Cadmium Modifies the Cell Cycle and Apoptotic Profiles of Human Breast Cancer Cells Treated with 5-Fluorouracil


 ,
,  ,
, 

 ,
, 
Abstract
:1. Introduction
2. Results
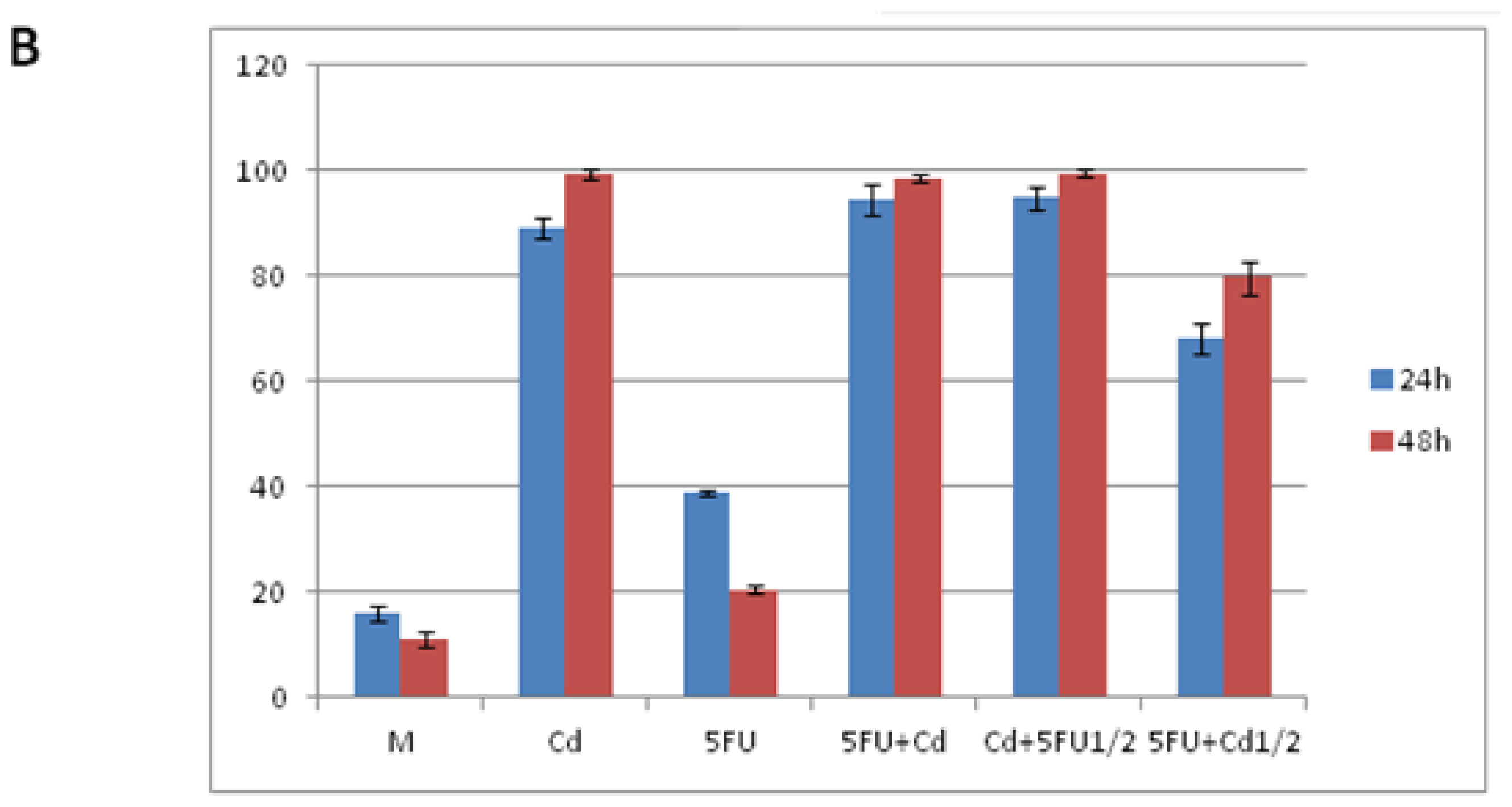
2.1. Effect of Cd and 5-FU on Cell Cycle Analysis and Apoptosis
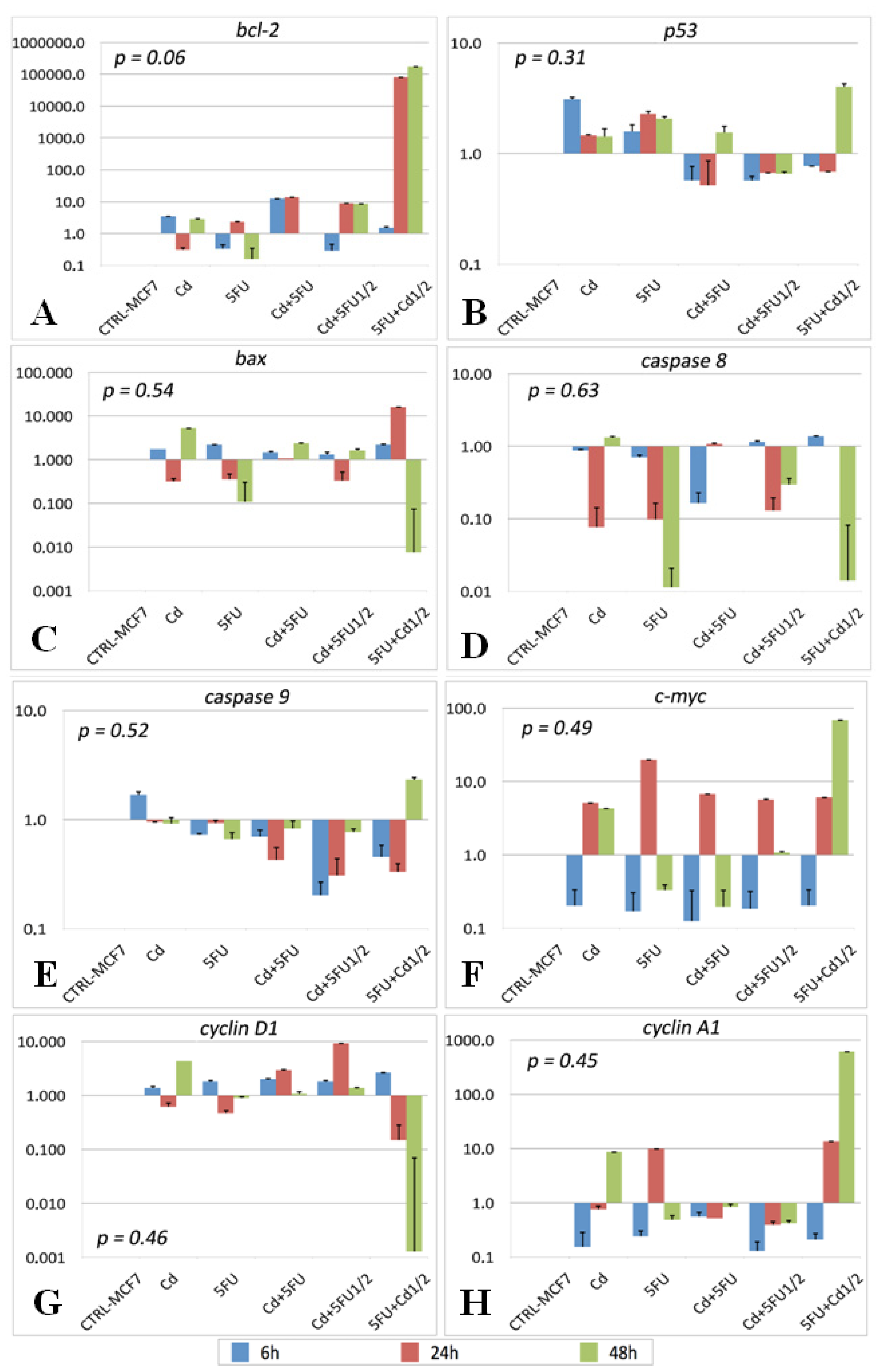
2.2. Gene Expression
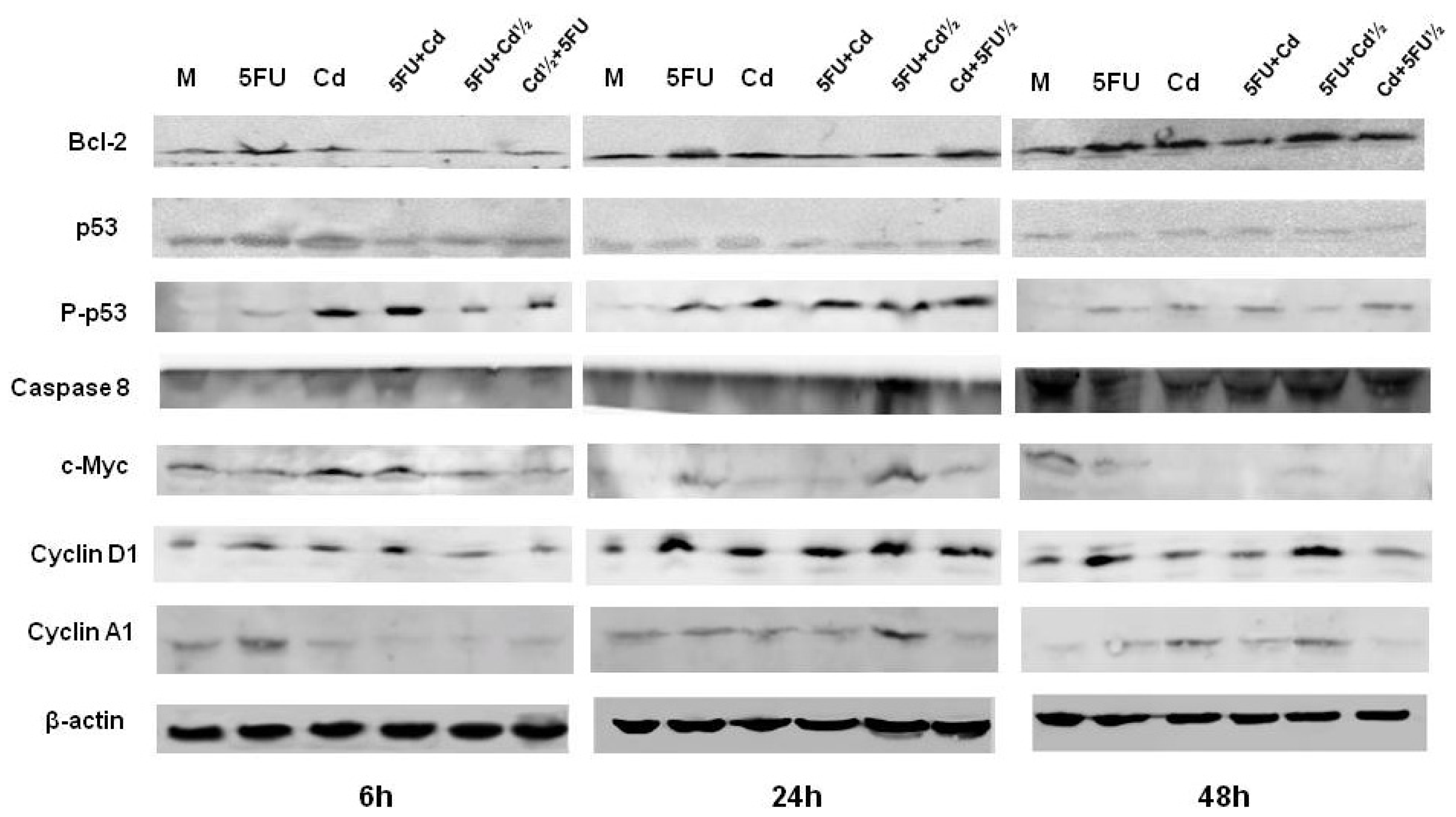
2.3. Protein Expression
3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. Drugs
4.3. Cell Cycle Distribution Analysis
4.4. Apoptosis Detection by Staining with Annexin V-FITC and Propidium Iodide
4.5. Gene Expression
4.6. Protein Expression
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Nadkarni, R.A. Some considerations of metal content of tobacco products. Chem. Ind 1974, 17, 693–696. [Google Scholar]
- Hartwig, A. Cadmium and cancer. Met. Ions Life Sci 2013, 11, 491–507. [Google Scholar]
- Liu, Y.; Templeton, D.M. Role of the cytoskeleton in Cd2+-induced death of mouse mesangial cells. Can. J. Physiol. Pharmacol 2010, 88, 341–352. [Google Scholar]
- Waalkes, M.P.; Rehm, S.; Riggs, C.W.; Bare, R.M.; Devor, D.E.; Poirier, L.A.; Wenk, M.L.; Henneman, J.R.; Balaschak, M.S. Cadmium carcinogenesis in male Wistar rats: Dose-response analysis of tumor induction in the prostate and testes and at the injection site. Cancer Res 1988, 48, 4656–4663. [Google Scholar]
- Waalkes, M.P. Cadmium carcinogenesis. Mutat. Res 2003, 533, 107–120. [Google Scholar]
- Lewis, G.P.; Coughlin, L.; Jusko, W.; Hartz, S. Contribution of cigarette smoking to cadmium accumulation in man. Lancet 1972, 1, 291–292. [Google Scholar]
- Madeddu, R.; Muresu, E.; Montella, A.; Chessa, G.; Cherchi, G.B.; Piras, P.; Vargiu, P.; Tolu, P.; Pirino, A.; Prados, J.C.; et al. Low cadmium concentration in whole blood from residents in Northern Sardinia (Italy) with special reference to smoking habits. JPMH 2009, 50, 46–52. [Google Scholar]
- Galal-Gorchev, H. Dietary intake, levels in food and estimated intake of lead, cadmium and mercury. Food Add. Contam 1993, 10, 115–128. [Google Scholar]
- Moy, G.; Käferstein, F.; Kim, Y.M.; Motarjemi, Y.; Quevedo, F. Dietary exposure to lead, cadmium, mercury and polychlorinated biphenyls. Archv. Lebensmittelhygiene 1993, 44, 45–51. [Google Scholar]
- Madeddu, R.; Tolu, P.; Asara, Y.; Farace, C.; Forte, G.; Bocca, B. Blood biomonitoring of metals in subjects living near abandoned mining and active industrial areas. Environ. Monit. Assess 2012, 185, 5837–5846. [Google Scholar]
- Matović, V.; Buha, A.; Bulat, Z.; Dukić-Ćosić, D. Cadmium toxicity revisited: Focus on oxidative stress induction and interactions with zinc and magnesium. Arh. Hig. Rada. Toksikol 2011, 62, 65–76. [Google Scholar]
- World Health Organization, Environmental Health Criteria 134–Cadmium International Programme on Chemical Safety (IPCS) Monograph; World Health Organization: Geneva, Switzerland, 1992.
- Benbrahim-Tallaa, L.; Tokar, E.J.; Diwan, B.E.; Dill, A.L.; Coppin, J.F.; Waalkes, M.P. Cadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotype. Environ. Health Perspect 2009, 117, 1847–1852. [Google Scholar]
- Geeth-Gunawardana, C.; Martinez, R.E.; Weiqun, X.; Templeton, D.M. Cadmium inhibits both intrinsic and extrinsic apoptotic pathways in renal mesanglial cells. Am. J. Physiol. Renal. Physiol 2005, 290, F1074–F1082. [Google Scholar]
- Itoh, H.; Iwasaki, M.; Sawada, N.; Takachi, R.; Kasuga, Y.; Yokoyama, S.; Onuma, H.; Nishimura, H.; Kusama, R.; Yokoyama, K.; et al. Dietary cadmium intake and breast cancer risk in Japanese women: A case-control study. Int. J. Hyg. Environ. Health 2013. [Google Scholar] [CrossRef]
- Nagata, C.; Nagao, Y.; Nakamura, K.V.; Wada, K.V.; Tamai, Y.; Tsuji, M.; Yamamoto, S.; Kashiki, Y. Cadmium exposure and the risk of breast cancer in Japanese women. Breast Cancer Res. Treat 2013, 138, 235–239. [Google Scholar]
- Byrne, C.; Divekar, S.D.; Storchan, G.B.; Parodi, D.A.; Martin, M.B. Metals and breast cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 63–73. [Google Scholar]
- Lacroix, M.; Leclercq, G. Relevance of breast cancer cell lines as models for breast tumours: An update. Breast Res. Treat 2004, 83, 249–289. [Google Scholar]
- Lacroix, M.; Toillon, R.A.; Leclercq, G. p53 and breast cancer, an update. Endocr.-Relat. Cancer 2006, 13, 293–325. [Google Scholar]
- Levenson, A.S.; Jordan, V.C. MCF-7: The first hormone-responsive breast cancer cell line. Cancer Res 1997, 57, 3071–3078. [Google Scholar]
- Charafe-Jauffret, E.; Ginestier, C.; Monville, F.; Finetti, P.; Adelaide, J.; Cervera, N.; Fekairi, S.; Xerri, L.; Jacquemier, J.; Birnbaum, D.; et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006, 25, 2273–2284. [Google Scholar]
- Jian, H.; Coleman, J.; Miskimins, R.; Miskimins, K. Expression of constituvely active 4EBP-I enhances p27kipI expression and inhibits proliferation of MCF7 breast cancer cells. Cancer Cell Int 2003, 3, 2. [Google Scholar]
- Lukyanova, N.Y.; Rusetskya, N.V.; Tregubova, N.A.; Chekhun, V.F. Molecular and cell cycle in MCF-7 cells resistant to cisplatin and doxorubicin. Exp. Oncol 2009, 31, 87–91. [Google Scholar]
- Soule, H.D.; Vazquez, J.; Long, A.; Albert, S.; Brennan, M. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst 1973, 51, 1409–1416. [Google Scholar]
- Zhang, Y.; Li, J.H.; Jiang, F.L.; Tian, F.F.; Liu, Y. Spectroscopic and microscopic studies on the mechanisms of mitochondrial toxicity induced by different concentrations of cadmium. J. Membr. Biol 2011, 241, 39–49. [Google Scholar]
- Lu, Z.; Zhang, R.; Diasio, R.B. Population characteristics of hepatic dihydropyrimidine dehydrogenase activity, a key metabolic enzyme in 5-fluorouracil chemotherapy. Clin. Pharmacol. Ther 1995, 58, 512–522. [Google Scholar]
- Marchal, J.A.; Melguizo, C.; Prados, J.; Aránega, A.E.; Gómez, J.A.; Campos, J.; Gallo, M.A.; Espinosa, A.; Arena, N.; Aránega, A. Modulation of myogenic differentiation in a human rhabdomyosarcoma cell line by a new derivative of 5-fluorouracil (QF-3602). Jpn. J. Cancer Res 2000, 91, 934–940. [Google Scholar]
- Asara, Y.; Marchal, J.A.; Bandiera, P.; Mazzarello, V.; Delogu, L.G.; Sotgiu, M.A.; Montella, A.; Madeddu, R. Cadmium influences the 5-Fluorouracil cytotoxic effects on breast cancer cells. Eur. J. Histochem 2012, 56, e1. [Google Scholar]
- Bocca, B.; Madeddu, R.; Asara, Y.; Tolu, P.; Marchal, J.A.; Forte, G. Assessment of reference ranges for blood Cu, Mn, Se and Zn in a selected Italian population. J. Trace Elem. Med. Biol 2011, 25, 19–26. [Google Scholar]
- Magné, N.; Fischel, J.L.; Tiffon, C.; Formento, P.; Dubreuil, A.; Renée, N.; Formento, J.L.; Francoual, M.; Ciccolini, J.; Etienne, M.C.; et al. Molecular mechanisms underlying the interaction between ZD1839 (“Iressa”) and cisplatin/5-fluorouracil. Br. J. Cancer 2003, 4, 89, , 585–592.. [Google Scholar]
- Brama, M.; Gnessi, L.; Basciani, S.; Cerulli, N.; Politi, L.; Spera, G.; Mariani, S.; Cherubini, S.; d’Abusco, A.S.; Scandurra, R.; et al. Cadmium induces mitogenic signaling in breast cancer cell by an ERalpha-dependent mechanism. Mol. Cell. Endocrinol 2007, 264, 102–108. [Google Scholar]
- Bertin, G.; Averbeck, D. Cadmium: Cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences. Biochimie 2006, 88, 1549–1559. [Google Scholar]
- Aimola, P.; Carmignani, M.; Volpe, A.R.; Di Benedetto, A.; Claudio, L.; Waalkes, M.P.; van Bokhoven, A.; Tokar, E.J.; Claudio, P.P. Cadmium induces p53-dependent apoptosis in human prostate epithelial cells. PLoS One 2012, 7, e33647. [Google Scholar]
- Vujčić, M.; Lazić, M.; Milenković, M.; Sladić, D.; Radulović, S.; Filipović, N.; Anđelković, K. A comparative study of DNA binding and cell cycle phase perturbation by the dinuclear complex of Cd(II) with the condensation product of 2-acetylpyridine and malonic acid dihydrazide N′,N′ (2)-bis[(1E)-1-(2-pyridyl)ethylidene]propanedihydrazide. J. Biochem. Mol. Toxicol 2011, 25, 175–182. [Google Scholar]
- Álvarez, P.; Marchal, J.A.; Boulaiz, H.; Carrillo, E.; Vélez, C.; Rodríguez-Serrano, F.; Melguizo, C.; Prados, J.; Madeddu, R.; Aranega, A. 5-Fluorouracil derivatives: A patent review. Expert Opin. Ther. Pat 2012, 22, 107–123. [Google Scholar]
- Wyatt, M.D.; Wilson, D.M., III. Participation of DNA repair in the response to 5-fluorouracil. Cell Mol. Life Sci. 2009, 66, 788–799. [Google Scholar]
- Achanzar, W.E.; Achanzar, K.B.; Lewis, J.G.; Webber, M.M.; Waalkes, M.P. Cadmium induces c-myc, p53, and c-jun expression in normal human prostate epithelial cells as a prelude to apoptosis. Toxicol. Appl. Pharmacol 2000, 164, 291–300. [Google Scholar]
- Waisberg, M.; Joseph, P.; Hale, B.; Beyersmann, D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 2003, 192, 95–117. [Google Scholar]
- Bursch, W.; Karwan, A.; Mayer, M.; Dornetshuber, J.; Fröhwein, U.; Schulte-Hermann, R.; Fazi, B.; Di Sano, F.; Piredda, L.; Piacentini, M.; et al. Cell death and autophagy: Cytokines, drugs, and nutritional factors. Toxicology 2008, 254, 147–157. [Google Scholar]
- Singh, R.; Saini, N. Downregulation of BCL2 by miRNAs augments drug-induced apoptosis—A combined computational and experimental approach. J. Cell Sci 2012, 125, 1568–1578. [Google Scholar]
- Matsuoka, M.; Igisu, H. Cadmium Induces Phosphorylation of p53 at Serine 15 in MCF-7 Cells. Biochem. Biophys. Res. Commun 2001, 282, 1120–1125. [Google Scholar]
- Cheok, C.F.; Verma, C.S.; Baselga, J.; Lane, D.P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol 2011, 8, 25–37. [Google Scholar]
- Cheok, C.F.; Lane, D.P. Seeking synergy in p53 transcriptional activation for cancer therapy. Discov. Med 2012, 14, 263–271. [Google Scholar]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar]
- Xue, C.; Haber, M.; Flemming, C.; Marshall, G.M.; Lock, R.B.; MacKenzie, K.L.; Gurova, K.V.; Norris, M.D.; Gudkov, A.V. p53 determines multidrug sensitivity of childhood neuroblastoma. Cancer Res 2007, 67, 10351–10360. [Google Scholar]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol 2008, 21, 1156–1167. [Google Scholar]
- Palaskas, N.; Larson, S.M.; Schultz, N.; Komisopoulou, E.; Wong, J.; Rohle, D.; Campos, C.; Yannuzzi, N.; Osborne, J.R.; Linkov, I.; et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res 2011, 71, 5164–5174. [Google Scholar]
- Chan, C.B.; Liu, X.; Jang, S.W.; Hsu, S.I.H.; Williams, I.; Kang, S.; Chen, J.; Ye, K. NGF inhibits human leukemia proliferation by downregulatingcyclin A1 expression through promoting acinus/CtBP2 association. Oncogene 2009, 28, 3825–3836. [Google Scholar]
- Marlow, L.A.; von Roemeling, C.A.; Cooper, S.J.; Zhang, Y.; Rohl, S.D.; Arora, S.; Gonzales, I.M.; Azorsa, D.O.; Reddi, H.V.; Tun, H.W.; et al. Foxo3a drives proliferation in anaplastic thyroid carcinoma through transcriptional regulation of cyclin A1: A paradigm shift that impacts current therapeutic strategies. J. Cell Sci 2012, 125, 4253–4263. [Google Scholar]
- Yang, Y.; Wang, F.; Shi, C.; Zou, Y.; Qin, H.; Ma, Y. Cyclin D1 G870A polymorphism contributes to colorectal cancer susceptibility: Evidence from a systematic review of 22 case-control studies. PLoS One 2012, 7, e36813. [Google Scholar]
- Marchal, J.A.; Boulaiz, H.; Suarez, I.; Saniger, I.; Campos, J.; Carillo, E.; Prados, J.; Gallo, M.A.; Espinosa, A.; Aránega, A. Growth inhibition, G1-arrest, and apoptosis in MCF-7 human breast cancer cells by novel highly lipophilic 5-fluorouracil derivatives. Investig. N. Drugs 2004, 22, 379–389. [Google Scholar]
- STATA/SE, version 12; software for statistical analyse; StataCorp LP: College Station, TX, USA, 2011.
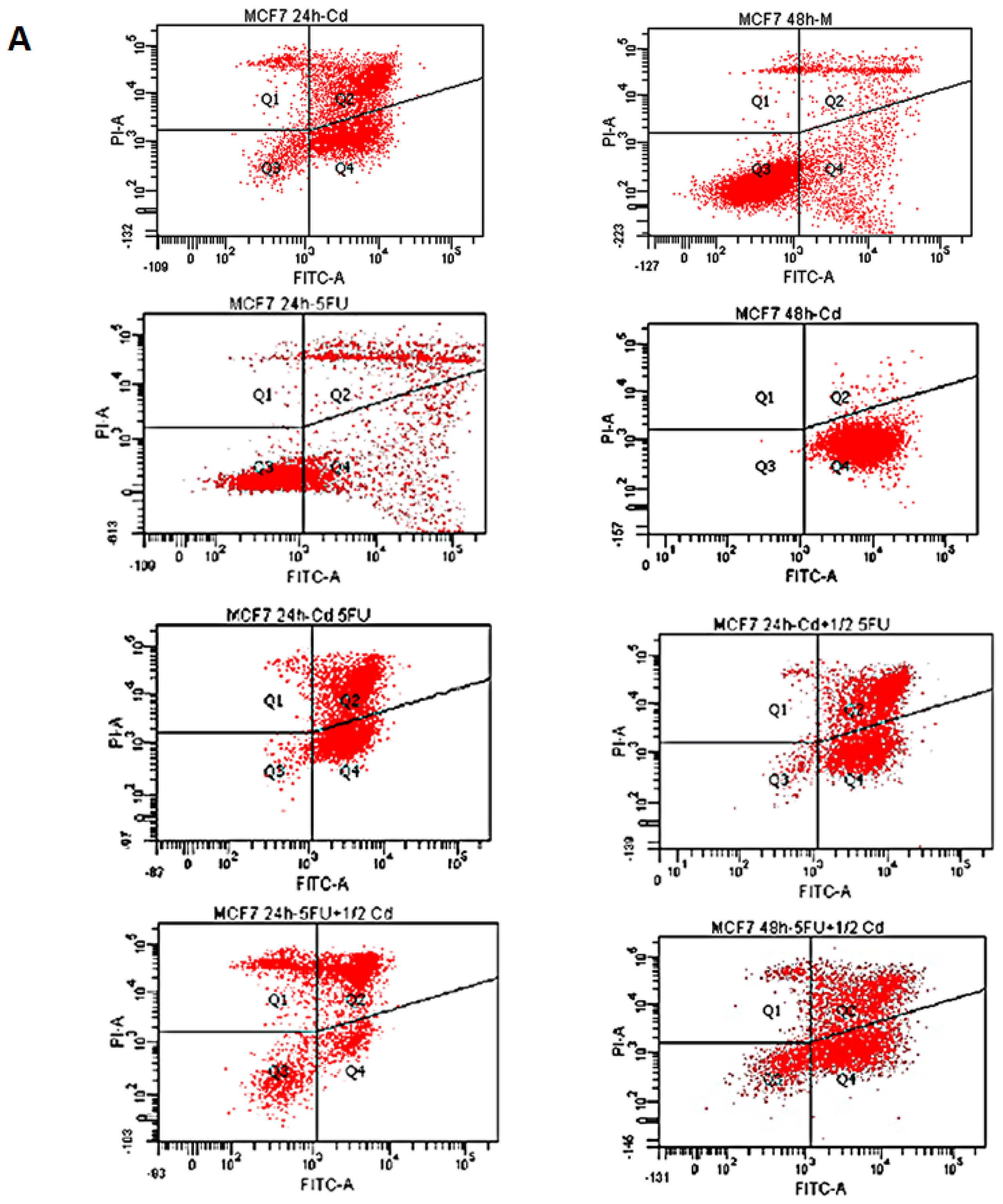

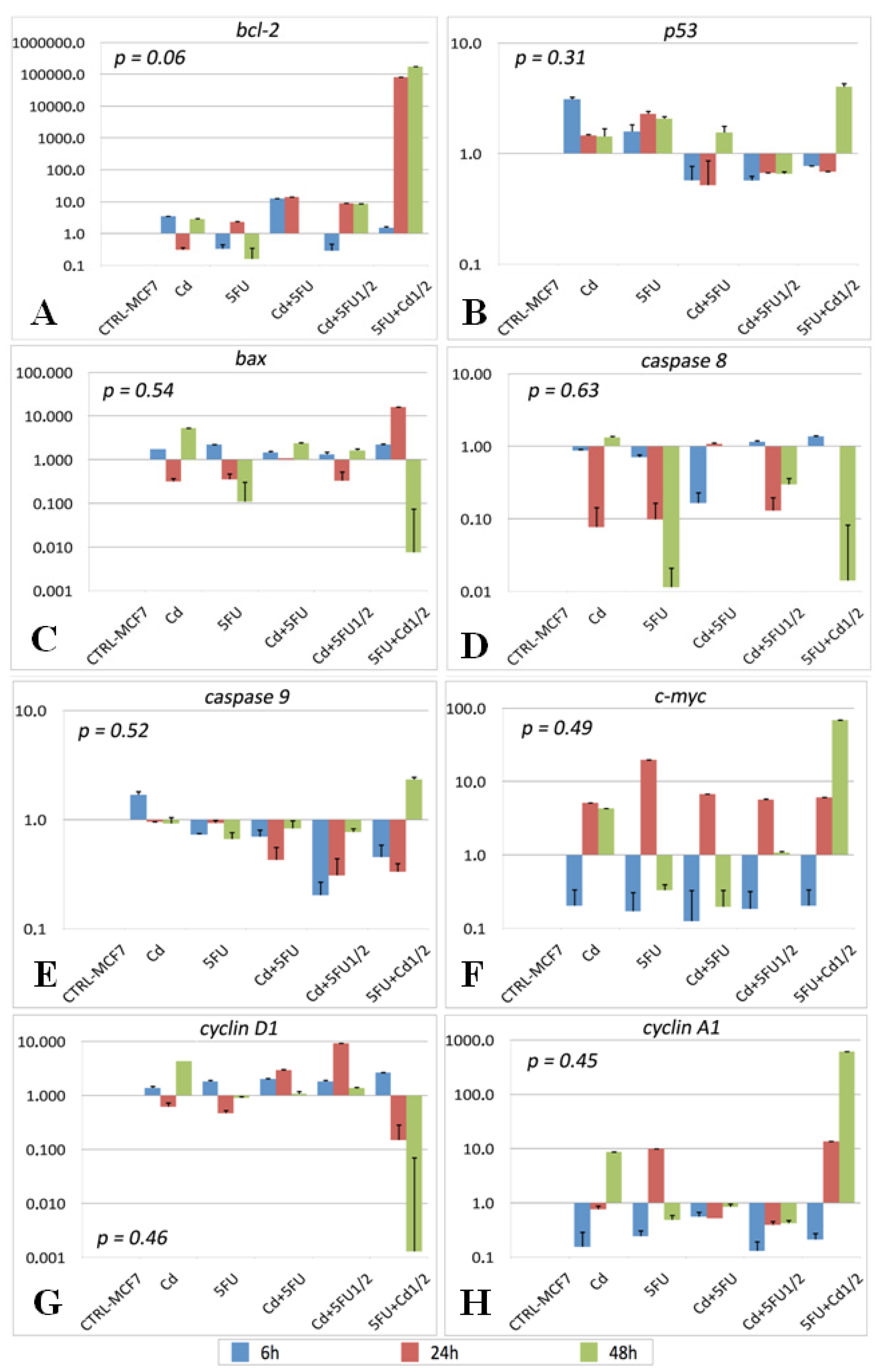
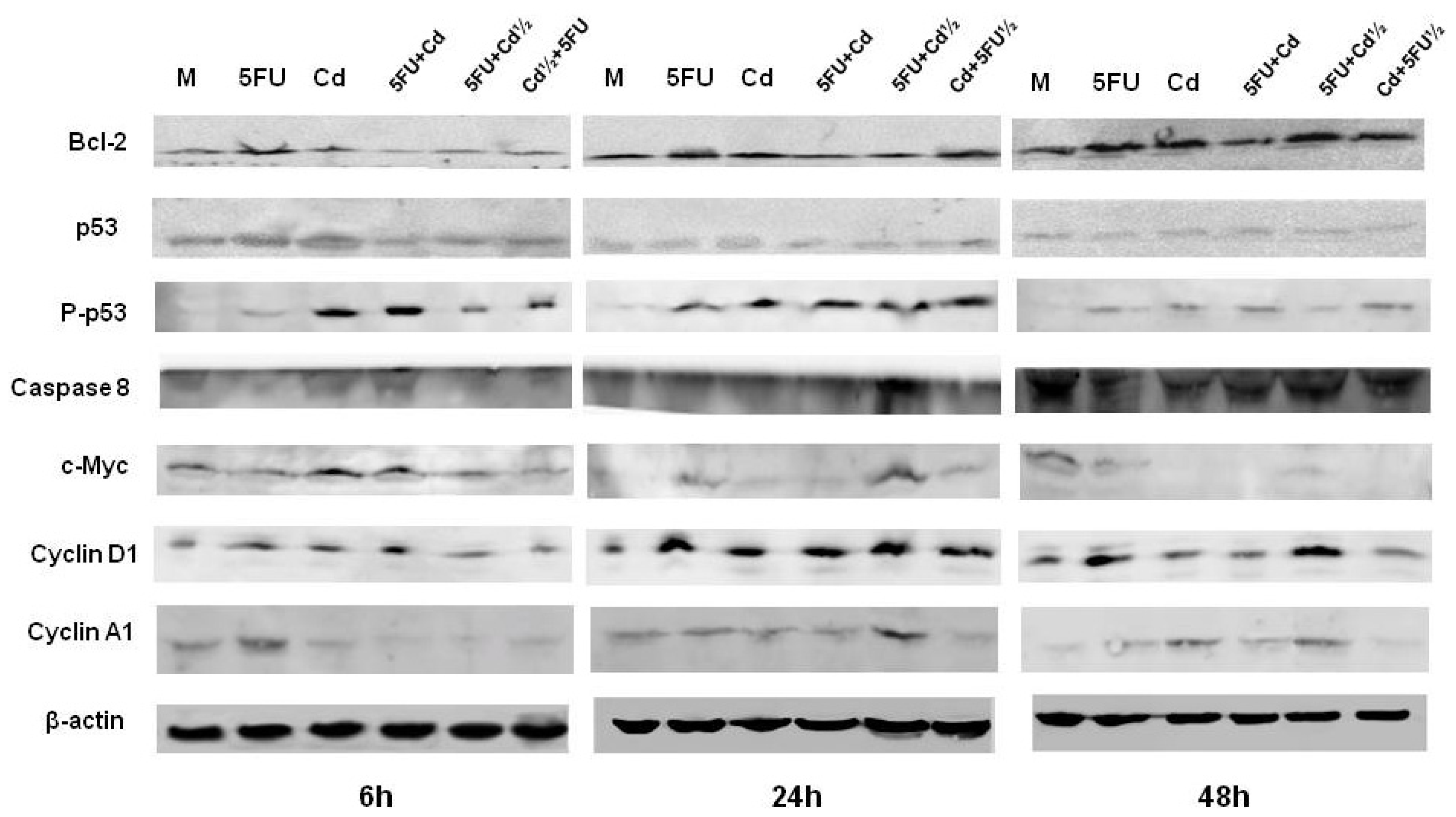

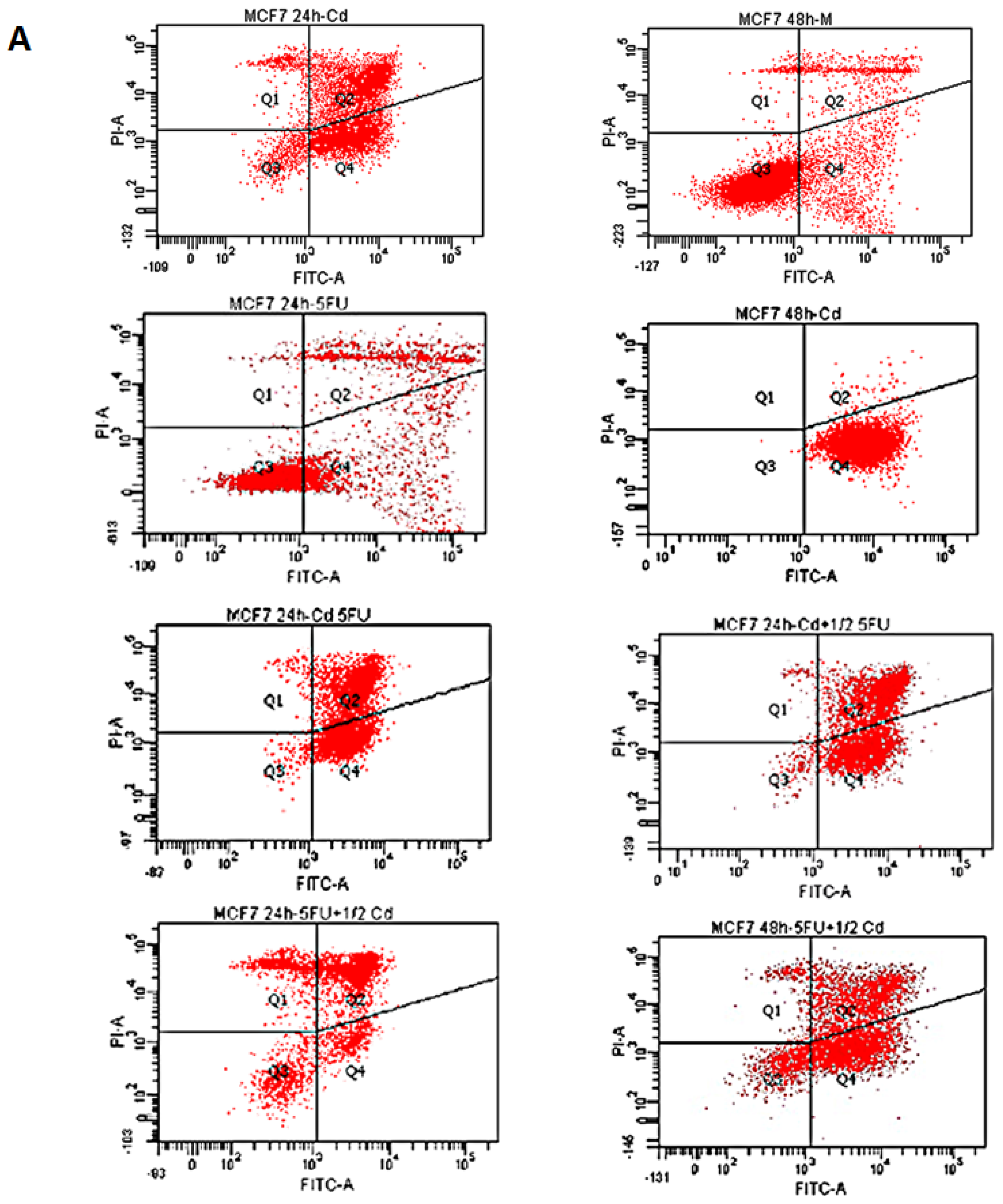{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | Cd | 5FU | 5FU + Cd | Cd + 5FU½ | 5FU + Cd½ | |
|---|---|---|---|---|---|---|
| 6 h | ||||||
| G0/G1 | 56.7 ± 1.2 | 61.1 ± 1.64 | 59.0 ± 0.85 | 61.5 ± 1.23 | 62.9 ± 0.86 | 61.1 ± 2.34 |
| S | 28.0 ± 0.95 | 23.0 ± 1.06 | 28.6 ± 0.19 | 24.2 ± 1.27 | 24.9 ± 0.56 | 26.8 ± 1.21 |
| G2/M | 14.0 ± 0.35 | 15.8 ± 0.57 | 12.2 ± 0.78 | 14.2 ± 0.19 | 11.3 ± 0.75 | 15.7 ± 0.1 |
| 12 h | ||||||
| G0/G1 | 61.6 ± 2.07 * | 49.0 ± 1.19 ** | 62.1 ± 1.27 * | 58.9 ± 1.21 | 61.8 ± 0.93 | 60.0 ± 1.54 |
| S | 21.7 ± 1.54 * | 26.1 ± 0.56 * | 24.6 ± 0.29 *** | 26.2 ± 0.31 | 22.2 ± 1.07 * | 26.9 ± 0.6 |
| G2/M | 16.2 ± 0.77 * | 6.9 ± 0.74 * | 12.9 ± 0.35 | 14.4 ± 0.43 | 14.6 ± 0.65 * | 12.6 ± 0.22 *** |
| 24 h | ||||||
| G0/G1 | 81.7 ± 2.88 ** | 66.9 ± 1.2 * | 75.2 ± 2.33 ** | 63.0 ± 1.54 | 74.9 ± 1.76 ** | 72.2 ± 2.81 |
| S | 9.5 ± 0.32 *** | 18.1 ± 1.35 * | 15.3 ± 0.88 *** | 15.7 ± 1.02 ** | 14.0 ± 0.58 *** | 15.2 ± 0.76 ** |
| G2/M | 8.5 ± 0.08 *** | 5.5 ± 0.1 ** | 8.8 ± 0.55 * | 2.3 ± 0.12 *** | 3.1 ± 0.05 *** | 9.5 ± 0.56 *** |
| 48 h | ||||||
| G0/G1 | 85.9 ± 3.21 ** | 60.5 ± 2.03 | 82.9 ± 3.08 ** | 72.3 ± 2.44 * | 75.5 ± 1.56 ** | 78.2 ± 1.8 ** |
| S | 5.8 ± 0.88 *** | 23.5 ± 1.1 | 9.2 ± 0.77 *** | 11.5 ± 1.1 ** | 12.3 ± 0.9 *** | 9.0 ± 1.3 ** |
| G2/M | 6.7 ± 0.5 *** | 12.3 ± 0.21 ** | 4.9 ± 0.4 ** | 0.3 ± 0.02 *** | 0.2 ± 0.01 *** | 0.9 ± 0.03 *** |
| Time points | ||
|---|---|---|
| M | Control non treated cells | 6 h, 24 h, 48 h |
| Cd | Cells treated with Cd | 6 h, 24 h, 48 h |
| 5FU | Cells treated with 5FU | 6 h, 24 h, 48 h |
| Cd + 5FU | Cells treated with both drugs | 6 h, 24 h, 48 h |
| Cd + 5FU½ | Cells treated with Cd plus 5-FU added after the half of time from the experiment started | 6 h, 24 h, 48 h |
| 5FU + Cd½ | Cells treated with 5-FU plus Cd added after the half time from the experiment started | 6 h, 24 h, 48 h |
| Primer | Annealing temperature (°C) | |
|---|---|---|
| bcl-2 | Forward: 5′-TGGTGGTTTGACCTTTAGAGA-3′ Reverse: 5′-AGGTCTGATCATTCTGTTC-3′ | 55 |
| p 53 | Forward: 5′-GGCATTCTGGGAGCTTCATCT-3′ Reverse: 5′-CCCAAGCAATGGATGATTTGA-3′ | 58.5 |
| bax | Forward: 5′-TGCTTCAGGGTTTCATCCAG-3′ Reverse: 5′-GGCGGCAATCATCCTCTG-3′ | 55 |
| caspase 8 | Forward: 5′-AGGAGGAGATGGAAAGGGAACTT-3′ Reverse: 5′-ACCTCAATTCTGATCTGCTCACTTCT-3′ | 55 |
| caspase 9 | Forward: 5′-CCTCAAACTCTCAAGAGCAC-3′ Reverse: 5′-GAGTCAGGCTCTTCCTTTG-3′ | 58.5 |
| c-myc | Forward: 5′-GGACGACGAGACCTTCATCAA-3′ Reverse: 5′-CCAGCTTCTCTGAGACGAGCTT-3′ | 55 |
| cyclin D1 | Forward: 5′-CCGTCCATGCGGAAGATC-3′ Reverse: 5′-ATGGCCAGCGGGAAGAC-3′ | 55 |
| cyclin A1: | Forward: 5′-GCACCCTGCTCGTCACTTG-3′ Reverse: 5′-AGCCCCCAATAAAAGATCCAG-3′ | 55 |
| GAPDH | Forward: 5′-CAAGGAGTAAGACCCCTGGAC-3′ Reverse: 5′-TCTACATGGCAACTGTGAGGAG-3′ | 58.5 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Asara, Y.; Marchal, J.A.; Carrasco, E.; Boulaiz, H.; Solinas, G.; Bandiera, P.; Garcia, M.A.; Farace, C.; Montella, A.; Madeddu, R. Cadmium Modifies the Cell Cycle and Apoptotic Profiles of Human Breast Cancer Cells Treated with 5-Fluorouracil. Int. J. Mol. Sci. 2013, 14, 16600-16616. https://doi.org/10.3390/ijms140816600
Asara Y, Marchal JA, Carrasco E, Boulaiz H, Solinas G, Bandiera P, Garcia MA, Farace C, Montella A, Madeddu R. Cadmium Modifies the Cell Cycle and Apoptotic Profiles of Human Breast Cancer Cells Treated with 5-Fluorouracil. International Journal of Molecular Sciences. 2013; 14(8):16600-16616. https://doi.org/10.3390/ijms140816600
Chicago/Turabian StyleAsara, Yolande, Juan A. Marchal, Esther Carrasco, Houria Boulaiz, Giuliana Solinas, Pasquale Bandiera, Maria A. Garcia, Cristiano Farace, Andrea Montella, and Roberto Madeddu. 2013. "Cadmium Modifies the Cell Cycle and Apoptotic Profiles of Human Breast Cancer Cells Treated with 5-Fluorouracil" International Journal of Molecular Sciences 14, no. 8: 16600-16616. https://doi.org/10.3390/ijms140816600
APA StyleAsara, Y., Marchal, J. A., Carrasco, E., Boulaiz, H., Solinas, G., Bandiera, P., Garcia, M. A., Farace, C., Montella, A., & Madeddu, R. (2013). Cadmium Modifies the Cell Cycle and Apoptotic Profiles of Human Breast Cancer Cells Treated with 5-Fluorouracil. International Journal of Molecular Sciences, 14(8), 16600-16616. https://doi.org/10.3390/ijms140816600