Oxidative Stress and Epigenetic Regulation in Ageing and Age-Related Diseases


 ,
,
Abstract
:1. Introduction
2. The Epigenetic Machinery
3. Epigenetic Traits of Ageing
3.1. Chromatin Alterations
3.2. miRNA Role in Ageing
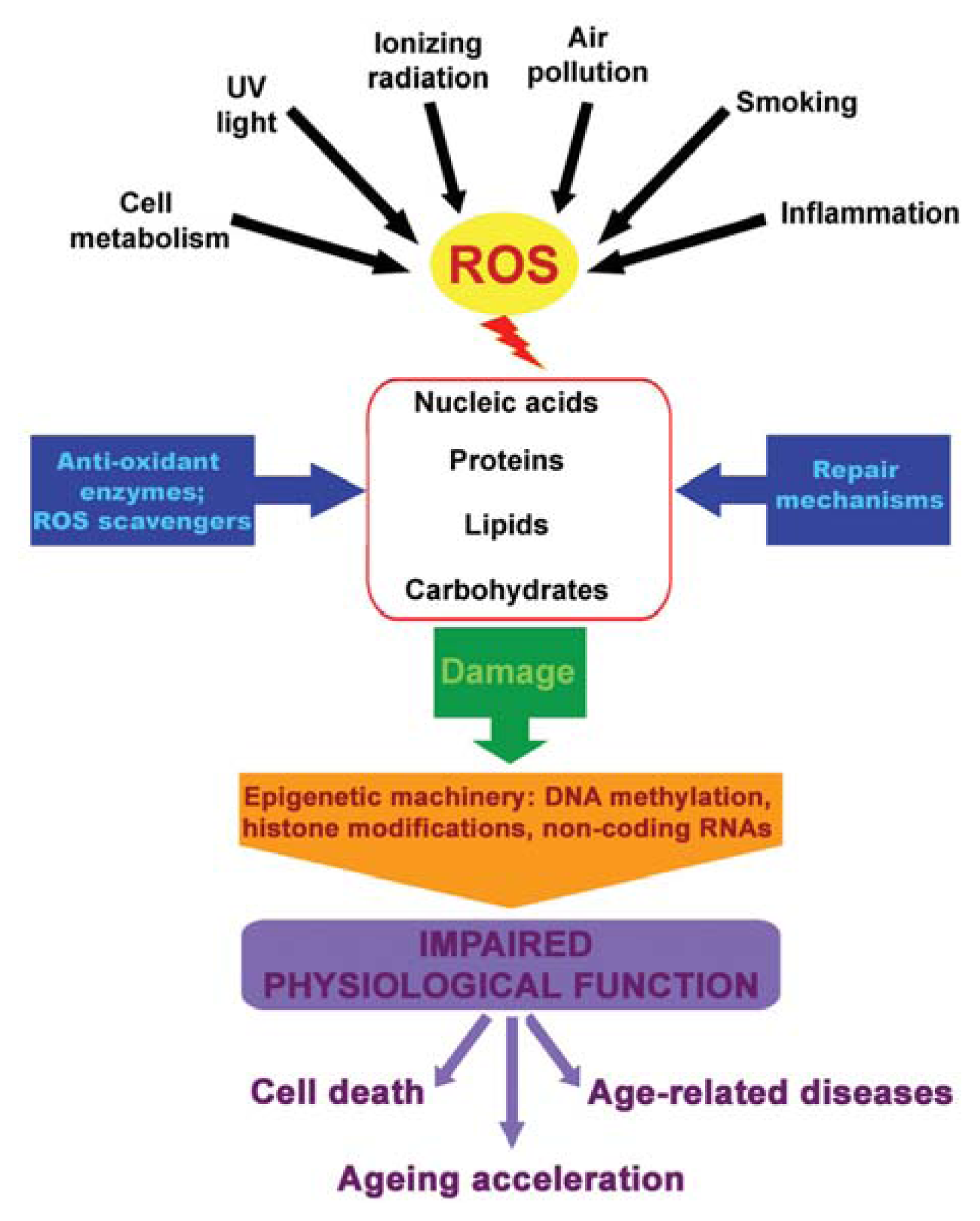
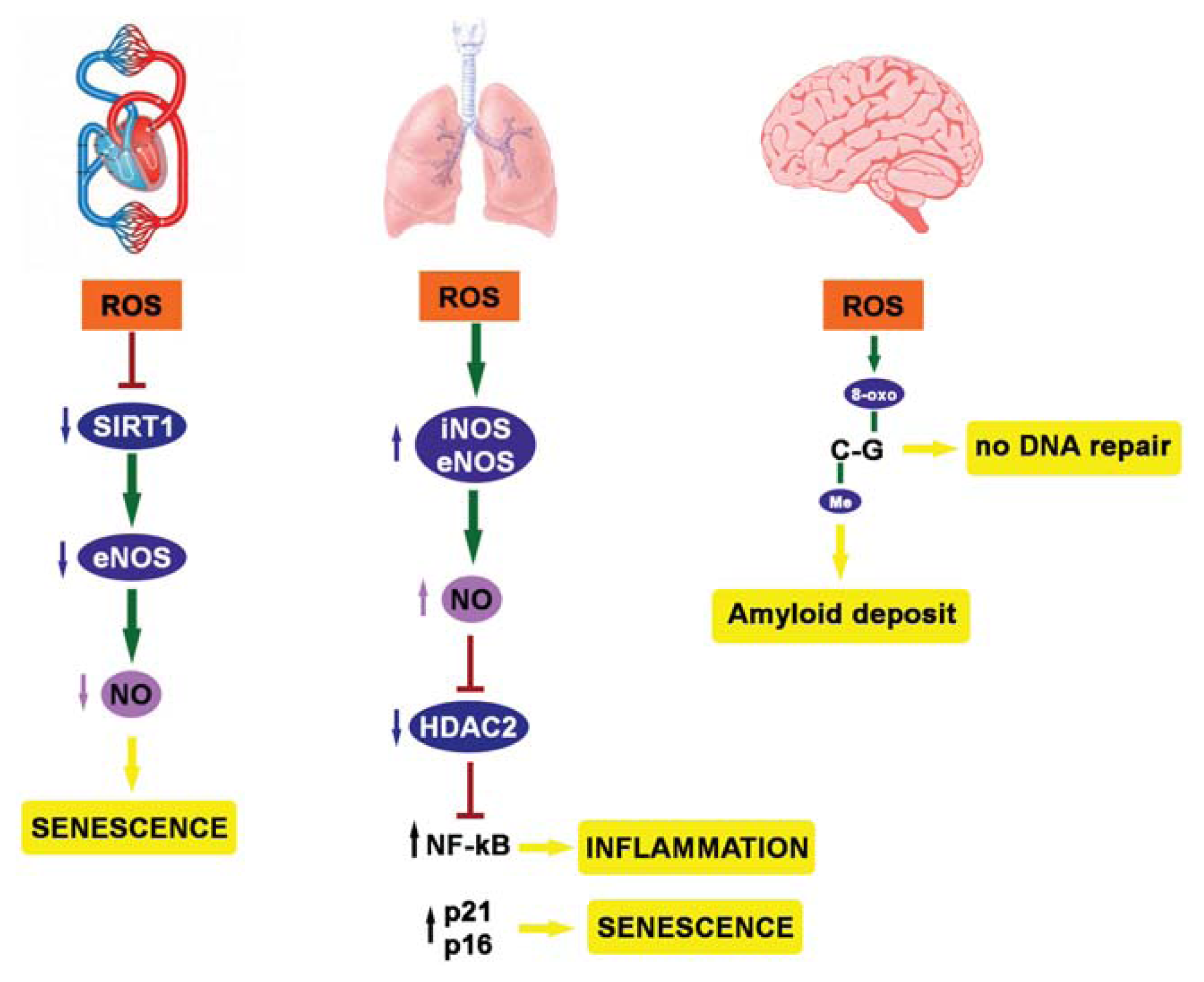
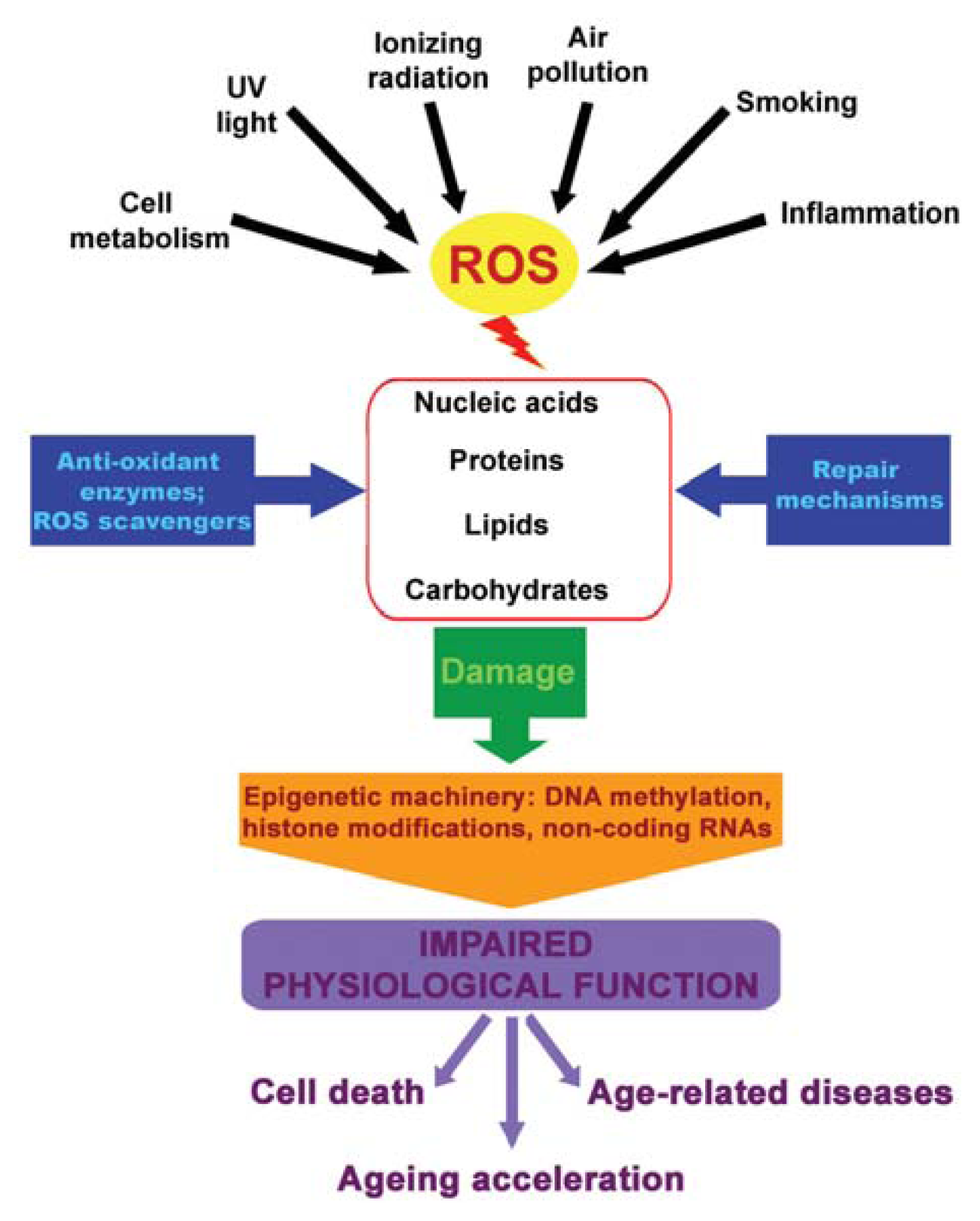
4. ROS, Epigenetics and Diseases
5. Youth Fountain: Struggle with ROS
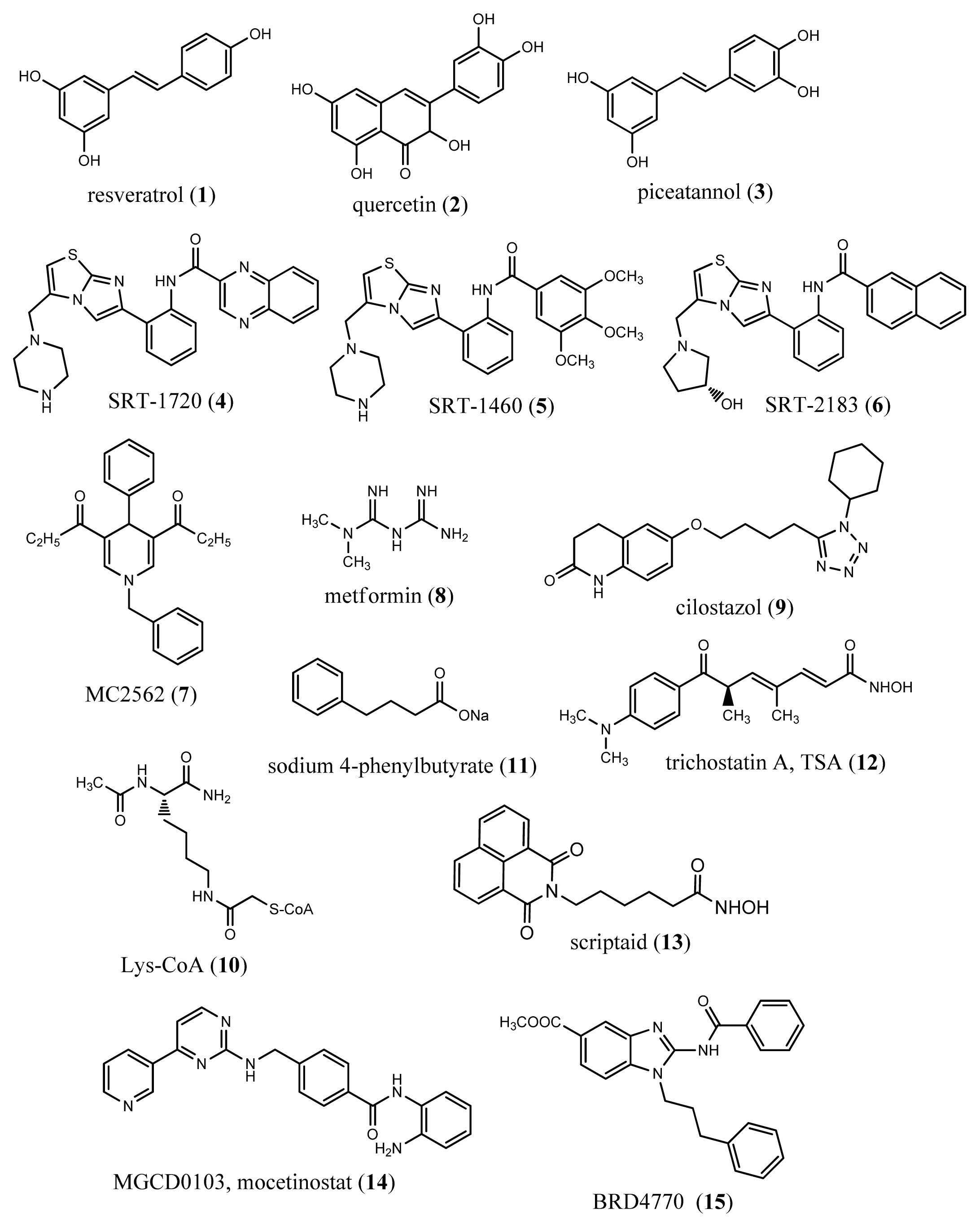
6. Epigenetic Drugs in Ageing and Age-Related Diseases
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Martin, G.M. The biology of aging: 1985–2010 and beyond. FASEB J 2011, 25, 3756–3762. [Google Scholar]
- Olshansky, S.J.; Goldman, D.P.; Zheng, Y.; Rowe, J.W. Aging in America in the twenty-first century: Demographic forecasts from the MacArthur foundation research network on an aging society. Milbank Q 2009, 87, 842–862. [Google Scholar]
- Hayflick, L. Biological aging is no longer an unsolved problem. Ann. N. Y. Acad. Sci 2007, 1100, 1–13. [Google Scholar]
- Kirkwood, T.B. Understanding the odd science of aging. Cell 2005, 120, 437–447. [Google Scholar]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev 1998, 78, 547–581. [Google Scholar]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene expression profile of aging and its retardation by caloric restriction. Science 1999, 285, 1390–1393. [Google Scholar]
- Stadtman, E.R. Protein oxidation and aging. Science 1992, 257, 1220–1224. [Google Scholar]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar]
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar]
- Stadtman, E.R. Protein oxidation in aging and age-related diseases. Ann. N. Y. Acad. Sci 2001, 928, 22–38. [Google Scholar]
- Ben-Avraham, D.; Muzumdar, R.H.; Atzmon, G. Epigenetic genome-wide association methylation in aging and longevity. Epigenomics 2012, 4, 503–509. [Google Scholar]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev 2009, 23, 781–783. [Google Scholar]
- Skinner, M.K. Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics 2011, 6, 838–842. [Google Scholar]
- Illi, B.; Colussi, C.; Grasselli, A.; Farsetti, A.; Capogrossi, M.C.; Gaetano, C. NO sparks off chromatin: Tales of a multifaceted epigenetic regulator. Pharmacol. Ther 2009, 123, 344–352. [Google Scholar]
- Williams, K.; Christensen, J.; Helin, K. DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep 2011, 13, 28–35. [Google Scholar]
- Cedar, H.; Bergman, Y. Programming of DNA methylation patterns. Annu. Rev. Biochem 2012, 81, 97–117. [Google Scholar]
- Gronbaek, K.; Hother, C.; Jones, P.A. Epigenetic changes in cancer. APMIS 2007, 115, 1039–1059. [Google Scholar]
- Guil, S.; Esteller, M. Cis-acting noncoding RNAs: Friends and foes. Nat. Struct. Mol. Biol 2011, 19, 1068–1075. [Google Scholar]
- Vanyushin, B.F.; Nemirovsky, L.E.; Klimenko, V.V.; Vasiliev, V.K.; Belozersky, A.N. The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents. Gerontologia 1973, 19, 138–152. [Google Scholar]
- Choi, E.K.; Uyeno, S.; Nishida, N.; Okumoto, T.; Fujimura, S.; Aoki, Y.; Nata, M.; Sagisaka, K.; Fukuda, Y.; Nakao, K.; et al. Alterations of c-fos gene methylation in the processes of aging and tumorigenesis in human liver. Mutat. Res 1996, 354, 123–128. [Google Scholar]
- Issa, J.P.; Vertino, P.M.; Boehm, C.D.; Newsham, I.F.; Baylin, S.B. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 11757–11762. [Google Scholar]
- So, K.; Tamura, G.; Honda, T.; Homma, N.; Waki, T.; Togawa, N.; Nishizuka, S.; Motoyama, T. Multiple tumor suppressor genes are increasingly methylated with age in non-neoplastic gastric epithelia. Cancer Sci 2006, 97, 1155–1158. [Google Scholar]
- Gonzalo, S. Epigenetic alterations in aging. J. Appl. Physiol 2010, 109, 586–597. [Google Scholar]
- Pruitt, K.; Zinn, R.L.; Ohm, J.E.; McGarvey, K.M.; Kang, S.H.; Watkins, D.N.; Herman, J.G.; Baylin, S.B. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet 2006, 2, e40. [Google Scholar]
- Vaquero, A.; Scher, M.; Erdjument-Bromage, H.; Tempst, P.; Serrano, L.; Reinberg, D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature 2007, 450, 440–444. [Google Scholar]
- Thakur, M.K.; Kanungo, M.S. Methylation of chromosomal proteins and DNA of rat brain and its modulation by estradiol and calcium during aging. Exp. Gerontol 1981, 16, 331–336. [Google Scholar]
- Wang, C.M.; Tsai, S.N.; Yew, T.W.; Kwan, Y.W.; Ngai, S.M. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology 2010, 11, 87–102. [Google Scholar]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet 2007, 23, 413–418. [Google Scholar]
- Sarg, B.; Koutzamani, E.; Helliger, W.; Rundquist, I.; Lindner, H.H. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J. Biol. Chem 2002, 277, 39195–39201. [Google Scholar]
- Agger, K.; Cloos, P.A.; Rudkjaer, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev 2009, 23, 1171–1176. [Google Scholar]
- Jung, J.W.; Lee, S.; Seo, M.S.; Park, S.B.; Kurtz, A.; Kang, S.K.; Kang, K.S. Histone deacetylase controls adult stem cell aging by balancing the expression of polycomb genes and jumonji domain containing 3. Cell. Mol. Life Sci 2010, 67, 1165–1176. [Google Scholar]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar]
- Zhang, R.; Adams, P.D. Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle 2007, 6, 784–789. [Google Scholar]
- Ye, X.; Zerlanko, B.; Zhang, R.; Somaiah, N.; Lipinski, M.; Salomoni, P.; Adams, P.D. Definition of pRB- and p53-dependent and -independent steps in HIRA/ASF1a-mediated formation of senescence-associated heterochromatin foci. Mol. Cell. Biol 2007, 27, 2452–2465. [Google Scholar]
- Boulias, K.; Horvitz, H.R. The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab 2012, 15, 439–450. [Google Scholar]
- Boon, R.A.; Seeger, T.; Heydt, S.; Fischer, A.; Hergenreider, E.; Horrevoets, A.J.; Vinciguerra, M.; Rosenthal, N.; Sciacca, S.; Pilato, M.; et al. MicroRNA-29 in aortic dilation: Implications for aneurysm formation. Circ. Res 2011, 109, 1115–1119. [Google Scholar]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar]
- Olivieri, F.; Lazzarini, R.; Recchioni, R.; Marcheselli, F.; Rippo, M.R.; di Nuzzo, S.; Albertini, M.C.; Graciotti, L.; Babini, L.; Mariotti, S.; et al. MiR-146a as marker of senescence-associated pro-inflammatory status in cells involved in vascular remodelling. Age 2012, 35, 1157–1172. [Google Scholar]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Treguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar]
- Ito, T.; Yagi, S.; Yamakuchi, M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys. Res. Commun 2010, 398, 735–740. [Google Scholar]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ 2011, 18, 1628–1639. [Google Scholar]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar]
- Sedding, D.G. FoxO transcription factors in oxidative stress response and ageing—A new fork on the way to longevity? Biol. Chem 2008, 389, 279–283. [Google Scholar]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 1999, 13, 2570–2580. [Google Scholar]
- Romanov, V.S.; Pospelov, V.A.; Pospelova, T.V. Cyclin-dependent kinase inhibitor p21Waf1: Contemporary view on its role in senescence and oncogenesis. Biochemistry (Mosc. ) 2012, 77, 575–584. [Google Scholar]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany N.Y.) 2009, 1, 402–411. [Google Scholar]
- Vasa-Nicotera, M.; Chen, H.; Tucci, P.; Yang, A.L.; Saintigny, G.; Menghini, R.; Mahe, C.; Agostini, M.; Knight, R.A.; Melino, G.; et al. miR-146a is modulated in human endothelial cell with aging. Atherosclerosis 2011, 217, 326–330. [Google Scholar]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res 2012, 110, 1097–1108. [Google Scholar]
- Oxenham, H.; Sharpe, N. Cardiovascular aging and heart failure. Eur. J. Heart Fail 2003, 5, 427–434. [Google Scholar]
- Versari, D.; Daghini, E.; Virdis, A.; Ghiadoni, L.; Taddei, S. The ageing endothelium, cardiovascular risk and disease in man. Exp. Physiol 2009, 94, 317–321. [Google Scholar]
- Strait, J.B.; Lakatta, E.G. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail. Clin 2012, 8, 143–164. [Google Scholar]
- Tsutsui, M.; Shimokawa, H.; Morishita, T.; Nakashima, Y.; Yanagihara, N. Development of genetically engineered mice lacking all three nitric oxide synthases. J. Pharmacol. Sci 2006, 102, 147–154. [Google Scholar]
- Hwang, J.W.; Yao, H.; Caito, S.; Sundar, I.K.; Rahman, I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med 2013, 61C, 95–110. [Google Scholar]
- Ota, H.; Tokunaga, E.; Chang, K.; Hikasa, M.; Iijima, K.; Eto, M.; Kozaki, K.; Akishita, M.; Ouchi, Y.; Kaneki, M. SIRT1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 2006, 25, 176–185. [Google Scholar]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. SIRT1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell. Cardiol 2007, 43, 571–579. [Google Scholar]
- Ota, H.; Eto, M.; Kano, M.R.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of SIRT1 in human endothelial cells. Arterioscler. Thromb. Vasc. Biol 2008, 28, 1634–1639. [Google Scholar]
- Ota, H.; Eto, M.; Kano, M.R.; Kahyo, T.; Setou, M.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler. Thromb. Vasc. Biol 2010, 30, 2205–2211. [Google Scholar]
- Rahman, I.; Marwick, J.; Kirkham, P. Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem. Pharmacol 2004, 68, 1255–1267. [Google Scholar]
- Repine, J.E.; Bast, A.; Lankhorst, I. The oxidative stress study group. Oxidative stress in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med 1997, 156, 341–357. [Google Scholar]
- Tuder, R.M.; Kern, J.A.; Miller, Y.E. Senescence in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc 2012, 9, 62–63. [Google Scholar]
- Yao, H.; Rahman, I. Role of histone deacetylase 2 in epigenetics and cellular senescence: Implications in lung inflammaging and COPD. Am. J. Physiol. Lung Cell. Mol. Physiol 2012, 303, L557–L566. [Google Scholar]
- Sundar, I.K.; Yao, H.; Rahman, I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxid. Redox. Signal 2012, 18, 1956–1971. [Google Scholar]
- Adenuga, D.; Yao, H.; March, T.H.; Seagrave, J.; Rahman, I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am. J. Respir. Cell. Mol. Biol 2009, 40, 464–473. [Google Scholar]
- Marwick, J.A.; Kirkham, P.A.; Stevenson, C.S.; Danahay, H.; Giddings, J.; Butler, K.; Donaldson, K.; Macnee, W.; Rahman, I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am. J. Respir. Cell. Mol. Biol 2004, 31, 633–642. [Google Scholar]
- Moodie, F.M.; Marwick, J.A.; Anderson, C.S.; Szulakowski, P.; Biswas, S.K.; Bauter, M.R.; Kilty, I.; Rahman, I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-κB activation and proinflammatory cytokine release in alveolar epithelial cells. Faseb. J 2004, 18, 1897–1899. [Google Scholar]
- Seimetz, M.; Parajuli, N.; Pichl, A.; Veit, F.; Kwapiszewska, G.; Weisel, F.C.; Milger, K.; Egemnazarov, B.; Turowska, A.; Fuchs, B.; et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 2011, 147, 293–305. [Google Scholar]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med 2008, 177, 861–870. [Google Scholar]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-κB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol 2006, 291, L46–L57. [Google Scholar]
- Kedar, N.P. Can we prevent Parkinson’s and Alzheimer’s disease? J. Postgrad. Med 2003, 49, 236–245. [Google Scholar]
- Coppede, F.; Migliore, L. Evidence linking genetics, environment, and epigenetics to impaired DNA repair in Alzheimer’s disease. J. Alzheimers Dis 2010, 20, 953–966. [Google Scholar]
- Dizdaroglu, M.; Jaruga, P.; Birincioglu, M.; Rodriguez, H. Free radical-induced damage to DNA: Mechanisms and measurement. Free Radic. Biol. Med 2002, 32, 1102–1115. [Google Scholar]
- Einolf, H.J.; Schnetz-Boutaud, N.; Guengerich, F.P. Steady-state and pre-steady-state kinetic analysis of 8-oxo-7,8-dihydroguanosine triphosphate incorporation and extension by replicative and repair DNA polymerases. Biochemistry 1998, 37, 13300–13312. [Google Scholar]
- Hayakawa, H.; Taketomi, A.; Sakumi, K.; Kuwano, M.; Sekiguchi, M. Generation and elimination of 8-oxo-7,8-dihydro-2′-deoxyguanosine 5′-triphosphate, a mutagenic substrate for DNA synthesis, in human cells. Biochemistry 1995, 34, 89–95. [Google Scholar]
- Myung, N.H.; Zhu, X.; Kruman, I.I.; Castellani, R.J.; Petersen, R.B.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Lee, H.G. Evidence of DNA damage in Alzheimer disease: Phosphorylation of histone H2AX in astrocytes. Age 2008, 30, 209–215. [Google Scholar]
- Zawia, N.H.; Lahiri, D.K.; Cardozo-Pelaez, F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic. Biol. Med 2009, 46, 1241–1249. [Google Scholar]
- Masoro, E.J. Caloric restriction and aging: An update. Exp. Gerontol 2000, 35, 299–305. [Google Scholar]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 2004, 306, 2105–2108. [Google Scholar]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar]
- Feige, J.N.; Auwerx, J. Transcriptional targets of sirtuins in the coordination of mammalian physiology. Curr. Opin. Cell Biol 2008, 20, 303–309. [Google Scholar]
- Chen, Z.; Peng, I.C.; Cui, X.; Li, Y.S.; Chien, S.; Shyy, J.Y. Shear stress, SIRT1, and vascular homeostasis. Proc. Natl. Acad. Sci. USA 2010, 107, 10268–10273. [Google Scholar]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar]
- Donato, A.J.; Magerko, K.A.; Lawson, B.R.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol 2011, 589, 4545–4554. [Google Scholar]
- Spallotta, F.; Cencioni, C.; Straino, S.; Nanni, S.; Rosati, J.; Artuso, S.; Manni, I.; Colussi, C.; Piaggio, G.; Martelli, F.; et al. A nitric oxide-dependent crosstalk between Class I and III histone deacetylases accelerates skin repair. J. Biol. Chem 2013, 288, 11004–11012. [Google Scholar]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar]
- Pelicci, G.; Lanfrancone, L.; Grignani, F.; McGlade, J.; Cavallo, F.; Forni, G.; Nicoletti, I.; Grignani, F.; Pawson, T.; Pelicci, P.G. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 1992, 70, 93–104. [Google Scholar]
- Ventura, A.; Luzi, L.; Pacini, S.; Baldari, C.T.; Pelicci, P.G. The p66Shc longevity gene is silenced through epigenetic modifications of an alternative promoter. J. Biol. Chem 2002, 277, 22370–22376. [Google Scholar]
- Chen, H.Z.; Wan, Y.Z.; Liu, D.P. Cross-talk between SIRT1 and p66Shc in vascular diseases. Trends Cardiovasc. Med. 2013, 14. [Google Scholar] [CrossRef]
- Zaccagnini, G.; Martelli, F.; Fasanaro, P.; Magenta, A.; Gaetano, C.; di Carlo, A.; Biglioli, P.; Giorgio, M.; Martin-Padura, I.; Pelicci, P.G.; et al. p66ShcA modulates tissue response to hindlimb ischemia. Circulation 2004, 109, 2917–2923. [Google Scholar]
- Zaccagnini, G.; Martelli, F.; Magenta, A.; Cencioni, C.; Fasanaro, P.; Nicoletti, C.; Biglioli, P.; Pelicci, P.G.; Capogrossi, M.C. p66(ShcA) and oxidative stress modulate myogenic differentiation and skeletal muscle regeneration after hind limb ischemia. J. Biol. Chem 2007, 282, 31453–31459. [Google Scholar]
- Di Stefano, V.; Cencioni, C.; Zaccagnini, G.; Magenta, A.; Capogrossi, M.C.; Martelli, F. p66ShcA modulates oxidative stress and survival of endothelial progenitor cells in response to high glucose. Cardiovasc. Res 2009, 82, 421–429. [Google Scholar]
- Giorgio, M.; Berry, A.; Berniakovich, I.; Poletaeva, I.; Trinei, M.; Stendardo, M.; Hagopian, K.; Ramsey, J.J.; Cortopassi, G.; Migliaccio, E.; et al. The p66Shc knocked out mice are short lived under natural condition. Aging Cell 2012, 11, 162–168. [Google Scholar]
- Satoh, A.; Stein, L.; Imai, S. The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. Handb. Exp. Pharmacol 2011, 206, 125–162. [Google Scholar]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov 2006, 5, 493–506. [Google Scholar]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch. Biochem. Biophys 2010, 501, 79–90. [Google Scholar]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar]
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004, 116, 551–563. [Google Scholar]
- Minor, R.K.; Baur, J.A.; Gomes, A.P.; Ward, T.M.; Csiszar, A.; Mercken, E.M.; Abdelmohsen, K.; Shin, Y.K.; Canto, C.; Scheibye-Knudsen, M.; et al. SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 2011, 1. [Google Scholar] [CrossRef]
- Mai, A.; Valente, S.; Meade, S.; Carafa, V.; Tardugno, M.; Nebbioso, A.; Galmozzi, A.; Mitro, N.; de Fabiani, E.; Altucci, L.; et al. Study of 1,4-dihydropyridine structural scaffold: Discovery of novel sirtuin activators and inhibitors. J. Med. Chem 2009, 52, 5496–5504. [Google Scholar]
- Caton, P.W.; Nayuni, N.K.; Kieswich, J.; Khan, N.Q.; Yaqoob, M.M.; Corder, R. Metformin suppresses hepatic gluconeogenesis through induction of SIRT1 and GCN5. J. Endocrinol 2010, 205, 97–106. [Google Scholar]
- Li, Q.; Xiao, H.; Isobe, K. Histone acetyltransferase activities of cAMP-regulated enhancer-binding protein and p300 in tissues of fetal, young, and old mice. J. Gerontol. A Biol. Sci. Med. Sci 2002, 57, B93–B98. [Google Scholar]
- Bandyopadhyay, D.; Okan, N.A.; Bales, E.; Nascimento, L.; Cole, P.A.; Medrano, E.E. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res 2002, 62, 6231–6239. [Google Scholar]
- Kang, H.L.; Benzer, S.; Min, K.T. Life extension in Drosophila by feeding a drug. Proc. Natl. Acad. Sci. USA 2002, 99, 838–843. [Google Scholar]
- Tao, D.; Lu, J.; Sun, H.; Zhao, Y.M.; Yuan, Z.G.; Li, X.X.; Huang, B.Q. Trichostatin A extends the lifespan of Drosophila melanogaster by elevating hsp22 expression. Acta Biochim. Biophys. Sin 2004, 36, 618–622. [Google Scholar]
- Zhao, Y.; Sun, H.; Lu, J.; Li, X.; Chen, X.; Tao, D.; Huang, W.; Huang, B. Lifespan extension and elevated hsp gene expression in Drosophila caused by histone deacetylase inhibitors. Exp. Biol 2005, 208, 697–705. [Google Scholar]
- Bush, E.W.; McKinsey, T.A. Protein acetylation in the cardiorenal axis: The promise of histone deacetylase inhibitors. Circ. Res 2010, 106, 272–284. [Google Scholar]
- McKinsey, T.A. Targeting inflammation in heart failure with histone deacetylase inhibitors. Mol. Med 2011, 17, 434–441. [Google Scholar]
- Ferguson, B.S.; Harrison, B.C.; Jeong, M.Y.; Reid, B.G.; Wempe, M.F.; Wagner, F.F.; Holson, E.B.; McKinsey, T.A. Signal-dependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 9806–9811. [Google Scholar]
- Yuan, Y.; Wang, Q.; Paulk, J.; Kubicek, S.; Kemp, M.M.; Adams, D.J.; Shamji, A.F.; Wagner, B.K.; Schreiber, S.L. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Biol 2012, 7, 1152–1157. [Google Scholar]

{kind=link}
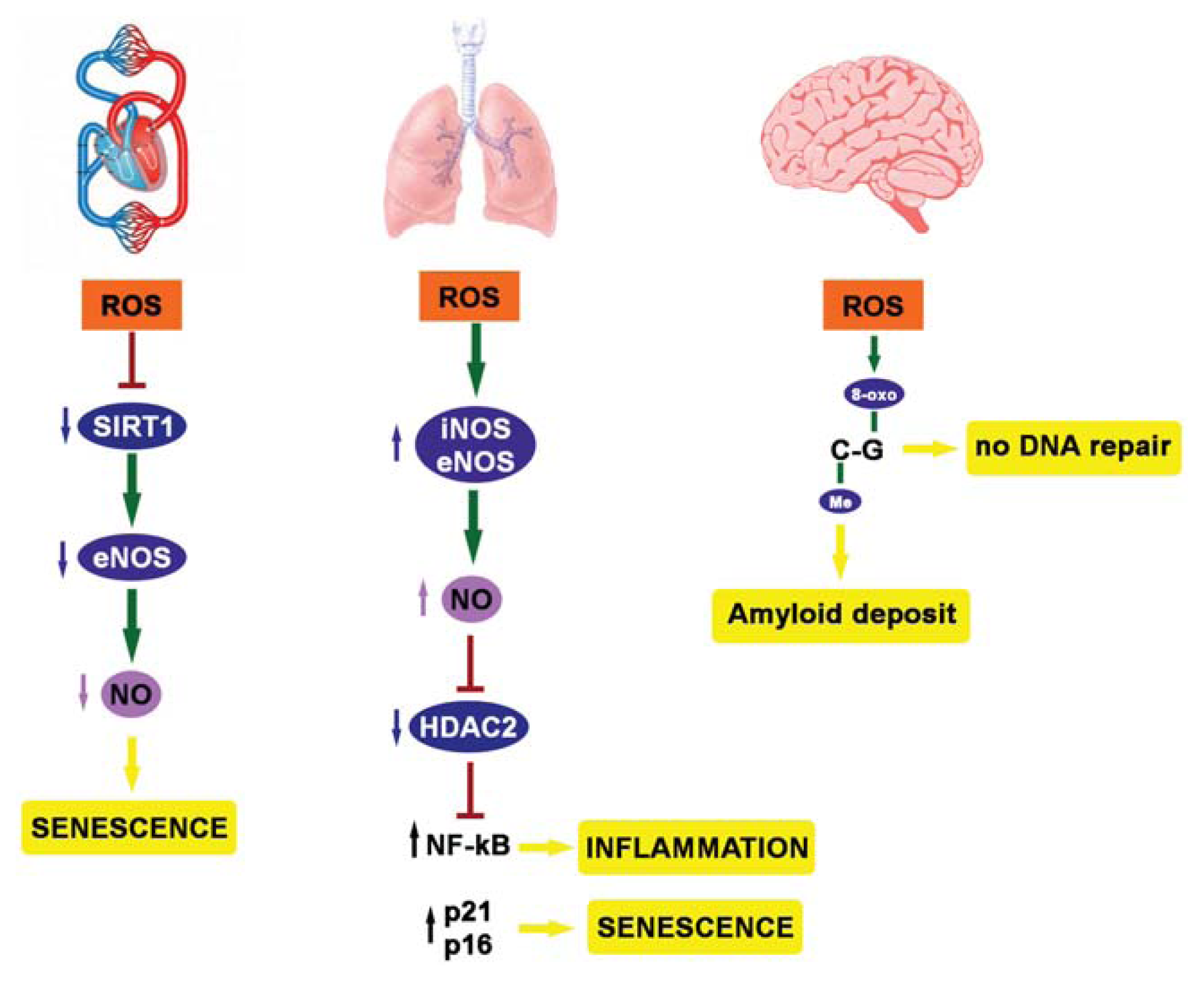{kind=link}
{kind=link}
| Epigenetic ageing marker | Regulation | Reference |
|---|---|---|
| Global DNA methylation | Decreased | [22] |
| DNA methylase activity | Decreased | [22] |
| PRC1, PRC2 | Decreased | [26] |
| SIRT1 | Decreased | [22,26,27] |
| H3K36me3, H3K9me3, H4K20me | Decreased | [30] |
| miR-71 | Decreased | [38] |
| c-fos, IGF-II, p16Ink4a methylation | Increased | [23–25] |
| H4K16ac | Increased | [22,26,27] |
| JMJD3 | Increased | [33,34] |
| H3K27me3, H3K79me/me2 | Increased | [30] |
| H4K20me3 | Increased | [31,32] |
| SAHFs | Increased | [35–37] |
| mir-29 | Increased | [39–41] |
| mir-34a | Increased | [40–43] |
| mir-200 family | Increased | [44] |
| Drugs | Condition | clinicaltrials.gov Identifier | Phase |
|---|---|---|---|
| Resveratrol | Type 2 diabetes | NCT01677611 | I, Completed |
| Resveratrol | Vascular resistance, aging, hypertension, antioxidants, aerobic capacity | NCT01842399 | II |
| Resveratrol | Healthy | NCT00996229 | III |
| Resveratrol | Alzheimer’s disease | NCT00678431 | III, completed |
| SRT-2104 | Type 2 diabetes | NCT00937872, NCT00933062, NCT00933530, NCT01018017 | I, II |
| SRT-2379 | Type 2 diabetes | NCT01018628 | I |
| Metformin | COPD | NCT01247870 | IV |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cencioni, C.; Spallotta, F.; Martelli, F.; Valente, S.; Mai, A.; Zeiher, A.M.; Gaetano, C. Oxidative Stress and Epigenetic Regulation in Ageing and Age-Related Diseases. Int. J. Mol. Sci. 2013, 14, 17643-17663. https://doi.org/10.3390/ijms140917643
Cencioni C, Spallotta F, Martelli F, Valente S, Mai A, Zeiher AM, Gaetano C. Oxidative Stress and Epigenetic Regulation in Ageing and Age-Related Diseases. International Journal of Molecular Sciences. 2013; 14(9):17643-17663. https://doi.org/10.3390/ijms140917643
Chicago/Turabian StyleCencioni, Chiara, Francesco Spallotta, Fabio Martelli, Sergio Valente, Antonello Mai, Andreas M. Zeiher, and Carlo Gaetano. 2013. "Oxidative Stress and Epigenetic Regulation in Ageing and Age-Related Diseases" International Journal of Molecular Sciences 14, no. 9: 17643-17663. https://doi.org/10.3390/ijms140917643
APA StyleCencioni, C., Spallotta, F., Martelli, F., Valente, S., Mai, A., Zeiher, A. M., & Gaetano, C. (2013). Oxidative Stress and Epigenetic Regulation in Ageing and Age-Related Diseases. International Journal of Molecular Sciences, 14(9), 17643-17663. https://doi.org/10.3390/ijms140917643