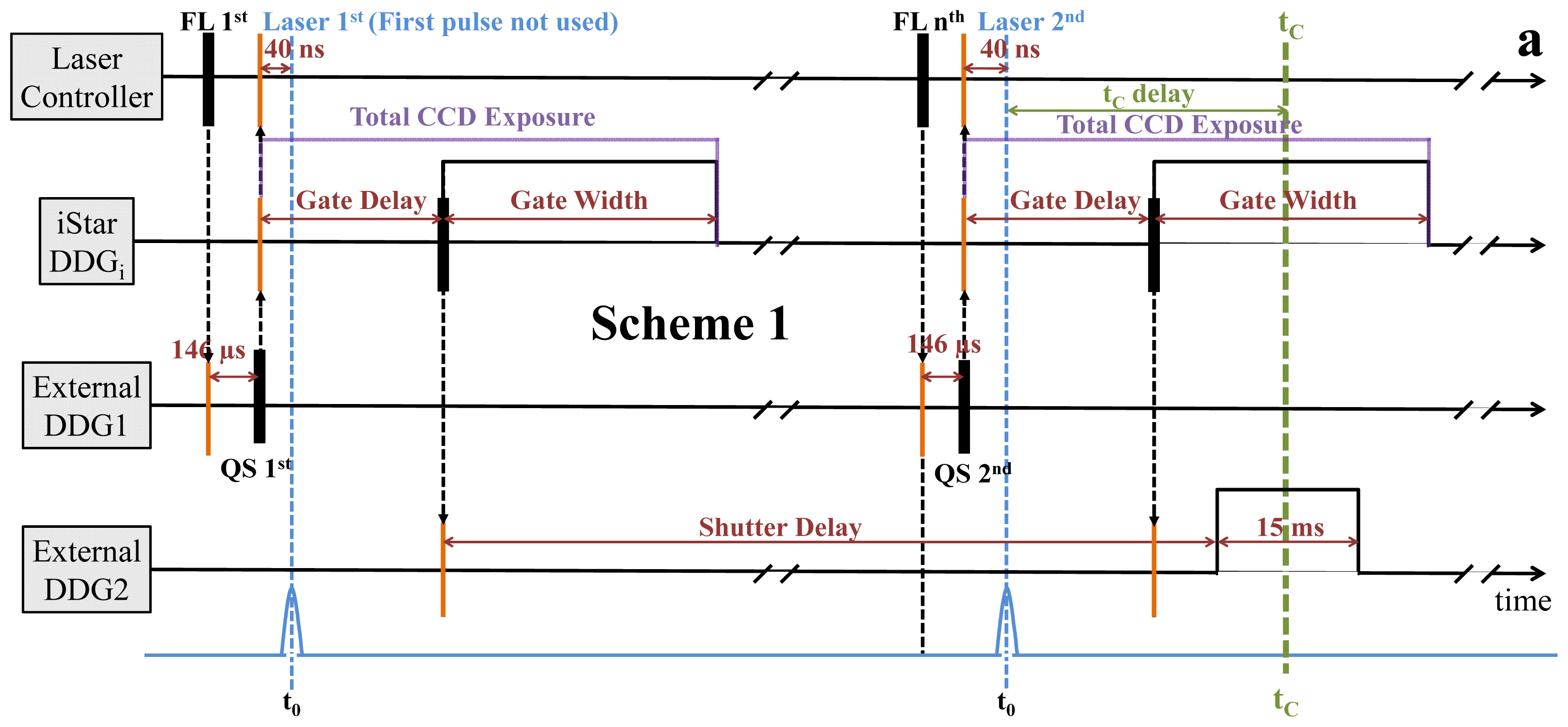
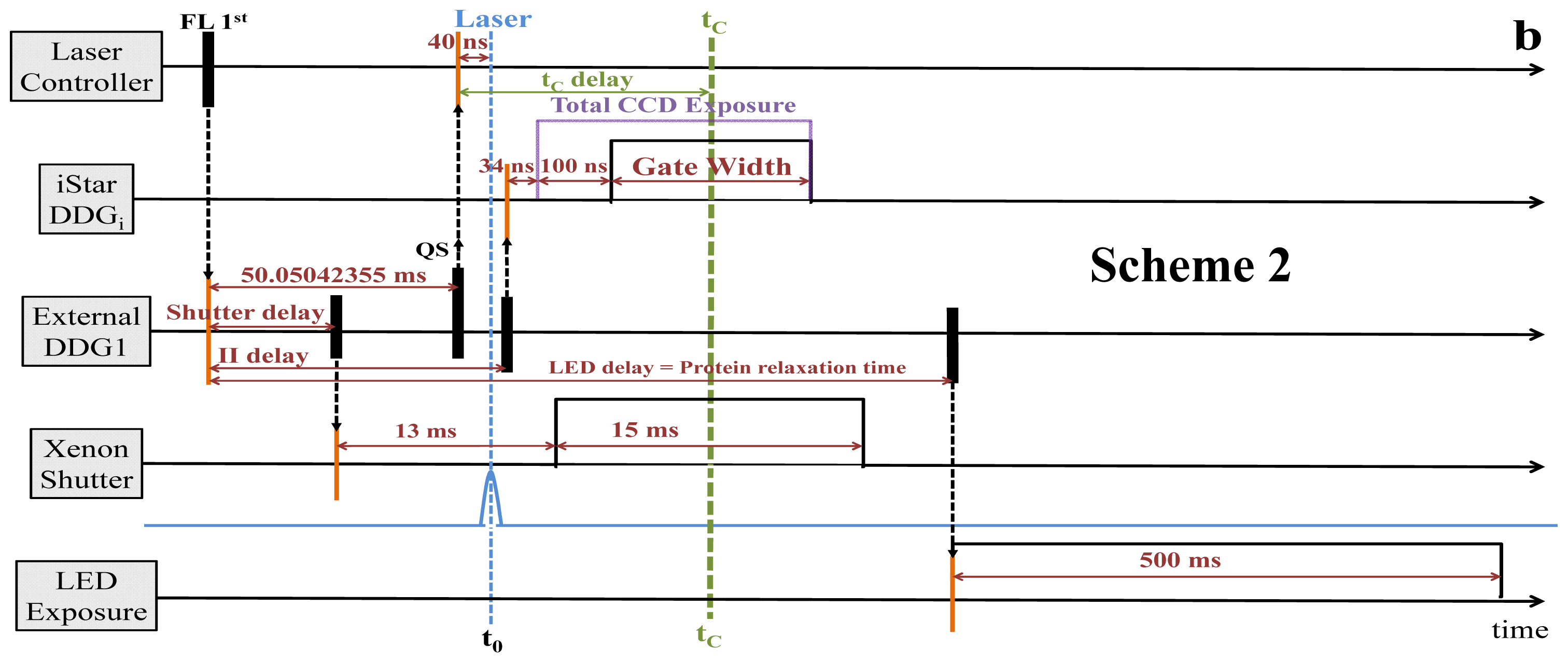
2.2. Synchronization and Computer Control
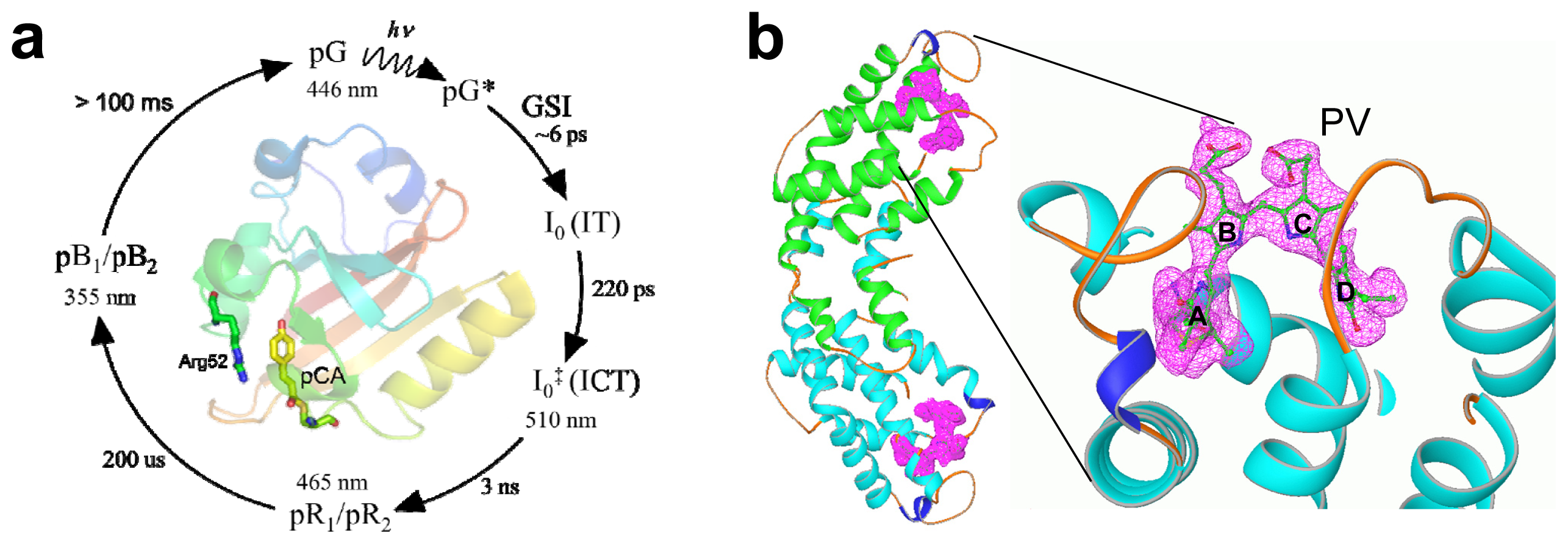
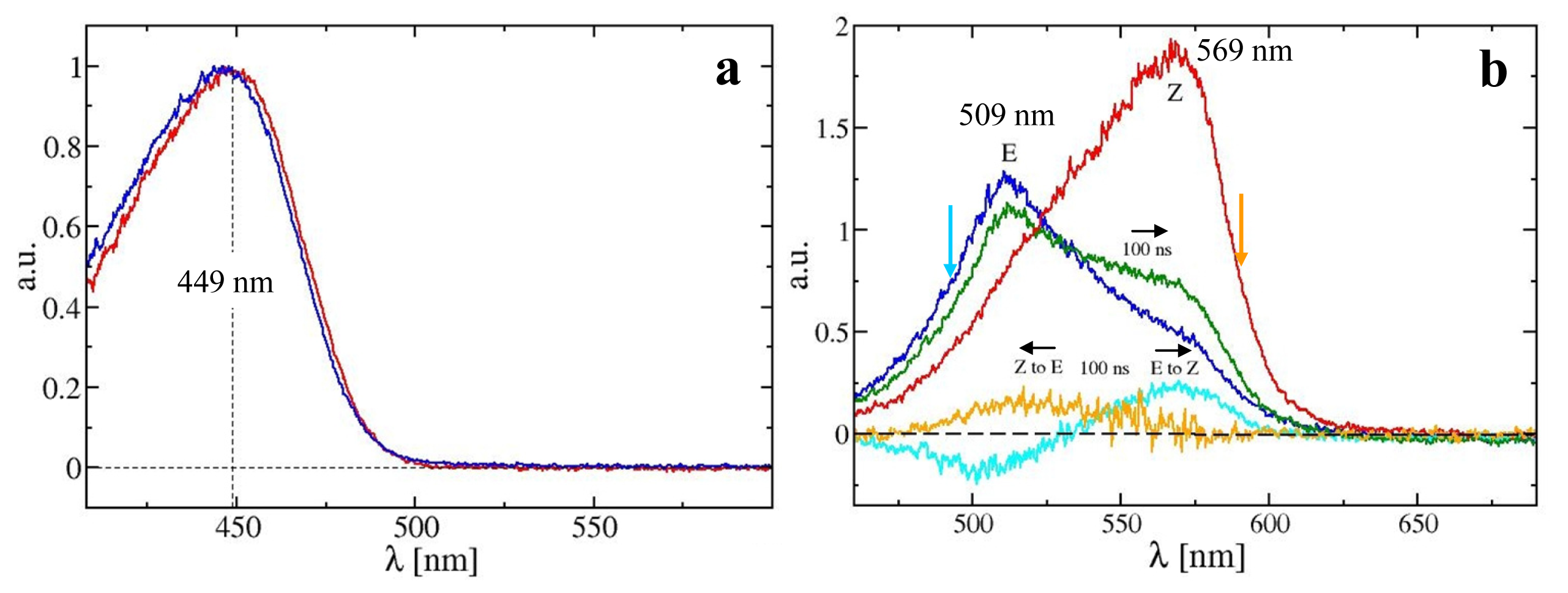
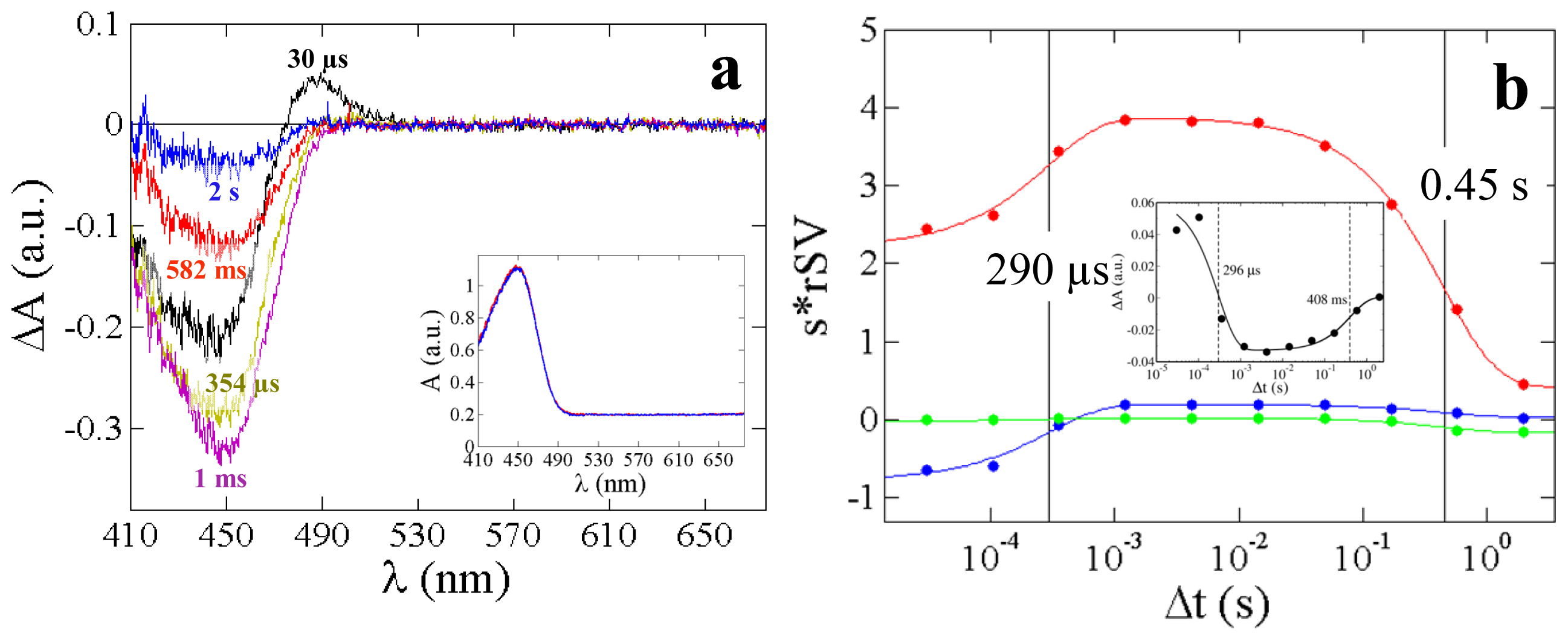
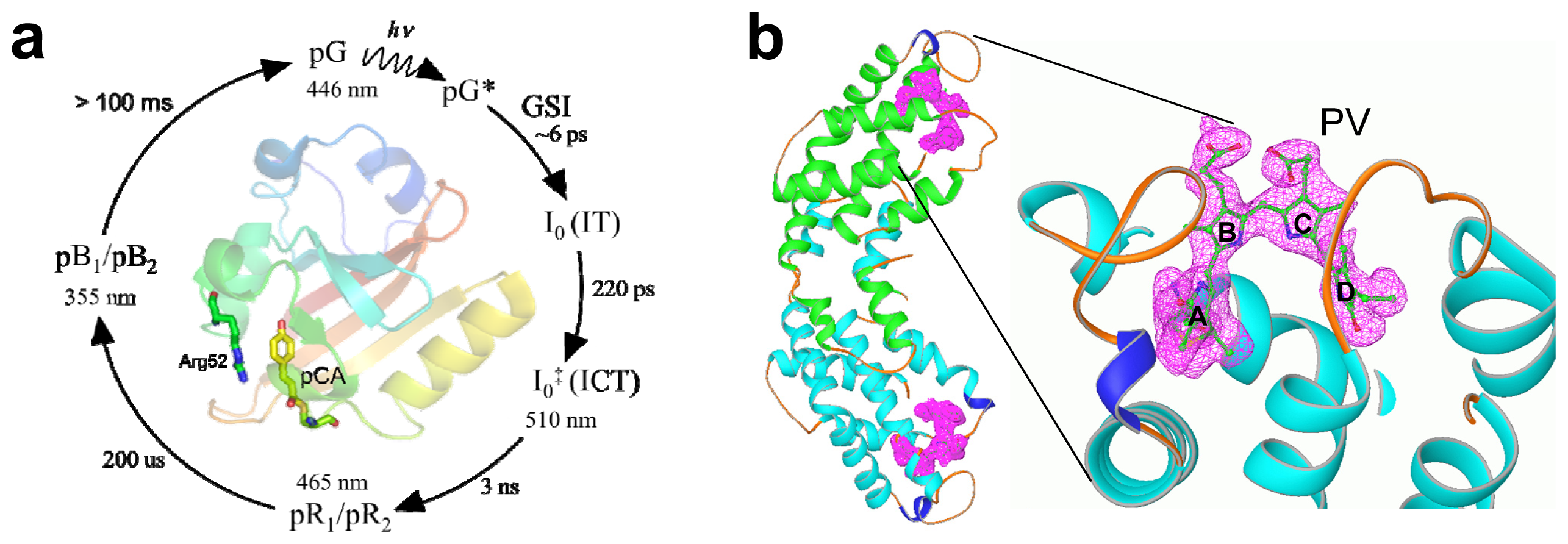
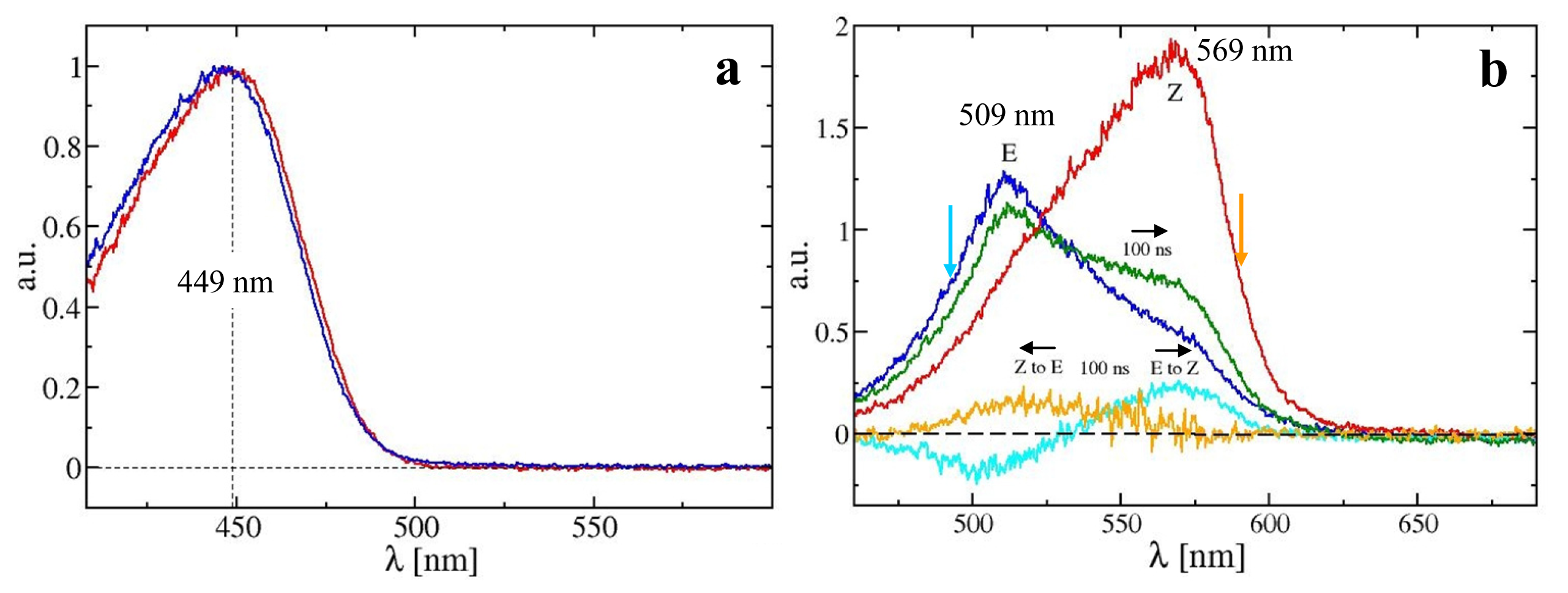
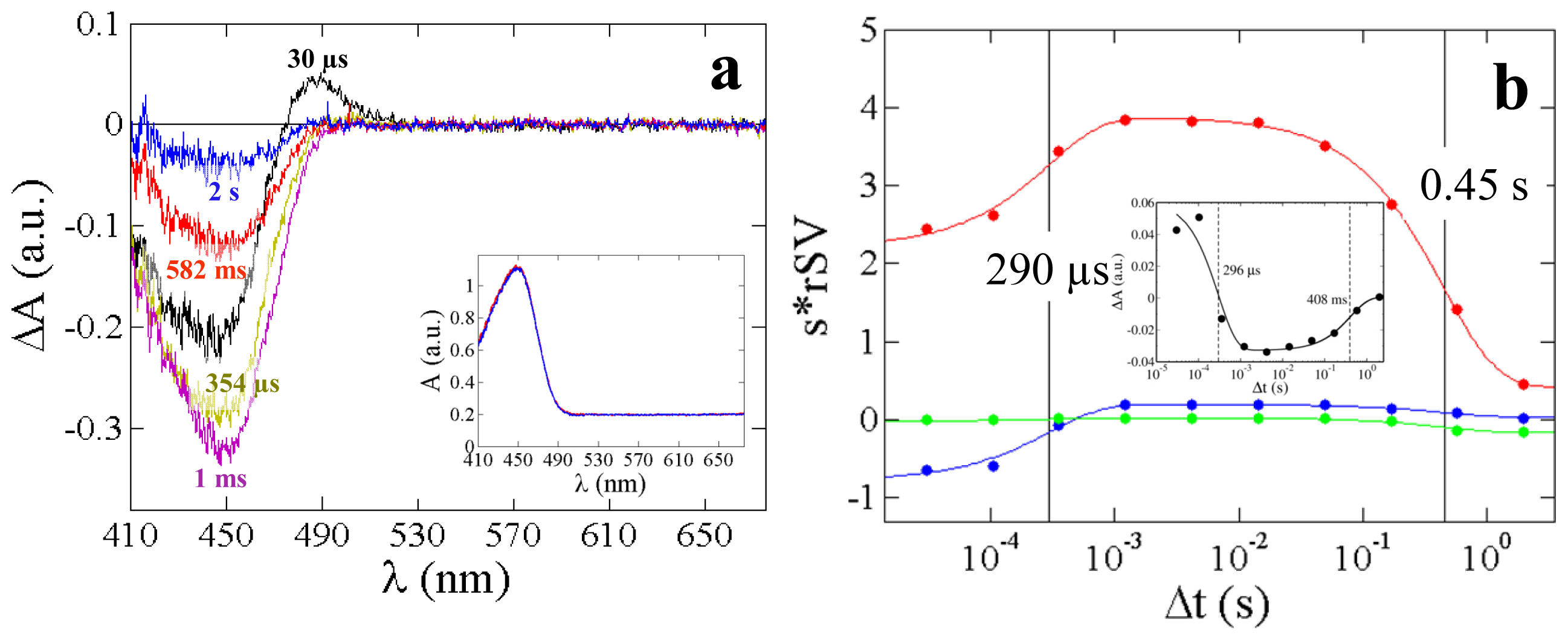
2.3. PYP Spectra
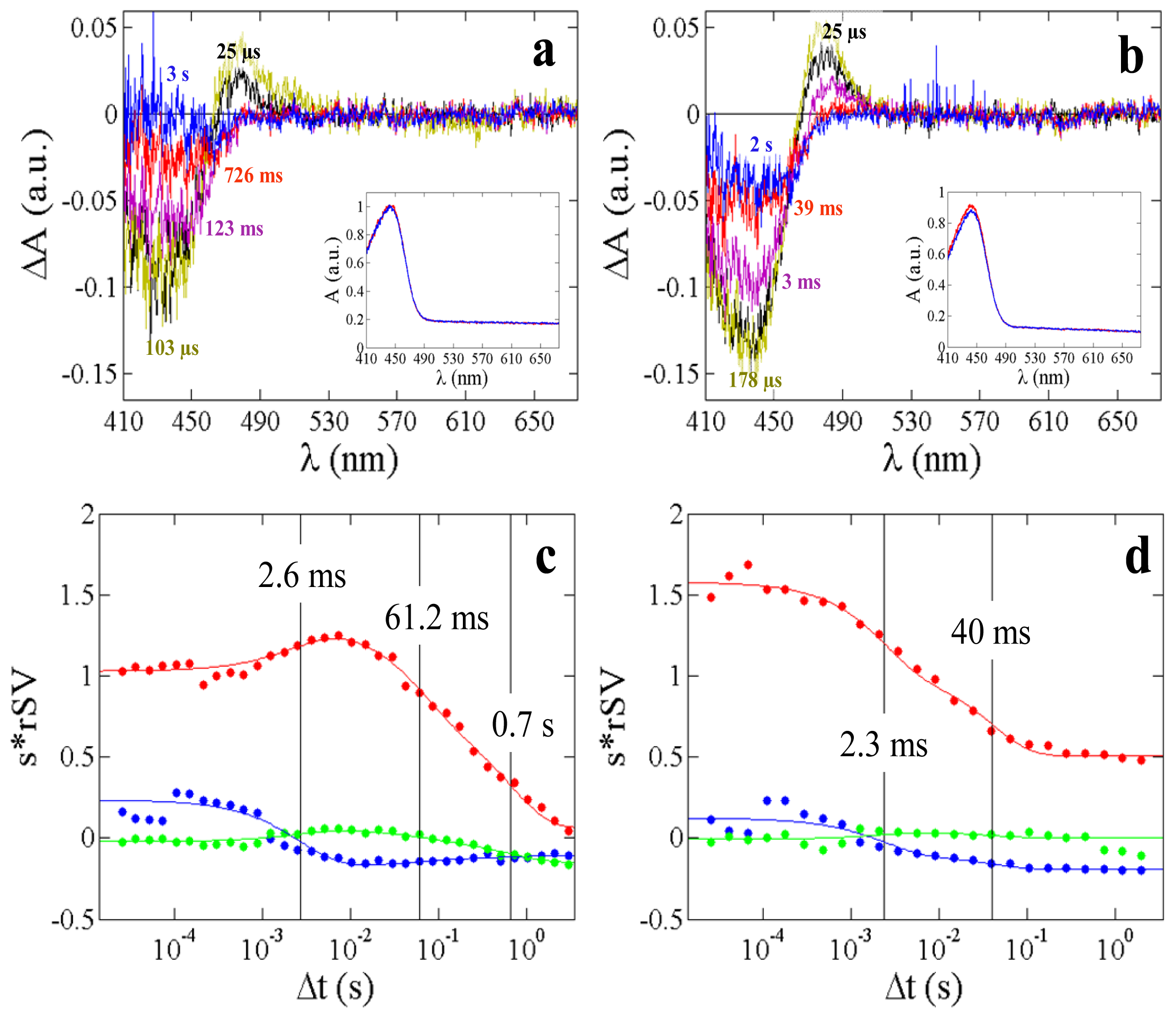
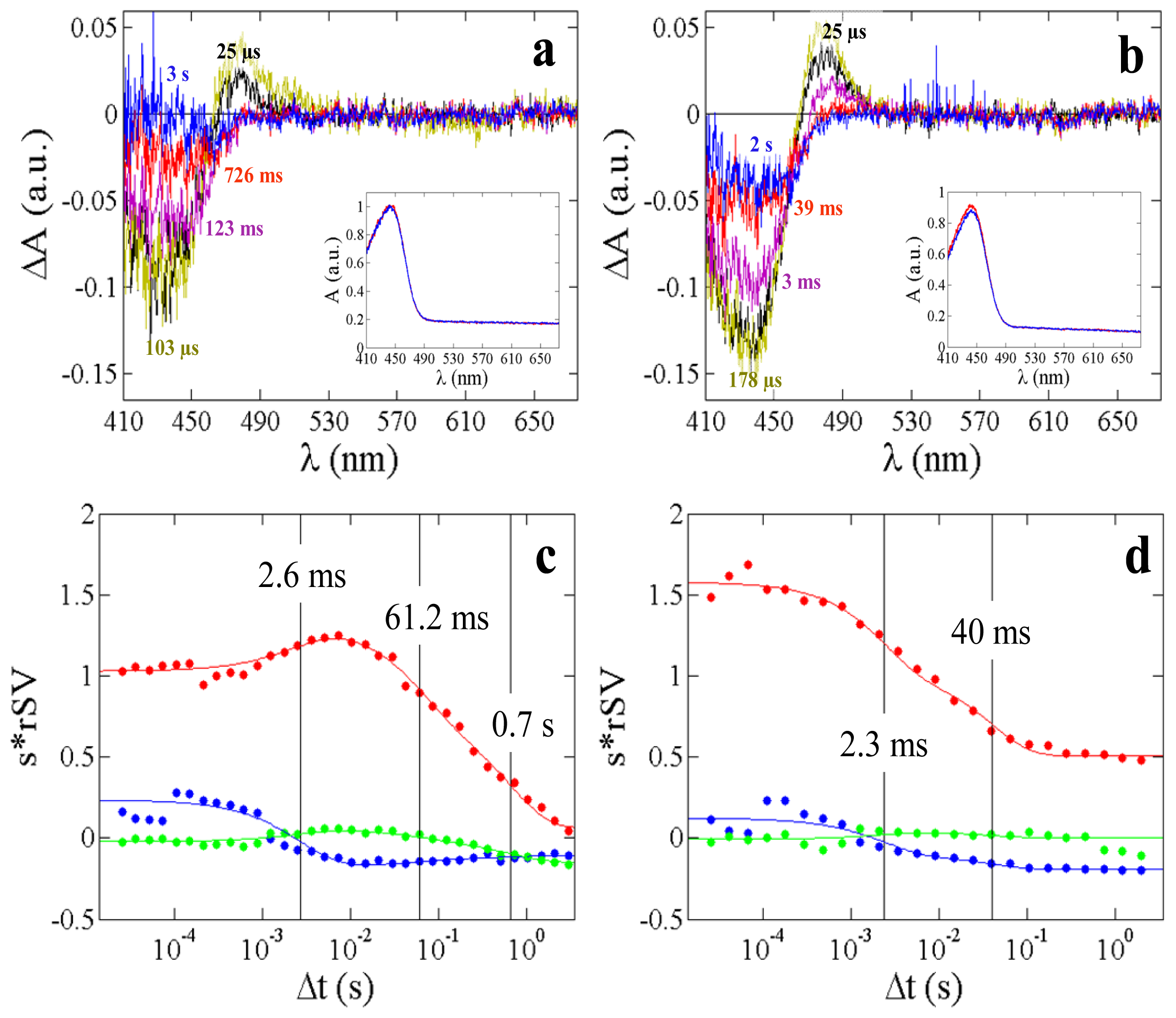
2.4. α-Phycoerythrocyanin (α-PEC) Spectra
2.5. Performance and Outlook
3. Experimental Section
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Fenimore, P.W.; Frauenfelder, H.; McMahon, B.H.; Parak, F.G. Slaving: Solvent fluctuations dominate protein dynamics and functions. Proc. Natl. Acad. Sci. USA 2002, 99, 16047–16051. [Google Scholar]
- Goelder, M.; Givens, R. Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules; Wiley VCH: Weinheim, Germany, 2005. [Google Scholar]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics, 4th ed.; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Rupp, B. Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology; Garland Science: New York, NY, USA; p. 2009.
- Henry, E.R.; Hofrichter, J. Singular value decomposition–application to analysis of experimental-data. Method Enzymol 1992, 210, 129–192. [Google Scholar]
- Van Stokkum, I.H.; Larsen, D.S.; van Grondelle, R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta 2004, 1657, 82–104. [Google Scholar]
- Van Amerongen, H.; van Grondelle, R. Transient absorption spectroscopy in study of processes and dynamics in biology. Methods Enzymol 1995, 246, 201–226. [Google Scholar]
- Moffat, K. Time-resolved biochemical crystallography: A mechanistic perspective. Chem. Rev 2001, 101, 1569–1581. [Google Scholar]
- Moffat, K.; Chen, Y.; Ng, K.M.; Mcree, D.; Getzoff, E.D. Time-resolved crystallography-principles, problems and practice. Philos. T. R. Soc. A 1992, 340, 175–189. [Google Scholar]
- Moffat, K. Time-resolved macromolecular crystallography. Annu. Rev. Biophys. Biophys. Chem 1989, 18, 309–332. [Google Scholar]
- Schmidt, M. Structure based enzyme kinetics by time-resolved X-ray crystallography. In Ultrashort Laser Pulses in Medicine and Biology; Springer-Verlag: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Schmidt, M.; Rajagopal, S.; Ren, Z.; Moffat, K. Application of singular value decomposition to the analysis of time-resolved macromolecular X-ray data. Biophys. J 2003, 84, 2112–2129. [Google Scholar]
- Schmidt, M.; Pahl, R.; Srajer, V.; Anderson, S.; Ren, Z.; Ihee, H.; Rajagopal, S.; Moffat, K. Protein kinetics: Structures of intermediates and reaction mechanism from time-resolved X-ray data. Proc. Natl. Acad. Sci. USA 2004, 101, 4799–4804. [Google Scholar]
- Schmidt, M.; Ihee, H.; Pahl, R.; Srajer, V. Protein-ligand interaction probed by time-resolved crystallography. Methods Mol. Biol 2005, 305, 115–154. [Google Scholar]
- Schmidt, M.; Srajer, V.; Henning, R.; Ihee, H.; Purwar, N.; Tenboer, J.; Tripathi, S. Protein energy landscapes determined by 5-dimensional crystallography. Acta Crystallogr. D Biol. Crystallogr 2013. submitted for publication. [Google Scholar]
- Schmidt, M.; Graber, T.; Henning, R.; Srajer, V. Five-dimensional crystallography. Acta Crystallogr. Sect. A Found. Crystallogr 2010, 66, 198–206. [Google Scholar]
- Ursby, T.; Weik, M.; Fioravanti, E.; Delarue, M.; Goeldner, M.; Bourgeois, D. Cryophotolysis of caged compounds: A technique for trapping intermediate states in protein crystals. Acta Crystallogr. Sect. D Biol. Crystallogr 2002, 58, 607–614. [Google Scholar]
- Bourgeois, D.; Royant, A. Advances in kinetic protein crystallography. Curr. Opin. Struct. Biol 2005, 15, 538–547. [Google Scholar]
- Bourgeois, D.; de Rosay, E.; Katona, G. Kinetic crystallography: A tool for filming proteins in action. Biofutur 2007, 26/280, 48–51. [Google Scholar]
- Vernede, X.; Lavault, B.; Ohana, J.; Nurizzo, D.; Joly, J.; Jacquamet, L.; Felisaz, F.; Cipriani, F.; Bourgeois, D. UV laser-excited fluorescence as a tool for the visualization of protein crystals mounted in loops. Acta Crystallogr. Sect. D Biol. Crystallogr 2006, 62, 253–261. [Google Scholar]
- Ostermann, A.; Waschipky, R.; Parak, F.G.; Nienhaus, G.U. Ligand binding and conformational motions in myoglobin. Nature 2000, 404, 205–208. [Google Scholar]
- Nienhaus, K.; Ostermann, A.; Nienhaus, G.U.; Parak, F.G.; Schmidt, M. Ligand migration and protein fluctuations in myoglobin mutant L29W. Biochemistry 2005, 44, 5095–5105. [Google Scholar]
- Yang, X.; Ren, Z.; Kuk, J.; Moffat, K. Temperature-scan cryocrystallography reveals reaction intermediates in bacteriophytochrome. Nature 2011, 479, 428–432. [Google Scholar]
- Weik, M.; Colletier, J.P. Temperature-dependent macromolecular X-ray crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66, 437–446. [Google Scholar]
- Sanchez-Weatherby, J.; Bowler, M.W.; Huet, J.; Gobbo, A.; Felisaz, F.; Lavault, B.; Moya, R.; Kadlec, J.; Ravelli, R.B.; Cipriani, F. Improving diffraction by humidity control: A novel device compatible with X-ray beamlines. Acta Crystallogr. Sect. D Biol. Crystallogr 2009, 65, 1237–1246. [Google Scholar]
- McGeehan, J.; Ravelli, R.B.G.; Murray, J.W.; Owen, R.L.; Cipriani, F.; McSweeney, S.; Weik, M.; Garman, E.F. Colouring cryo-cooled crystals: Online microspectrophotometry. J. Synchrotron Radiat 2009, 16, 163–172. [Google Scholar]
- Owen, R.L.; Axford, D.; Nettleship, J.E.; Owens, R.J.; Robinson, J.I.; Morgan, A.W.; Dore, A.S.; Lebon, G.; Tate, C.G.; Fry, E.E.; et al. Outrunning free radicals in room-temperature macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 2012, 68, 810–818. [Google Scholar]
- Gibson, Q.H.; Meda, E.; Roughton, F.J. Studies on the kinetics of the reaction of human hemoglobin with oxygen. I. Boll. Della Soc. Italiana Biol. Sper 1953, 29, 1471–1472. [Google Scholar]
- Austin, R.H.; Beeson, K.W.; Eisenstein, L.; Frauenfelder, H.; Gunsalus, I.C. Dynamics of ligand binding to myoglobin. Biochemistry 1975, 14, 5355–5373. [Google Scholar]
- Chen, Y.; Srajer, V.; Ng, K.; Legrand, A.; Moffat, K. Optical monitoring of protein crystals in time-resolved X-ray experiments-microspectrophotometer design and performance. Rev. Sci. Instrum 1994, 65, 1506–1511. [Google Scholar]
- Bourgeois, D.; Vernede, X.; Adam, V.; Fioravanti, E.; Ursby, T. A microspectrophotometer for UV-visible absorption and fluorescence studies of protein crystals. J. Appl. Crystallogr 2002, 35, 319–326. [Google Scholar]
- Hadfield, A.; Hajdu, J. A fast and portable microspectrophotometer for protein crystallography. J. Appl. Crystallogr 1993, 26, 839–842. [Google Scholar]
- Owen, R.L.; Pearson, A.R.; Meents, A.; Boehler, P.; Thominet, V.; Schulze-Briese, C. A new on-axis multimode spectrometer for the macromolecular crystallography beamlines of the Swiss Light Source. J. Synchrotron Radiat 2009, 16, 173–182. [Google Scholar]
- Pearson, A.R.; Pahl, R.; Kovaleva, E.G.; Davidson, V.L.; Wilmot, C.M. Tracking X-ray-derived redox changes in crystals of a methylamine dehydrogenase/amicyanin complex using single-crystal UV/Vis microspectrophotometry. J. Synchrotron Radiat 2007, 14, 92–98. [Google Scholar]
- Genick, U.K.; Soltis, S.M.; Kuhn, P.; Canestrelli, I.L.; Getzoff, E.D. Structure at 0.85 A resolution of an early protein photocycle intermediate. Nature 1998, 392, 206–209. [Google Scholar]
- Ihee, H.; Rajagopal, S.; Srajer, V.; Pahl, R.; Anderson, S.; Schmidt, M.; Schotte, F.; Anfinrud, P.A.; Wulff, M.; Moffat, K. Visualizing reaction pathways in photoactive yellow protein from nanoseconds to seconds. Proc. Natl. Acad. Sci. USA 2005, 102, 7145–7150. [Google Scholar]
- Jung, Y.O.; Lee, J.H.; Kim, J.; Schmidt, M.; Moffat, K.; Srajer, V.; Ihee, H. Volume-conserving trans-cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography. Nat. Chem 2013, 5, 212–220. [Google Scholar]
- Yeremenko, S.; van Stokkum, I.H.; Moffat, K.; Hellingwerf, K.J. Influence of the crystalline state on photoinduced dynamics of photoactive yellow protein studied by ultraviolet-visible transient absorption spectroscopy. Biophys. J 2006, 90, 4224–4235. [Google Scholar]
- Fitzpatrick, A.E.; Lincoln, C.N.; van Wilderen, L.J.; van Thor, J.J. Pump-dump-probe and pump-repump-probe ultrafast spectroscopy resolves cross section of an early ground state intermediate and stimulated emission in the photoreactions of the Pr ground state of the cyanobacterial phytochrome Cph1. J. Phys. Chem. B 2012, 116, 1077–1088. [Google Scholar]
- Ujj, L.; Devanathan, S.; Meyer, T.E.; Cusanovich, M.A.; Tollin, G.; Atkinson, G.H. New photocycle intermediates in the photoactive yellow protein from Ectothiorhodospira halophila: Picosecond transient absorption spectroscopy. Biophys. J 1998, 75, 406–412. [Google Scholar]
- Kim, T.W.; Lee, J.H.; Choi, J.; Kim, K.H.; van Wilderen, L.J.; Guerin, L.; Kim, Y.; Jung, Y.O.; Yang, C.; Kim, J.; et al. Protein structural dynamics of photoactive yellow protein in solution revealed by pump-probe X-ray solution scattering. J. Am. Chem. Soc 2012, 134, 3145–3153. [Google Scholar]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr 2002, 58, 899–907. [Google Scholar]
- Sidler, W.A. Phycobilisome and phycobiliprotein structures. In The Molecular Biology of Cyanobacteria; Bryant, D.A., Ed.; Kluwer Academic Publishers: Dortrecht, The Netherlands, 1994; pp. 139–216. [Google Scholar]
- Wiegand, G.; Parbel, A.; Seifert, M.H.; Holak, T.A.; Reuter, W. Purification, crystallization, NMR spectroscopy and biochemical analyses of alpha-phycoerythrocyanin peptides. Eur. J. Biochem. /FEBS 2002, 269, 5046–5055. [Google Scholar]
- Schmidt, M.; Krasselt, A.; Reuter, W. Local protein flexibility as a prerequisite for reversible chromophore isomerization in alpha-phycoerythrocyanin. Biochim. Biophys. Acta 2006, 1764, 55–62. [Google Scholar]
- Schmidt, M.; Patel, A.; Zhao, Y.; Reuter, W. Structural basis for the photochemistry of alpha-phycoerythrocyanin. Biochemistry 2007, 46, 416–423. [Google Scholar]
- Duerring, M.; Huber, R.; Bode, W.; Ruembeli, R.; Zuber, H. Refined 3-dimensional structure of phycoerythrocyanin from the cyanobacterium mastigocladus-laminosus at 2.7-A. J. Mol. Biol 1990, 211, 633–644. [Google Scholar]
- Hong, Q.; Zhao, K.H.; Scheer, H. Two different types of photochemistry in phycoerythrocyanin alpha-subunit. Photochem. Photobiol 1993, 58, 745–747. [Google Scholar]
- Kneip, C.; Parbel, A.; Foerstendorf, H.; Scheer, H.; Siebert, F.; Hildebrandt, P. Fourier transform near-infrared resonance Raman spectroscopic study of the alpha-subunit of phycoerythrocyanin and phycocyanin from the cyanobacterium Mastigocladus laminosus. J. Raman Spectrosc 1998, 29, 939–944. [Google Scholar]
- Zehetmayer, P.; Kupka, M.; Scheer, H.; Zumbusch, A. Energy transfer in monomeric phycoerythrocyanin. Bba-Bioenergetics 2004, 1608, 35–44. [Google Scholar]
- Ng, K.; Getzoff, E.D.; Moffat, K. Optical studies of a bacterial photoreceptor protein, photoactive yellow protein, in single crystals. Biochemistry 1995, 34, 879–890. [Google Scholar]
- Batschauer, A. Photoreceptors of higher plants. Planta 1998, 206, 479–492. [Google Scholar]
- Davis, S.J.; Vener, A.V.; Vierstra, R.D. Bacteriophytochromes: Phytochrome-like photoreceptors from nonphotosynthetic eubacteria. Science 1999, 286, 2517–2520. [Google Scholar]
- DePonte, D.P.; Doak, R.B.; Hunter, M.; Liu, Z.; Weierstall, U.; Spence, J.C. SEM imaging of liquid jets. Micron 2009, 40, 507–509. [Google Scholar]
- Park, H.Y.; Qiu, X.Y.; Rhoades, E.; Korlach, J.; Kwok, L.W.; Zipfel, W.R.; Webb, W.W.; Pollack, L. Achieving uniform mixing in a microfluidic device: Hydrodynamic focusing prior to mixing. Anal. Chem 2006, 78, 4465–4473. [Google Scholar]
- Schmidt, M. Mix and inject, reaction initiation by diffusion for time-resolved macromolecular crystallography. Adv. Condens. Matter Phys. 2013, 2013. [Google Scholar] [CrossRef]
- Poon, H.C.; Schmidt, M.; Saldin, D.K. Extraction of fast changes in the structure of a disordered ensemble of photoexcited biomolecules. Adv. Condens. Matter Phys. 2013, 2013. [Google Scholar] [CrossRef]
- Borgstahl, G.E.; Williams, D.R.; Getzoff, E.D. 1.4 A structure of photoactive yellow protein, a cytosolic photoreceptor: Unusual fold, active site, and chromophore. Biochemistry 1995, 34, 6278–6287. [Google Scholar]
- Fuglistaller, P.; Suter, F.; Zuber, H. The complete amino-acid sequence of both subunits of phycoerythrocyanin from the thermophilic cyanobacterium Mastigocladus laminosus. Hoppe-Seyler’s Z. Physiol. Chem 1983, 364, 691–712. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution | Crystal | ||||||
|---|---|---|---|---|---|---|---|
| Temperature | 295 K | 273 K | 303 K | ||||
| Macroscopic rates Λi (s−1) This work | 3.4 × 103 | 2.2 | 3.8 × 102 | 16.4 | 1.4 | 4.3 × 102 | 25 |
| Relaxation times τi (ms) This work | 0.29 | 450 | 2.6 | 61 | 700 | 2.3 | 40 |
| Relaxation times from 5D-crystallography (ms) [15] | n.a. | n.a. | 0.7 | 73 | n.o. | 6 | n.o. |
| Relaxation times from TR-spectroscopy (ms), 293 K [38] | 0.26 | 360 | n.d. | n.d. | n.d. | 4 | 20 |
| Sample | Spectrometer | |
|---|---|---|
| Xenon | 500 μW | 50 μW |
| Laser (per pulse) | 2 mJ | n. a. |
| a 380 μJ | n. a. | |
| LED | 36 mW | n. a. |
| 1 μs | 250 ns | 10 ns | |
|---|---|---|---|
| total number of photons | 1.7 × 108 | 4.3 × 107 | 1.7 × 106 |
| photons/channel, 400 μm slit | 1.7 × 105 | 4.1 × 104 | 1660 |
| photons/channel, 50 μm slit | 2 × 104 | 5 × 103 | 200 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Purwar, N.; Tenboer, J.; Tripathi, S.; Schmidt, M. Spectroscopic Studies of Model Photo-Receptors: Validation of a Nanosecond Time-Resolved Micro-Spectrophotometer Design Using Photoactive Yellow Protein and α-Phycoerythrocyanin. Int. J. Mol. Sci. 2013, 14, 18881-18898. https://doi.org/10.3390/ijms140918881
Purwar N, Tenboer J, Tripathi S, Schmidt M. Spectroscopic Studies of Model Photo-Receptors: Validation of a Nanosecond Time-Resolved Micro-Spectrophotometer Design Using Photoactive Yellow Protein and α-Phycoerythrocyanin. International Journal of Molecular Sciences. 2013; 14(9):18881-18898. https://doi.org/10.3390/ijms140918881
Chicago/Turabian StylePurwar, Namrta, Jason Tenboer, Shailesh Tripathi, and Marius Schmidt. 2013. "Spectroscopic Studies of Model Photo-Receptors: Validation of a Nanosecond Time-Resolved Micro-Spectrophotometer Design Using Photoactive Yellow Protein and α-Phycoerythrocyanin" International Journal of Molecular Sciences 14, no. 9: 18881-18898. https://doi.org/10.3390/ijms140918881
APA StylePurwar, N., Tenboer, J., Tripathi, S., & Schmidt, M. (2013). Spectroscopic Studies of Model Photo-Receptors: Validation of a Nanosecond Time-Resolved Micro-Spectrophotometer Design Using Photoactive Yellow Protein and α-Phycoerythrocyanin. International Journal of Molecular Sciences, 14(9), 18881-18898. https://doi.org/10.3390/ijms140918881