Neuroprotective Effect of Arctigenin via Upregulation of P-CREB in Mouse Primary Neurons and Human SH-SY5Y Neuroblastoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
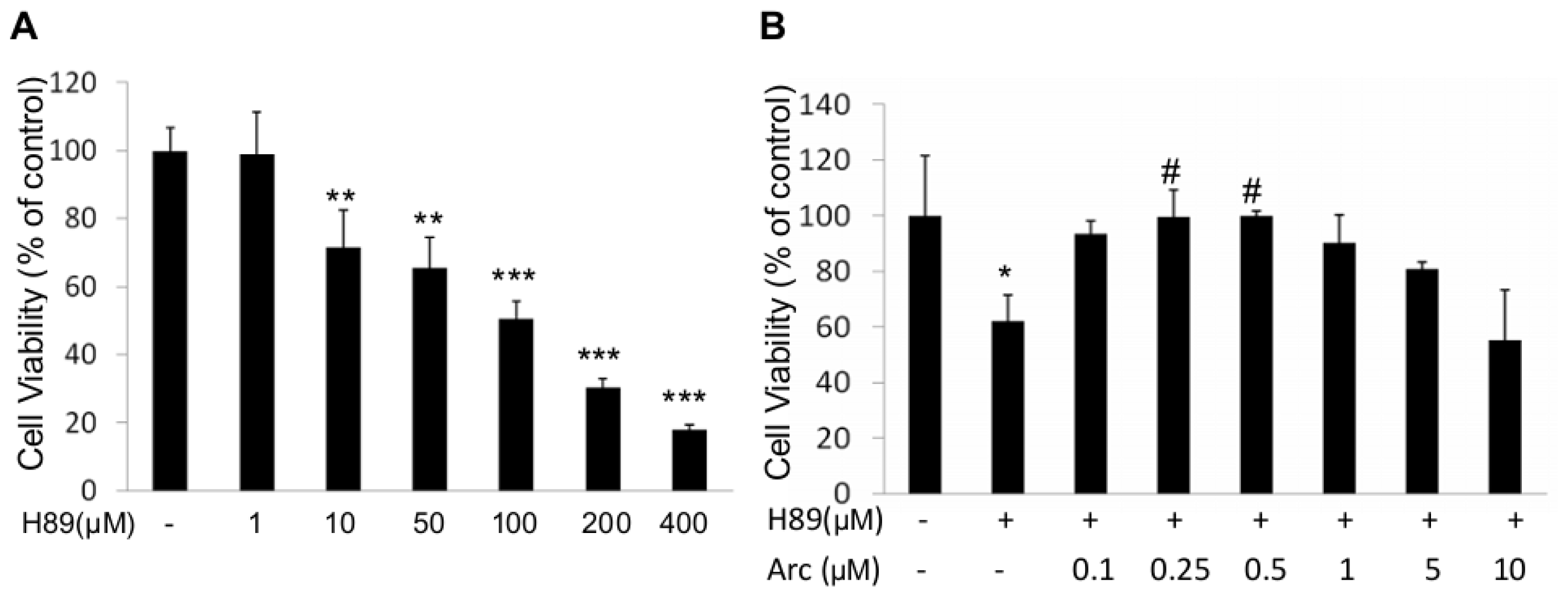
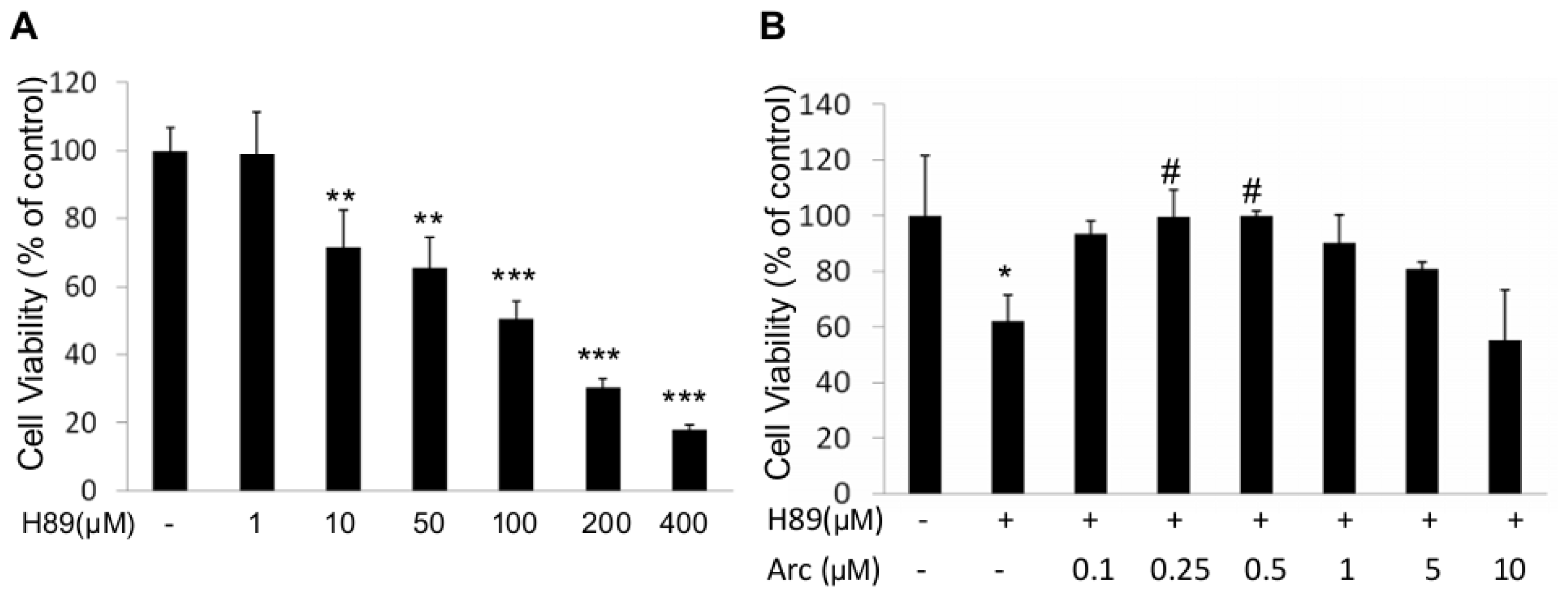
2.1. Arc Protects Human SH-SY5Y Cells from H89-Induced Reduction of Cell Viability
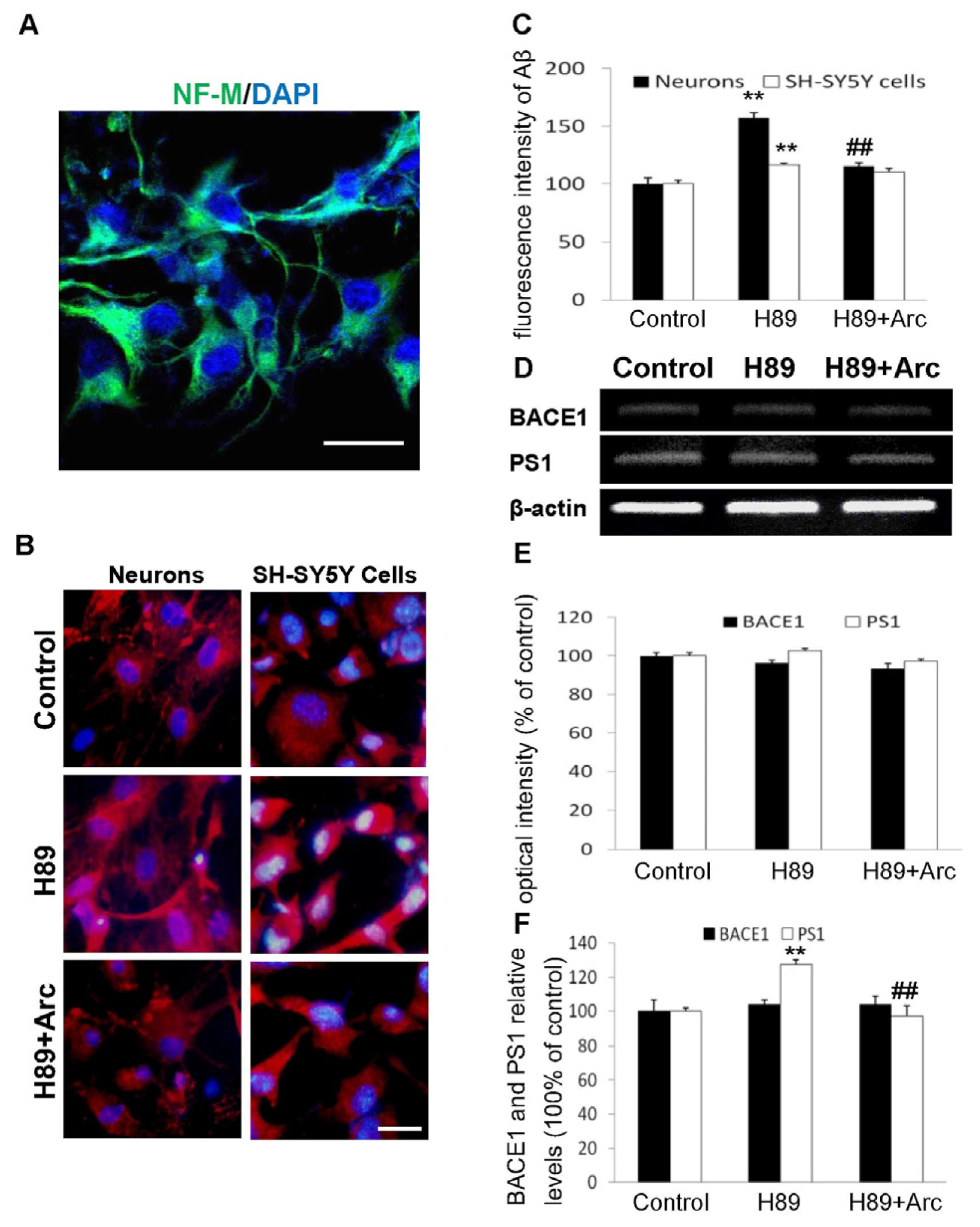
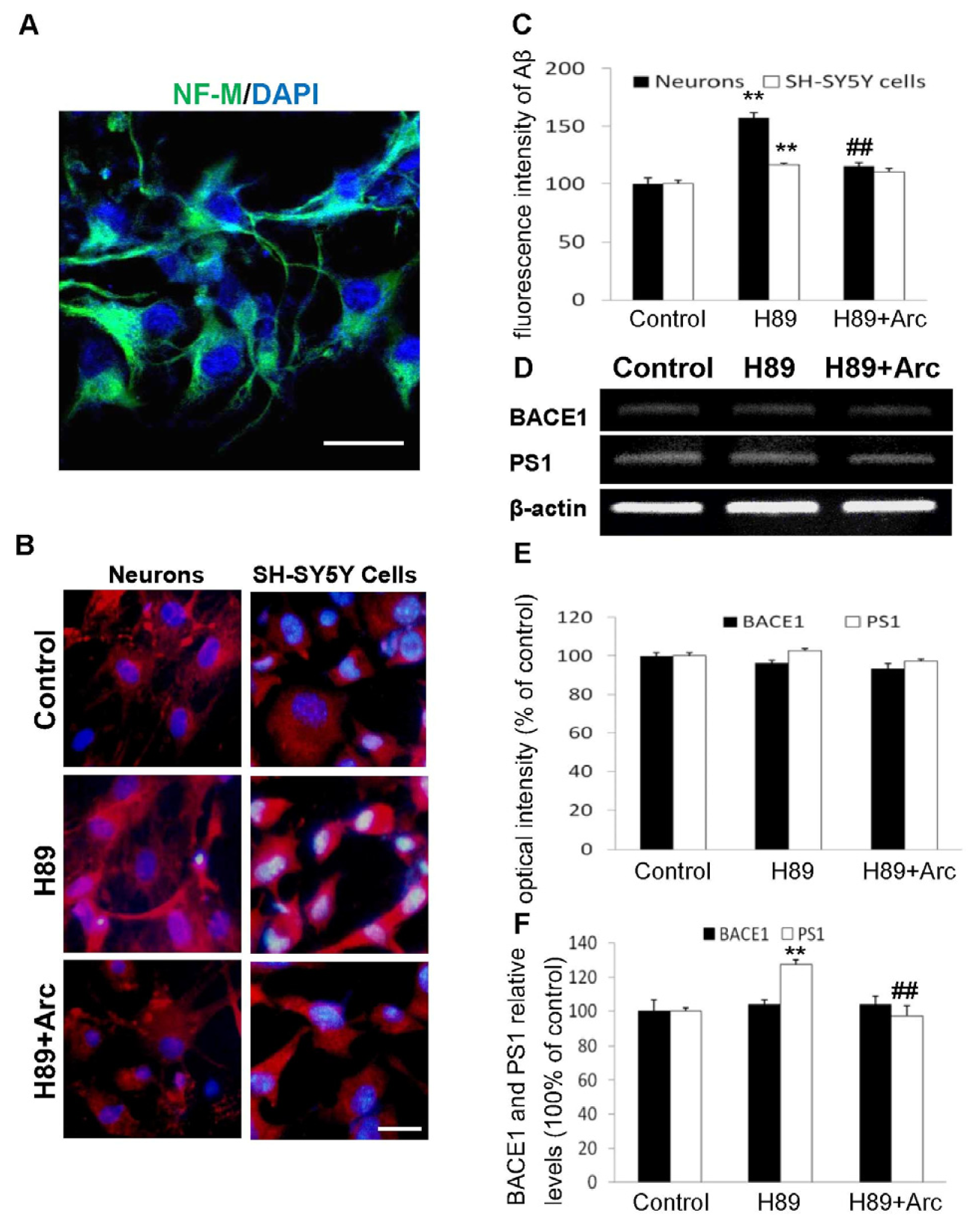
2.2. Arc Attenuates Aβ Production Induced by H89
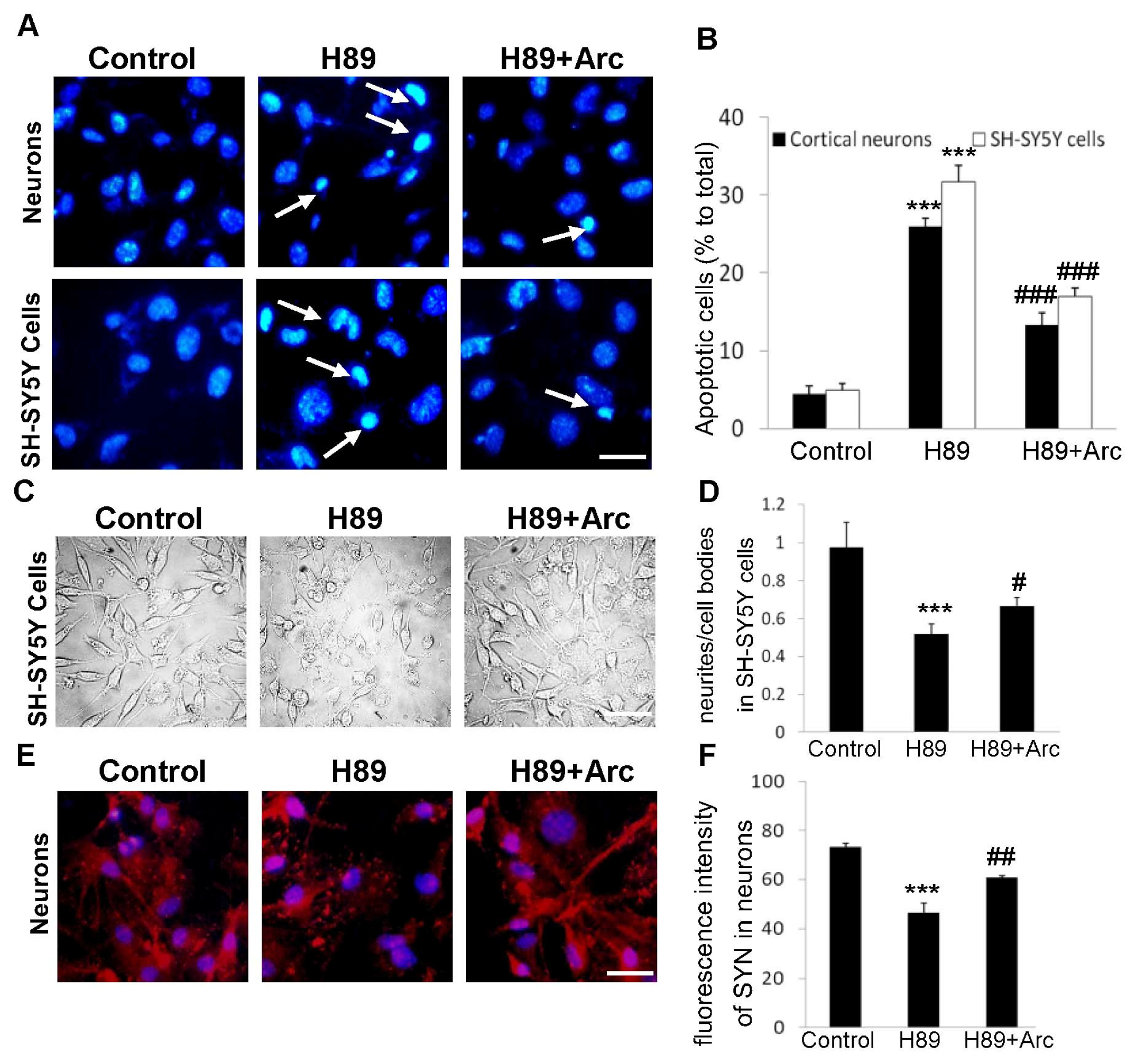
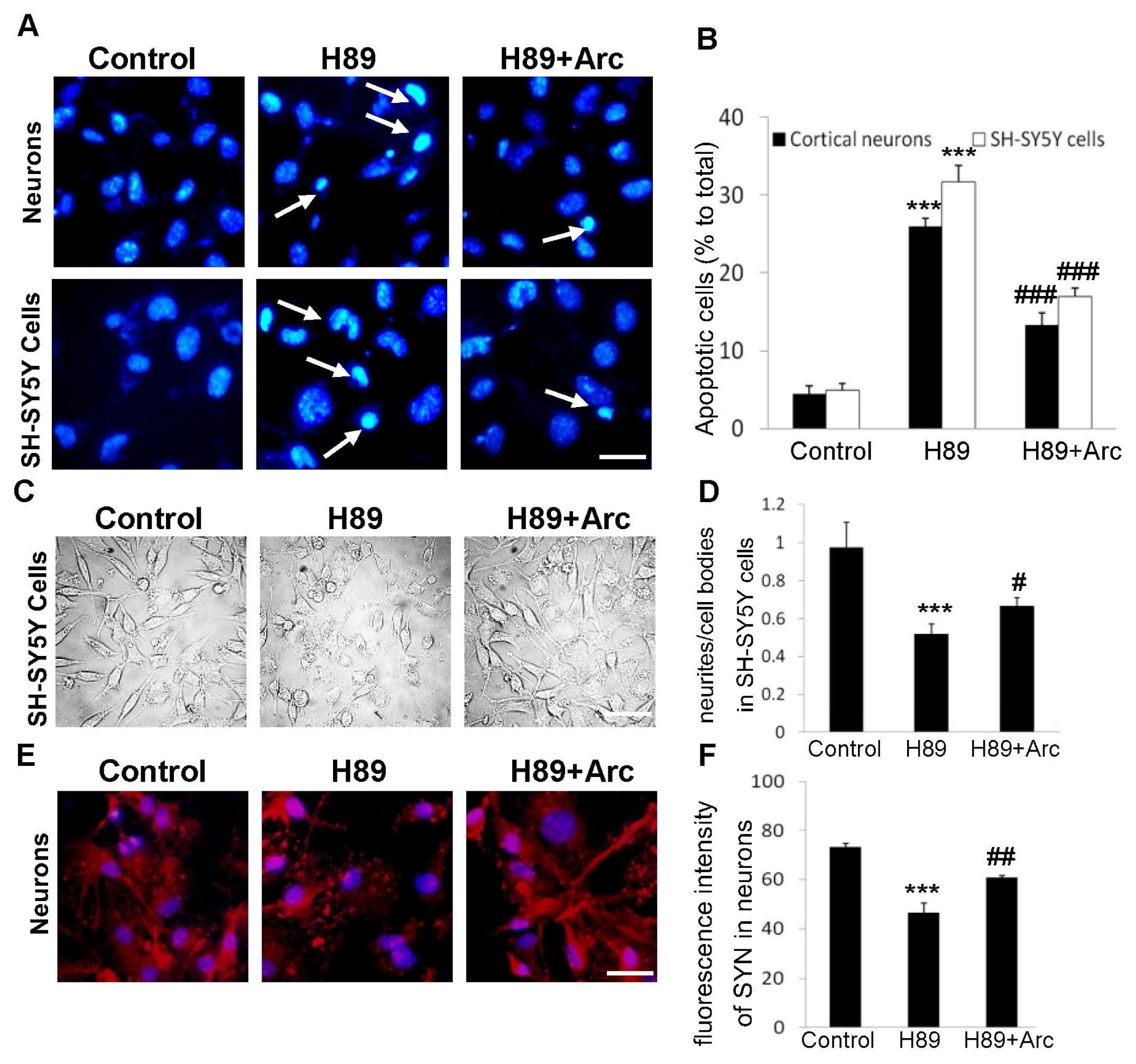
2.3. Arc Protects Neuron-Like Cells and Neurons against Apoptosis Induced by H89
2.4. Arc Restores Neurite Outgrowth and SYN Expression against H89-Induced Disorders
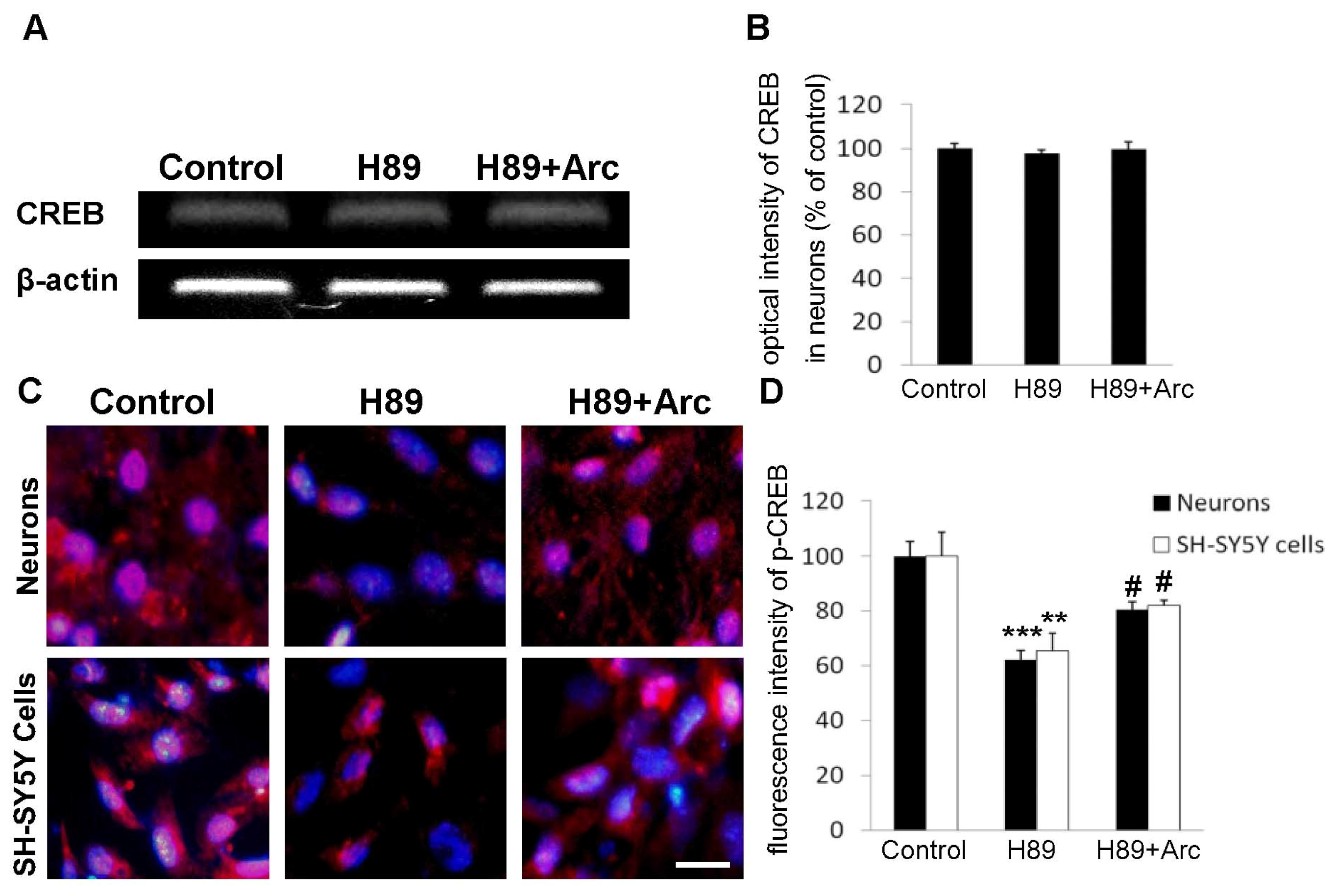
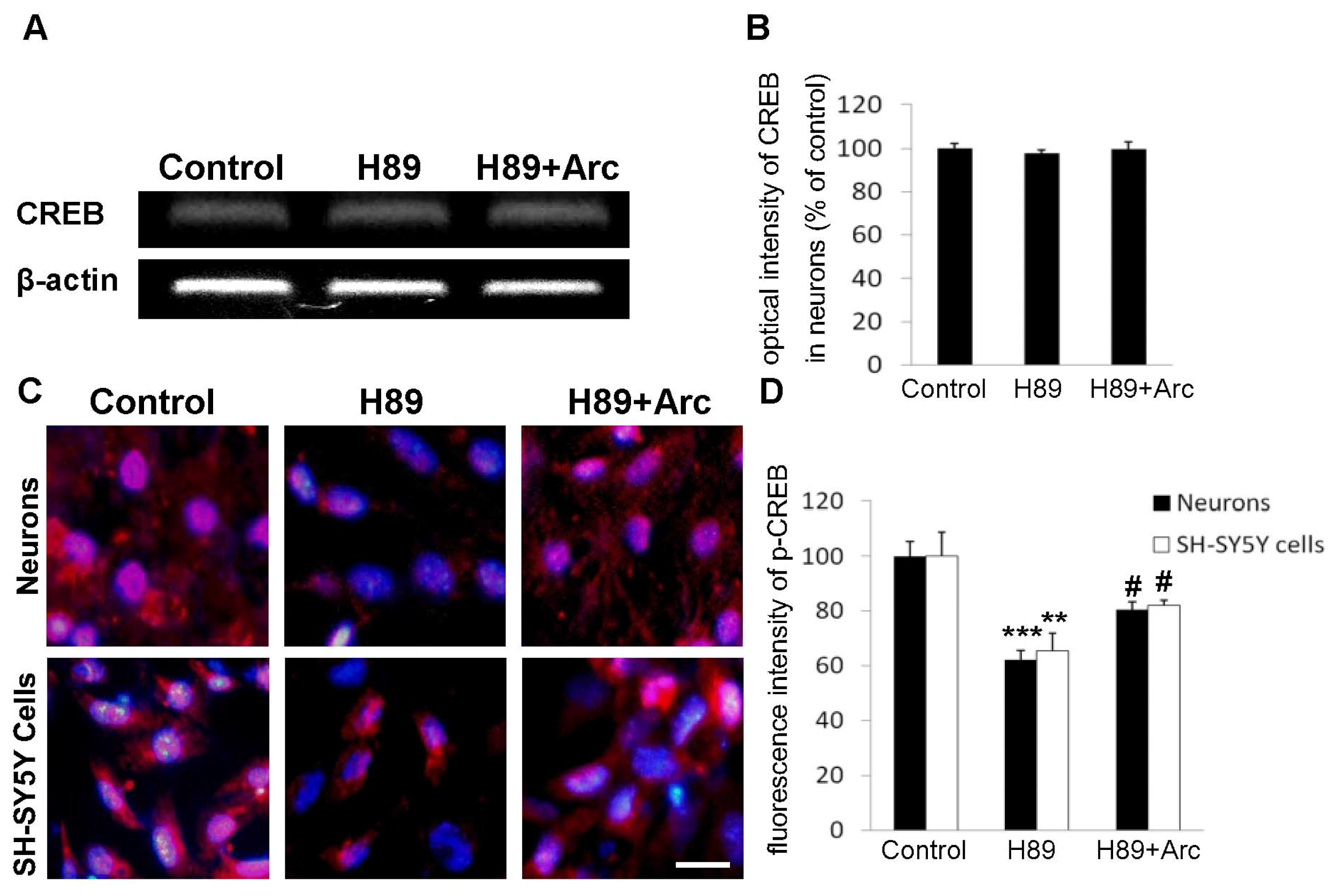
2.5. CREB May Be Involved in the Neuroprotection of Arc against H89-Induced Cell Injury
2.6. Discussion
3. Experimental Section
3.1. Cell Culture
3.2. Preparation of Arc
3.3. Cell Viability
3.4. Immunofluorescence, Hoechst33258 Staining and Neuritogenesis
3.5. Quantitative PCR with Reverse Transcription
3.6. Quantification of BACE1 and PS1 by ELISA
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lee, J.Y.; Cho, B.J.; Park, T.W.; Park, B.E.; Kim, S.J.; Sim, S.S.; Kim, C.J. Dibenzylbutyrolactone lignans from Forsythia koreana fruits attenuate lipopolysaccharide-induced inducible nitric oxide synthetase and cyclooxygenase-2 expressions through activation of nuclear factor-kappab and mitogen-activated protein kinase in RAW264.7 cells. Biol. Pharm. Bull 2010, 33, 1847–1853. [Google Scholar]
- Tsai, W.J.; Chang, C.T.; Wang, G.J.; Lee, T.H.; Chang, S.F.; Lu, S.C.; Kuo, Y.C. Arctigenin from Arctium lappa inhibits interleukin-2 and interferon gene expression in primary human T lymphocytes. Chin. Med 2011, 6, 12. [Google Scholar]
- Gu, Y.; Qi, C.; Sun, X.; Ma, X.; Zhang, H.; Hu, L.; Yuan, J.; Yu, Q. Arctigenin preferentially induces tumor cell death under glucose deprivation by inhibiting cellular energy metabolism. Biochem. Pharmacol 2012, 84, 468–476. [Google Scholar]
- Jang, Y.P.; Kim, S.R.; Choi, Y.H.; Kim, J.; Kim, S.G.; Markelonis, G.J.; Oh, T.H.; Kim, Y.C. Arctigenin protects cultured cortical neurons from glutamate-induced neurodegeneration by binding to kainate receptor. J. Neurosci. Res 2002, 68, 233–240. [Google Scholar]
- Lee, I.A.; Joh, E.H.; Kim, D.H. Arctigenin isolated from the seeds of Arctium lappa ameliorates memory deficits in mice. Planta Med 2011, 77, 1525–1527. [Google Scholar]
- Jawhar, S.; Wirths, O.; Bayer, T.A. Pyroglutamate amyloid-beta (Abeta): A hatchet man in Alzheimer disease. J. Biol. Chem 2011, 286, 38825–38832. [Google Scholar]
- Shaywitz, A.J.; Greenberg, M.E. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem 1999, 68, 821–861. [Google Scholar]
- Silva, A.J.; Kogan, J.H.; Frankland, P.W.; Kida, S. CREB and memory. Annu. Rev. Neurosci 1998, 21, 127–148. [Google Scholar]
- Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002, 35, 605–623. [Google Scholar]
- Pugazhenthi, S.; Wang, M.; Pham, S.; Sze, C.I.; Eckman, C.B. Downregulation of CREB expression in Alzheimer’s brain and in Abeta-treated rat hippocampal neurons. Mol. Neurodegener 2011, 6, 60. [Google Scholar]
- Dineley, K.T.; Westerman, M.; Bui, D.; Bell, K.; Ashe, K.H.; Sweatt, J.D. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci 2001, 21, 4125–4133. [Google Scholar]
- Funaki, C.; Hodges, R.R.; Dartt, D.A. Identification of the Raf-1 signaling pathway used by cAMP to inhibit p42/p44 MAPK in rat lacrimal gland acini: Role in potentiation of protein secretion. Invest. Ophthalmol. Visual Sci 2010, 51, 6321–6328. [Google Scholar]
- Lai, H.C.; Wu, M.J.; Chen, P.Y.; Sheu, T.T.; Chiu, S.P.; Lin, M.H.; Ho, C.T.; Yen, J.H. Neurotrophic effect of citrus 5-hydroxy-3,6,7,8,3′,4′-hexamethoxyflavone: Promotion of neurite outgrowth via cAMP/PKA/CREB pathway in PC12 cells. PLoS One 2011, 6, e28280. [Google Scholar]
- Chu, J.; Pratico, D. 5-lipoxygenase as an endogenous modulator of amyloid beta formation in vivo. Annu. Neurol 2011, 69, 34–46. [Google Scholar]
- Cho, I.J.; Woo, N.R.; Shin, I.C.; Kim, S.G. H89, an inhibitor of PKA and MSK, inhibits cyclic-AMP response element binding protein-mediated MAPK phosphatase-1 induction by lipopolysaccharide. Inflamm. Res 2009, 58, 863–872. [Google Scholar]
- Costes, S.; Broca, C.; Bertrand, G.; Lajoix, A.D.; Bataille, D.; Bockaert, J.; Dalle, S. ERK1/2 control phosphorylation and protein level of cAMP-responsive element-binding protein: A key role in glucose-mediated pancreatic beta-cell survival. Diabetes 2006, 55, 2220–2230. [Google Scholar]
- Markou, T.; Hadzopoulou-Cladaras, M.; Lazou, A. Phenylephrine induces activation of CREB in adult rat cardiac myocytes through MSK1 and PKA signaling pathways. J. Mol. Cell. Cardiol 2004, 37, 1001–1011. [Google Scholar]
- Wang, M.D.; Huang, Y.; Zhang, G.P.; Mao, L.; Xia, Y.P.; Mei, Y.W.; Hu, B. Exendin-4 improved rat cortical neuron survival under oxygen/glucose deprivation through PKA pathway. Neuroscience 2012, 226, 388–396. [Google Scholar]
- Farrow, B.; Rychahou, P.; Murillo, C.; O’Connor, K.L.; Iwamura, T.; Evers, B.M. Inhibition of pancreatic cancer cell growth and induction of apoptosis with novel therapies directed against protein kinase A. Surgery 2003, 134, 197–205. [Google Scholar]
- Hou, Y.; Aboukhatwa, M.A.; Lei, D.L.; Manaye, K.; Khan, I.; Luo, Y. Anti-depressant natural flavonols modulate BDNF and beta amyloid in neurons and hippocampus of double TgAD mice. Neuropharmacology 2010, 58, 911–920. [Google Scholar]
- Li, S.; Reinprecht, I.; Fahnestock, M.; Racine, R.J. Activity-dependent changes in synaptophysin immunoreactivity in hippocampus, piriform cortex, and entorhinal cortex of the rat. Neuroscience 2002, 115, 1221–1229. [Google Scholar]
- Butterfield, D.A. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radical Res 2002, 36, 1307–1313. [Google Scholar]
- Zhu, X.; Chen, C.; Ye, D.; Guan, D.; Ye, L.; Jin, J.; Zhao, H.; Chen, Y.; Wang, Z.; Wang, X.; et al. Diammonium glycyrrhizinate upregulates PGC-1alpha and protects against Abeta1–42-induced neurotoxicity. PLoS One 2012, 7, e35823. [Google Scholar]
- Vetrivel, K.S.; Thinakaran, G. Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology 2006, 66, S69–S73. [Google Scholar]
- Heredia, L.; Helguera, P.; de Olmos, S.; Kedikian, G.; Sola Vigo, F.; LaFerla, F.; Staufenbiel, M.; de Olmos, J.; Busciglio, J.; Caceres, A.; et al. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: A potential mechanism of neuronal dystrophy in Alzheimer’s disease. J. Neurosci 2006, 26, 6533–6542. [Google Scholar]
- Kudo, T.A.; Kanetaka, H.; Mizuno, K.; Ryu, Y.; Miyamoto, Y.; Nunome, S.; Zhang, Y.; Kano, M.; Shimizu, Y.; Hayashi, H. Dorsomorphin stimulates neurite outgrowth in PC12 cells via activation of a protein kinase A-dependent MEK-ERK1/2 signaling pathway. Genes Cells 2011, 16, 1121–1132. [Google Scholar]
- Zheng, Z.; Keifer, J. PKA has a critical role in synaptic delivery of GluR1- and GluR4-containing AMPARs during initial stages of acquisition of in vitro classical conditioning. J. Neurophysiol 2009, 101, 2539–2549. [Google Scholar]
- Delghandi, M.P.; Johannessen, M.; Moens, U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell. Signal 2005, 17, 1343–1351. [Google Scholar]
- Tan, Y.W.; Zhang, S.J.; Hoffmann, T.; Bading, H. Increasing levels of wild-type CREB up-regulates several activity-regulated inhibitor of death (AID) genes and promotes neuronal survival. BMC Neurosci 2012, 13, 48. [Google Scholar]
- Nie, B.M.; Jiang, X.Y.; Cai, J.X.; Fu, S.L.; Yang, L.M.; Lin, L.; Hang, Q.; Lu, P.L.; Lu, Y. Panaxydol and panaxynol protect cultured cortical neurons against Abeta25–35-induced toxicity. Neuropharmacology 2008, 54, 845–853. [Google Scholar]
- Kwon, M.; Fernandez, J.R.; Zegarek, G.F.; Lo, S.B.; Firestein, B.L. BDNF-promoted increases in proximal dendrites occur via CREB-dependent transcriptional regulation of cypin. J. Neurosci 2011, 31, 9735–9745. [Google Scholar]
- Saeki, K.; Nose, Y.; Hirao, N.; Takasawa, R.; Tanuma, S. Amyloid precursor protein binding protein Fe65 is cleaved by caspases during DNA damage-induced apoptosis. Biol. Pharm. Bull 2011, 34, 290–294. [Google Scholar]
- Dedoni, S.; Olianas, M.C.; Ingianni, A.; Onali, P. Type I interferons impair BDNF-induced cell signaling and neurotrophic activity in differentiated human SH-SY5Y neuroblastoma cells and mouse primary cortical neurons. J. Neurochem 2012, 122, 58–71. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, N.; Wen, Q.; Ren, L.; Liang, W.; Xia, Y.; Zhang, X.; Zhao, D.; Sun, D.; Hu, Y.; Hao, H.; et al. Neuroprotective Effect of Arctigenin via Upregulation of P-CREB in Mouse Primary Neurons and Human SH-SY5Y Neuroblastoma Cells. Int. J. Mol. Sci. 2013, 14, 18657-18669. https://doi.org/10.3390/ijms140918657
Zhang N, Wen Q, Ren L, Liang W, Xia Y, Zhang X, Zhao D, Sun D, Hu Y, Hao H, et al. Neuroprotective Effect of Arctigenin via Upregulation of P-CREB in Mouse Primary Neurons and Human SH-SY5Y Neuroblastoma Cells. International Journal of Molecular Sciences. 2013; 14(9):18657-18669. https://doi.org/10.3390/ijms140918657
Chicago/Turabian StyleZhang, Nan, Qingping Wen, Lu Ren, Wenbo Liang, Yang Xia, Xiaodan Zhang, Dan Zhao, Dong Sun, Yv Hu, Haiguang Hao, and et al. 2013. "Neuroprotective Effect of Arctigenin via Upregulation of P-CREB in Mouse Primary Neurons and Human SH-SY5Y Neuroblastoma Cells" International Journal of Molecular Sciences 14, no. 9: 18657-18669. https://doi.org/10.3390/ijms140918657
APA StyleZhang, N., Wen, Q., Ren, L., Liang, W., Xia, Y., Zhang, X., Zhao, D., Sun, D., Hu, Y., Hao, H., Yan, Y., Zhang, G., Yang, J., & Kang, T. (2013). Neuroprotective Effect of Arctigenin via Upregulation of P-CREB in Mouse Primary Neurons and Human SH-SY5Y Neuroblastoma Cells. International Journal of Molecular Sciences, 14(9), 18657-18669. https://doi.org/10.3390/ijms140918657