Exposure to Environmental Toxicants and Pathogenesis of Amyotrophic Lateral Sclerosis: State of the Art and Research Perspectives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
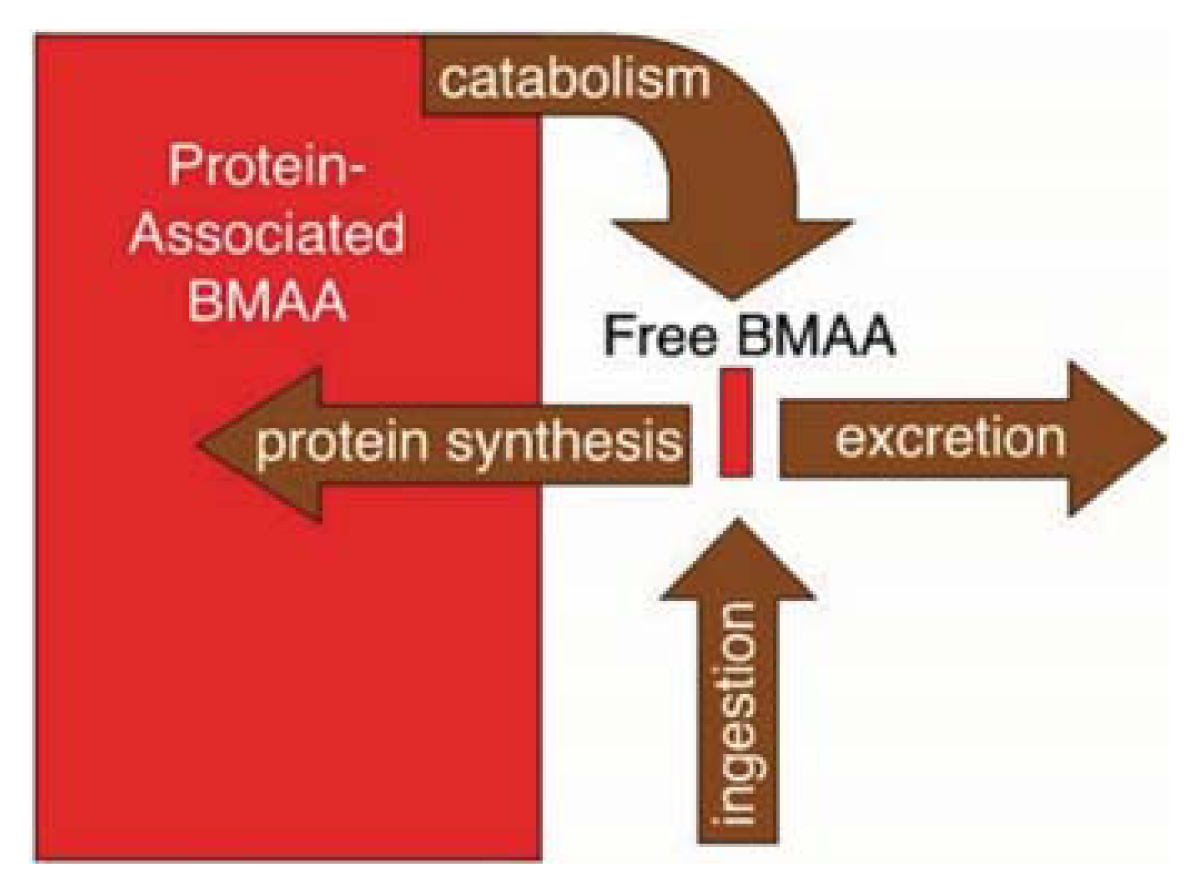
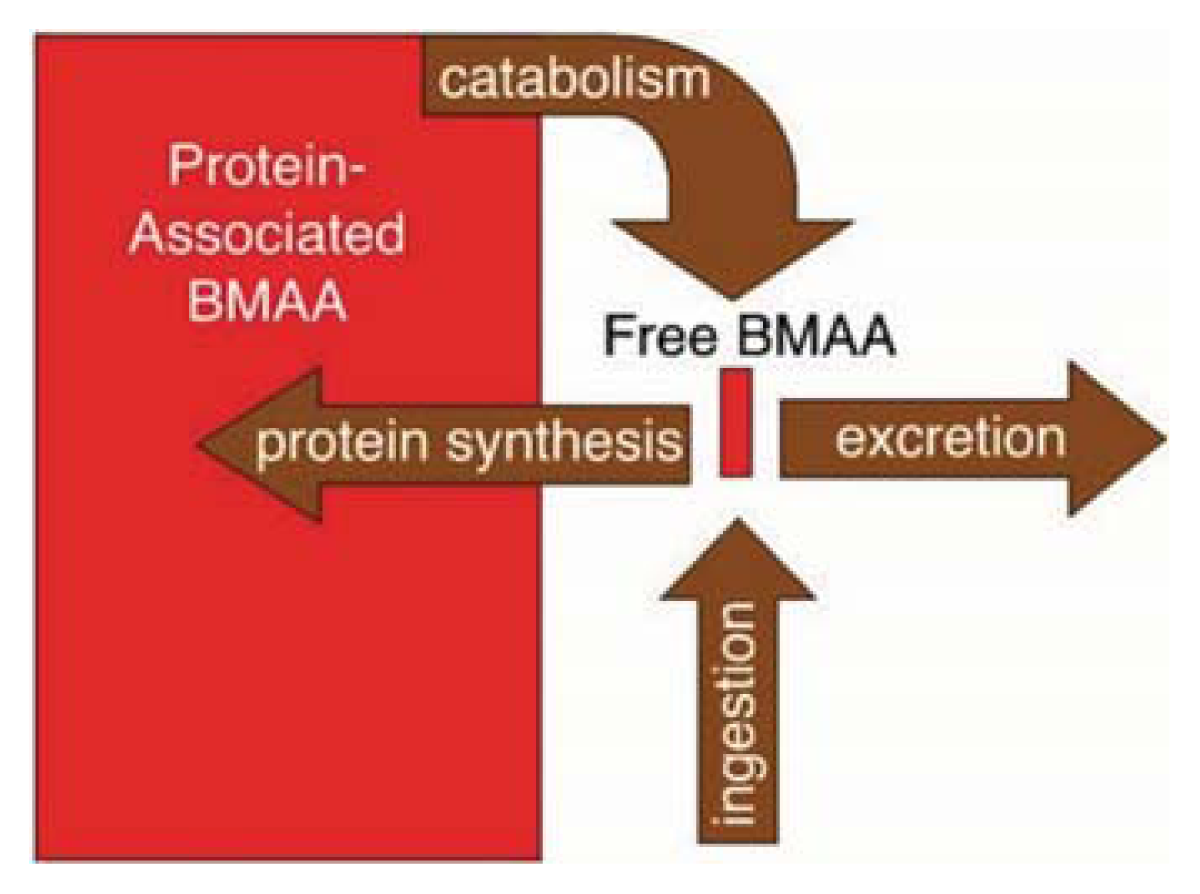
2. BMAA and Cyanobacteria
3. Heavy Metals
3.1. Lead
3.2. Mercury
3.3. Selenium
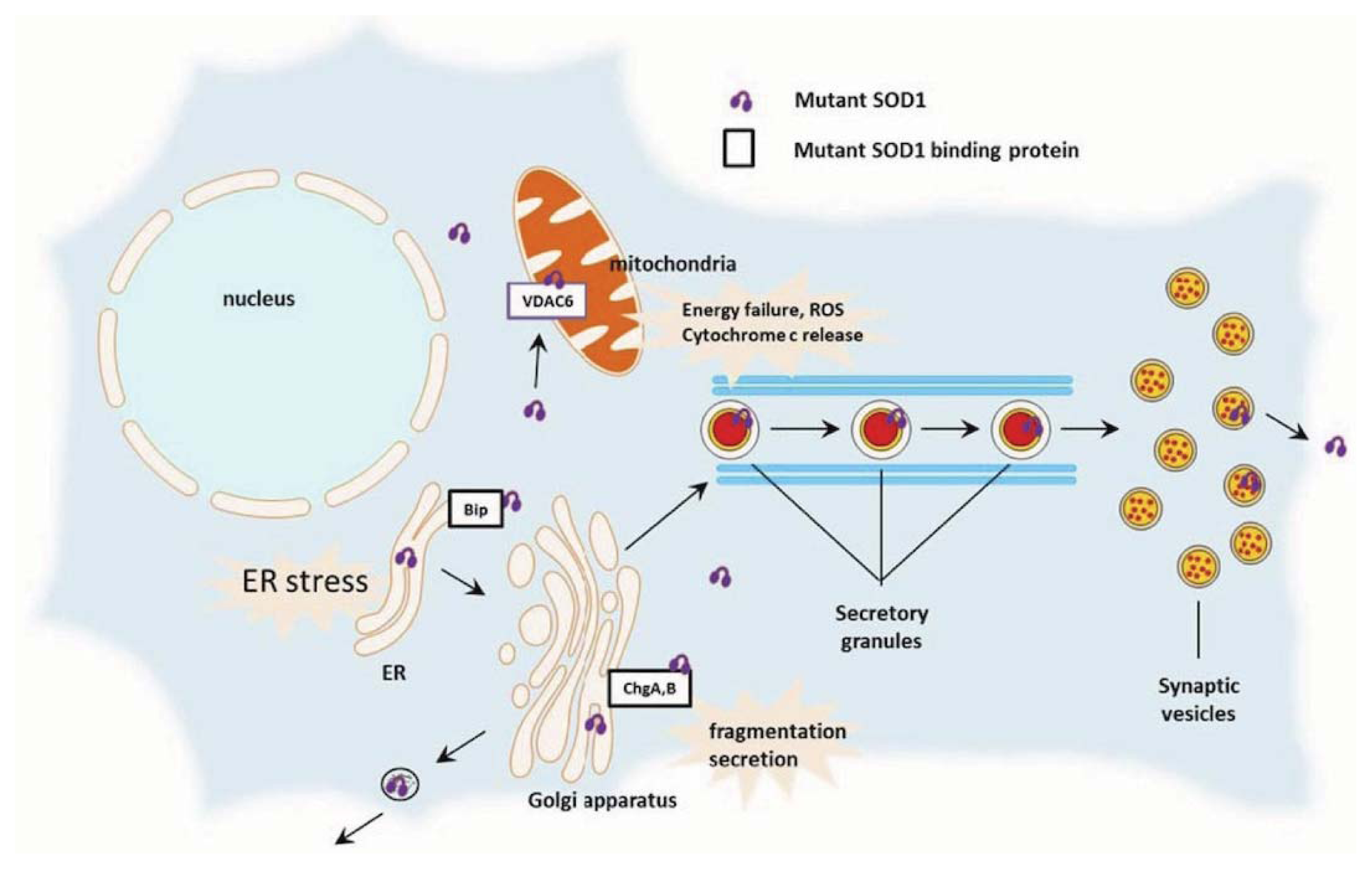
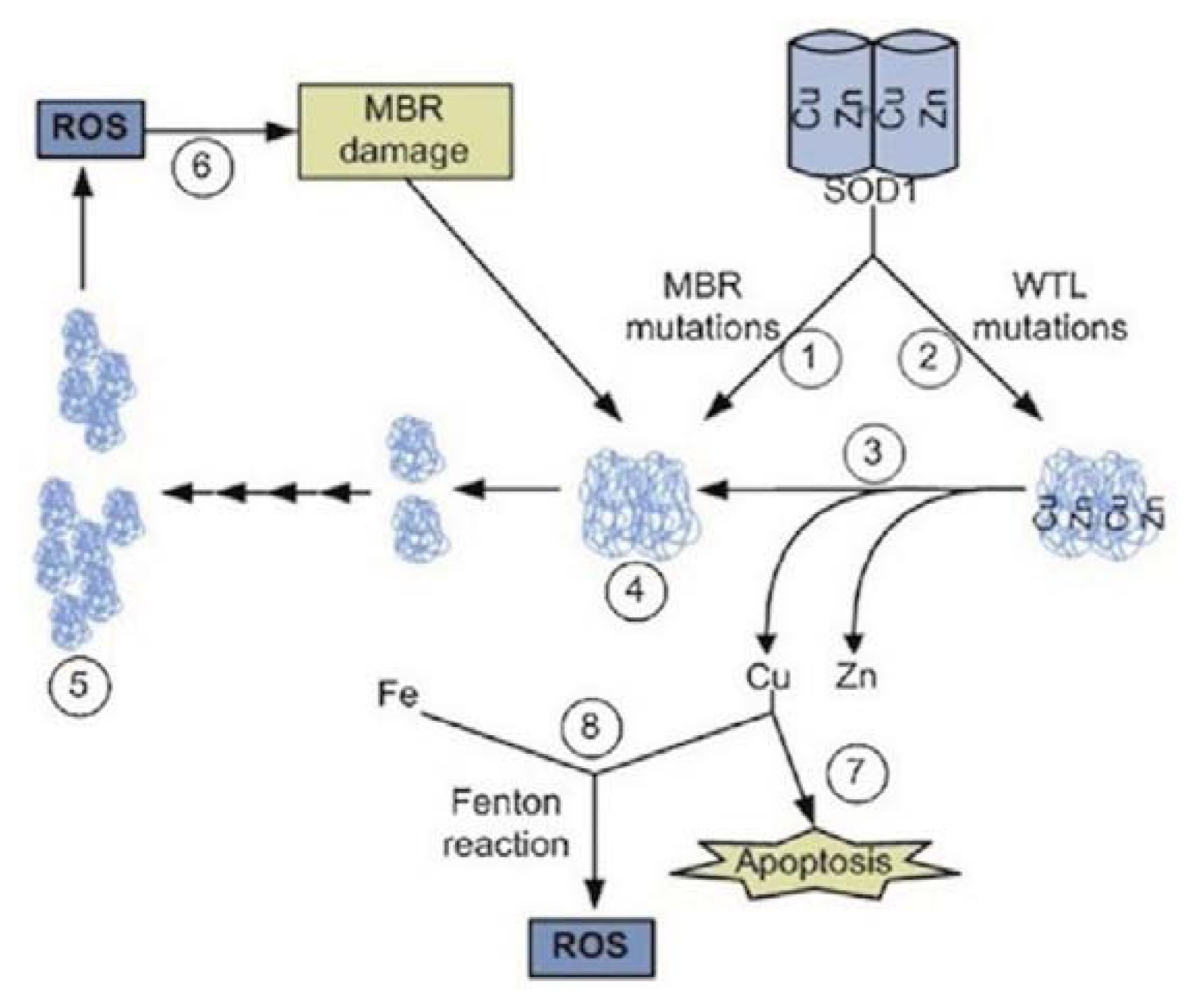
3.4. Zinc and Copper
3.5. Heavy Metals and other Chemical Compounds Implicated in Cigarette Smoking Neurotoxicity
3.6. Co-Exposure to Lead, Electrical Shocks and Electromagnetic Fields in “Electrical and Electronic” Workers
4. Pesticides
5. Concluding Remarks and Future Perspectives
Acknowledgements
Conflict of Interest
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 6736, 61156–61157. [Google Scholar]
- Ludolph, A.C.; Brettschneider, J.; Weishaupt, J.H. Amyotrophic lateral sclerosis. Curr. Opin. Neurol 2012, 25, 530–535. [Google Scholar]
- Soo, K.Y.; Farg, M.; Atkin, J.D. Molecular motor proteins and amyotrophic lateral sclerosis. Int. J. Mol. Sci 2011, 12, 9057–9082. [Google Scholar]
- Ido, A.; Fukuyama, H.; Urushitani, M. Protein misdirection inside and outside motor neurons in amyotrophic lateral sclerosis (ALS): A possible clue for therapeutic strategies. Int. J. Mol. Sci 2011, 12, 6980–7003. [Google Scholar]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar]
- Neumann, M. Molecular neuropathology of TDP-43 proteinopathies. Int. J. Mol. Sci 2009, 10, 232–246. [Google Scholar]
- Gawel, M.; Zaiwalla, Z.; Rose, C.F. Antecedent events in motor neuron disease. J. Neurol. Neurosurg. Psychiatry 1983, 46, 1041–1043. [Google Scholar]
- Deapen, D.M.; Henderson, B.E. A case-control study of amyotrophic lateral sclerosis. Am. J. Epidemiol 1986, 123, 790–799. [Google Scholar]
- Johansen, C. Exposure to electromagnetic fields and risk of central nervous system disease in utility workers. Epidemiology 2000, 11, 539–543. [Google Scholar]
- Hakansson, N.; Gustavsson, P.; Johansen, C.; Floderus, B. Neurodegenerative diseases in welders and other workers exposed to high levels of magnetic fields. Epidemiology 2003, 14, 420–426. [Google Scholar]
- Qureshi, M.M.; Hayden, D.; Urbinelli, L.; Ferrante, K.; Newhall, K.; Myers, D.; Hilgenberg, S.; Smart, R.; Brown, R.H.; Cudkowicz, M.E. Analysis of factors that modify susceptibility and rate of progression in amyotrophic lateral sclerosis (ALS). Amyotroph. Lateral Scler 2006, 7, 173–182. [Google Scholar]
- Morahan, J.M.; Pamphlett, R. Amyotrophic lateral sclerosis and exposure to environmental toxins: An Australian case-control study. Neuroepidemiology 2006, 27, 130–135. [Google Scholar]
- Steenland, K.; Hein, M.J.; Cassinelli, R.T., 2nd; Prince, M.M.; Nilsen, N.B.; Whelan, E.A.; Waters, M.A.; Ruder, A.M.; Schnorr, T.M. Polychlorinated biphenyls and neurodegenerative disease mortality in an occupational cohort. Epidemiology 2006, 17, 8–13. [Google Scholar]
- Weisskopf, M.G.; Morozova, N.; O’Reilly, E.J.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Prospective study of chemical exposures and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2009, 80, 558–561. [Google Scholar]
- Sutedja, N.A.; Veldink, J.H.; Fischer, K.; Kromhout, H.; Heederik, D.; Huisman, M.H.; Wokke, J.H.; van den Berg, L.H. Exposure to chemicals and metals and risk of amyotrophic lateral sclerosis: A systematic review. Amyotroph. Lateral Scler 2009, 10, 302–309. [Google Scholar]
- Johnson, F.O.; Atchison, W.D. The role of environmental mercury, lead and pesticide exposure in development of amyotrophic lateral sclerosis. Neurotoxicology 2009, 30, 761–765. [Google Scholar]
- Vanacore, N.; Cocco, P.; Fadda, D.; Dosemeci, M. Job strain, hypoxia and risk of amyotrophic lateral sclerosis: Results from a death certificate study. Amyotroph. Lateral Scler 2010, 11, 430–434. [Google Scholar]
- Callaghan, B.; Feldman, D.; Gruis, K.; Feldman, E. The association of exposure to lead, mercury, and selenium and the development of amyotrophic lateral sclerosis and the epigenetic implications. Neurodegener. Dis 2011, 8, 1–8. [Google Scholar]
- Bradley, W.G.; Borenstein, A.R.; Nelson, L.M.; Codd, G.A.; Rosen, B.H.; Stommel, E.W.; Cox, P.A. Is exposure to cyanobacteria an environmental risk factor for amyotrophic lateral sclerosis and other neurodegenerative diseases? Amyotroph. Lateral Scler. Frontotemporal Degener. 2013. [Google Scholar] [CrossRef]
- Cox, P.A.; Sacks, O.W. Cycad neurotoxin, consumption of fruit bats, and ALS/PDC disease in Guam. Neurology 2002, 59, 1664–1665. [Google Scholar]
- Miranda, M.L.; Galeano, M.A.O.; Tassone, E.; Allen, K.D.; Horner, R.D. Spatial analysis of the etiology of ALS among 1991 Gulf War veterans. Neurotoxicology 2008, 29, 964–970. [Google Scholar]
- Horner, R.D.; Grambow, S.C.; Coffman, C.J.; Lindquist, J.H.; Oddone, E.Z.; Allen, K.D.; Kasarskis, E.J. Amyotrophic lateral sclerosis among 1991 Gulf War veterans: Evidence for a time-limited outbreak. Neuroepidemiology 2008, 31, 28–32. [Google Scholar]
- Kisby, G.E.; Ellison, M.; Spencer, P.S. Content of the neurotoxins cycasin (methylazoxy-methanol beta-d-glucoside) and BMAA (beta-N-methylaminol-alanine) in cycad flour prepared by Guam Chamorros. Neurology 1992, 42, 1336–1340. [Google Scholar]
- Murch, S.J.; Cox, P.A.; Banack, S.A. A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc. Natl. Acad. Sci. USA 2004, 101, 12228–12231. [Google Scholar]
- Kuzuhara, S. ALS/Parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease). Historical review, epidemiology and concept. Rinsho Shinkeigaku 2007, 47, 962–965. [Google Scholar]
- Bradley, W.G.; Cox, P.A. Beyond Guam: BMAA and sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler 2009, 10, 5–6. [Google Scholar]
- Haley, R.W. Excess incidence of ALS in young Gulf War veterans. Neurology 2003, 61, 750–756. [Google Scholar]
- Kasarskis, E.J.; Lindquist, J.H.; Coffman, C.J.; Grambow, S.C.; Feussner, J.R.; Allen, K.D.; Oddone, E.Z.; Kamins, K.A.; Horner, R.D. Als Gulf War Clinical Review Team. Clinical aspects of ALS in Gulf War veterans. Amyotroph. Lateral Scler. 2009, 10, 35–41. [Google Scholar]
- Haley, R.W.; Billecke, S.; La Du, B.N. Association of low PON1 type Q (type A) arylesterase activity with neurologic symptom complexes in Gulf War veterans. Toxicol. Appl. Pharmacol 1996, 157, 227–233. [Google Scholar]
- Mackness, B.; Durrington, P.N.; Mackness, M.I. Low paraoxonase in Persian Gulf War Veterans self-reporting Gulf War Syndrome. Biochem. Biophys. Res. Commun 2000, 276, 729–733. [Google Scholar]
- Page, W.F.; Mahan, C.M.; Bullman, T.A.; Kang, H.K. Health effects in Army Gulf War veterans possibly exposed to chemical munitions destruction at Khamisiyah, Iraq. Part I. Morbidity associated with potential exposure. Mil. Med 2005, 170, 935–944. [Google Scholar]
- Bullman, T.A.; Mahan, C.M.; Kang, H.K.; Page, W.F. Mortality in US Army Gulf War veterans exposed to 1991 Khamisiyah chemical munitions destruction. Am. J. Public. Health 2005, 95, 1382–1388. [Google Scholar]
- Proctor, S.P.; Heaton, K.J.; Heeren, T.; White, R.F. Effects of sarin and cyclosarin exposure during the 1991 Gulf War on neurobehavioral functioning in US army veterans. Neurotoxicology 2006, 27, 931–939. [Google Scholar]
- Heaton, K.J.; Palumbo, C.L.; Proctor, S.P.; Killiany, R.J.; Yurgelun-Todd, D.A.; White, R.F. Quantitative magnetic resonance brain imaging in US army veterans of the 1991 Gulf War potentially exposed to sarin and cyclosarin. Neurotoxicology 2007, 28, 761–769. [Google Scholar]
- Cox, P.A.; Richer, R.; Metcalf, J.S.; Banack, S.A.; Codd, G.A.; Bradley, W.G. Cyanobacteria and BMAA exposure from desert dust: A possible link to sporadic ALS among Gulf War veterans. Amyotroph. Lateral Scler 2009, 10, 109–117. [Google Scholar]
- Metcalf, J.S.; Richer, R.; Cox, P.A.; Codd, G.A. Cyanotoxins in desert environments may present a risk to human health. Sci. Total Environ. 2012, 421–422, 118–123. [Google Scholar]
- Cox, P.A.; Banack, S.A. A non-protein amino acid and neurodegeneration. Science 2006, 314, 1242. [Google Scholar]
- Bell, E.A. The discovery of BMAA, and examples of biomagnification and protein incorporation involving other non-protein amino acids. Amyotroph. Lateral Scler 2009, 10, 21–25. [Google Scholar]
- Karlsson, O.; Roman, E.; Berg, A.L.; Brittebo, E.B. Early hippocampal cell death, and late learning and memory deficits in rats exposed to the environmental toxin BMAA (beta-N-methylamino-l-alanine) during the neonatal period. Behav. Brain Res 2011, 219, 310–320. [Google Scholar]
- Karlsson, O.; Berg, A.L.; Lindström, A.K.; Hanrieder, J.; Arnerup, G.; Roman, E.; Bergquist, J.; Lindquist, N.G.; Brittebo, E.B.; Andersson, M. Neonatal exposure to the cyanobacterial toxin BMAA induces changes in protein expression and neurodegeneration in adult hippocampus. Toxicol. Sci 2012, 130, 391–404. [Google Scholar]
- Blake, N.M.; Kirk, R.L.; Wilson, S.R.; Garruto, R.M.; Gajdusek, D.C.; Gibbs, C.J., Jr; Hoffman, P. Search for a red cell enzyme or serum protein marker in amyotrophic lateral sclerosis and parkinsonism-dementia of Guam. Am. J. Med. Genet. 1983, 14, 299–305. [Google Scholar]
- Garruto, R.M.; Plato, C.C.; Myrianthopoulos, N.C.; Schanfield, M.S.; Gajdusek, D.C. Blood groups, immunoglobulin allotypes and dermatoglyphic features of patients with amyotrophic lateral sclerosis and parkinsonism-dementia of Guam. Am. J. Med. Genet 1983, 14, 289–298. [Google Scholar]
- Plato, C.C.; Garruto, R.M.; Galasko, D.; Craig, U.K.; Plato, M.; Gamst, A.; Torres, J.M.; Wiederholt, W. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam: Changing incidence rates during the past 60 years. Am. J. Epidemiol 2003, 157, 149–157. [Google Scholar]
- Kurland, L.T.; Mulder, D.W. Epidemiologic investigations of amyotrophic lateral sclerosis. I. Preliminary report on geographic distribution, with particular reference to the Mariana Islands, including clinical and pathologic observations. Neurology 1954, 4, 338–348. [Google Scholar]
- Whiting, M.G. Food practices in ALS foci in Japan, the Marianas, and New Guinea. Fed. Proc 1964, 23, 1343–1345. [Google Scholar]
- Whiting, M.; Spatz, M.; Matsumoto, H. Research progress on cycads. Econ. Bot 1966, 20, 98–102. [Google Scholar]
- Duncan, M.W.; Kopin, I.J.; Garruto, R.M.; Lavine, L.; Markey, S.P. 2-Amino-3 (methylamino)-propionic acid in cycad-derived foods is an unlikely cause of amyotrophic lateral sclerosis/Parkinsonism. Lancet 1988, 2, 631–632. [Google Scholar]
- Banack, S.A.; Cox, P.A. Biomagnification of cycad neurotoxins in flying foxes: Implications for ALS-PDC in Guam. Neurology 2003, 61, 387–389. [Google Scholar]
- Cox, P.A.; Banack, S.A.; Murch, S.J. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc. Natl. Acad. Sci. USA 2003, 100, 13380–13383. [Google Scholar]
- Banack, S.A.; Murch, S.J.; Cox, P.A. Neurotoxic flying foxes as dietary items for the Chamorro people, Marianas Islands. J. Ethnopharmacol 2006, 106, 97–104. [Google Scholar]
- Xie, X.; Mondo, K.; Basile, M.J.; Bradley, W.G.; Mash, D.C. Tracking Brain Uptake and Protein Incorporation of Cyanobacterial Toxin BMAA. Proceedings of the 22nd International Symposium on ALS/MND, Sydney, Australia, 30 November–2 December 2011; Informa Healthcare: Stockholm, Sweden, 2011. [Google Scholar]
- Rodgers, K.; Dunlop, R. The Cyanobacteria-Derived BMAA Can Be Incorporated into Cell Proteins and Ccould thus Be an Environmental Trigger for ALS and Other Neurological Diseases Associated with Protein Misfolding. Proceedings of the 22nd International Symposium on ALS/MND, Sydney, Australia, 30 November–2 December 2011; Informa Healthcare: Stockholm, Sweden, 2011. [Google Scholar]
- Pablo, J.; Banack, S.A.; Cox, P.A.; Johnson, T.E.; Papapetropoulos, S.; Bradley, W.G.; Buck, A.; Mash, D.C. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol. Scand 2009, 120, 216–225. [Google Scholar]
- Esterhuizen, M.; Downing, T.G. Beta-N-methylamino-l-alanine (BMAA) in novel South African cyanobacterial isolates. Ecotoxicol. Environ. Saf 2008, 71, 309–313. [Google Scholar]
- Banack, S.A.; Johnson, H.E.; Cheng, R.; Cox, P.A. Production of the neurotoxin BMAA by a marine cyanobacterium. Mar. Drugs 2007, 5, 180–196. [Google Scholar]
- Jonasson, S.; Eriksson, J.; Berntzon, L.; Spácil, Z.; Ilag, L.L.; Ronnevi, L.O.; Rasmussen, U.; Bergman, B. Transfer of a cyanobacterial neurotoxin within a temperate aquatic ecosystem suggests pathways for human exposure. Proc. Natl. Acad. Sci. USA 2010, 18, 9252–9257. [Google Scholar]
- Brand, L.E.; Pablo, J.; Compton, A.; Hammerschlag, N.; Mash, D.C. Cyanobacterial blooms and the occurrence of the neurotoxin, beta-N-methylamino-l-alanine (BMAA), in South Florida aquatic food webs. Harmful Algae 2010, 9, 620–635. [Google Scholar]
- Li, A.; Tian, Z.; Li, J.; Yu, R.; Banack, S.A.; Wang, Z. Detection of the neurotoxin BMAA within cyanobacteria isolated from freshwater in China. Toxicon 2010, 55, 947–953. [Google Scholar]
- Sienko, D.G.; Davis, J.P.; Taylor, J.A.; Brooks, B.R. Amyotrophic lateral sclerosis. A case-control study following detection of a cluster in a small Wisconsin community. Arch. Neurol 1990, 47, 38–41. [Google Scholar]
- Sabel, C.E.; Boyle, P.J.; Löytönen, M.; Gatrell, A.C.; Jokelainen, M.; Flowerdew, R.; Maasilta, P. Spatial clustering of ALS in Finland at place of birth and place of death. Am. J. Epidemiol 2003, 157, 898–905. [Google Scholar]
- Caller, T.A.; Doolin, J.W.; Haney, J.F.; Murby, A.J.; West, K.G.; Farrar, H.E.; Ball, A.; Harris, B.T.; Stommel, E.W. A cluster of amyotrophic lateral sclerosis in New Hampshire: A possible role for toxic cyanobacteria blooms. Amyotroph. Lateral Scler 2009, 10, 101–108. [Google Scholar]
- Sieh, W.; Choi, Y.; Chapman, N.H.; Craig, U.K.; Steinbart, E.J.; Rothstein, J.H.; Garruto, R.M.; Bird, T.D.; Galasko, D.R.; Schellenberg, G.D.; et al. Identification of novel susceptibility loci for Guam neurodegenerative disease: Challenges of genome scans in genetic isolates. Hum. Mol. Genet 2009, 18, 3725–3738. [Google Scholar]
- Wilson, J.M.; Petrik, M.S.; Moghadasian, M.H.; Shaw, C.A. Examining the interaction of apo E and neurotoxicity on a murine model of ALS-PDC. Can. J. Physiol. Pharm 2005, 83, 131–141. [Google Scholar]
- Sundar, P.D.; Yu, C.E.; Sieh, W.; Steinbart, E.; Garruto, R.M.; Oyanagi, K.; Craig, U.K.; Bird, T.D.; Wijsman, E.M.; Galasko, D.R.; et al. Two sites in the MAPT region confer genetic risk for Guam ALS/PDC and dementia. Hum. Mol. Genet 2007, 16, 295–306. [Google Scholar]
- Smith, Q.R.; Nagura, H.; Takada, Y.; Duncan, M.W. Facilitated transport of the neurotoxin, β-N-methylamino-l-alanine, across the blood-brain barrier. J. Neurochem 1992, 58, 1330–1337. [Google Scholar]
- Lee, J.W.; Beebe, K.; Nangle, L.A.; Jang, J.; Longo-Guess, C.M.; Cook, S.A.; Davisson, M.T.; Sundberg, J.P.; Schimmel, P.; Ackerman, S.L. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 2006, 443, 50–55. [Google Scholar]
- Roos, P.M.; Vesterberg, O.; Syversen, T.; Peder Flaten, T.; Nordberg, M. Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol. Trace Elem. Res 2013, 151, 159–170. [Google Scholar]
- Bergomi, M.; Vinceti, M.; Nacci, G.; Pietrini, V.; Brätter, P.; Alber, D.; Ferrari, A.; Vescovi, L.; Guidetti, D.; Sola, P.; et al. Environmental exposure to trace elements and risk of amyotrophic lateral sclerosis: A population-based case-control study. Environ. Res 2002, 89, 116–123. [Google Scholar]
- Vinceti, M.; Guidetti, D.; Bergomi, M.; Caselgrandi, E.; Vivoli, R.; Olmi, M.; Rinaldi, L.; Rovesti, S.; Solime, F. Lead, cadmium, and selenium in the blood of patients with sporadic amyotrophic lateral sclerosis. Ital. J. Neurol. Sci 1997, 18, 87–92. [Google Scholar]
- Campbell, A.M.; Williams, E.R.; Barltrop, D. Motor neurone disease and exposure to lead. J. Neurol. Neurosurg. Psychiatry 1970, 33, 877–885. [Google Scholar]
- Kamel, F.; Umbach, D.M.; Hu, H.; Munsat, T.L.; Shefner, J.M.; Taylor, J.A.; Sandler, D.P. Lead exposure as a risk factor for amyotrophic lateral sclerosis. Neurodegener. Dis 2005, 2, 195–201. [Google Scholar]
- Barbeito, A.G.; Guelfi, N.; Varga, M.R.; Pehar, M.; Beckman, J.; Barbeito, L.; Cassina, P. Chronic low-level lead exposure increases survival of G93A SOD-1 transgenic mice. In Amyotrophic lateral sclerosis: Beyond the motor neuron, Proceedings of the International Symposium, CasaPueblo/Punta del Este, Uruguay, 30 March–2 April 2005.
- Kamel, F.; Umbach, D.M.; Stallone, L.; Richards, M.; Hu, H.; Sandler, D.P. Association of lead exposure with survival in amyotrophic lateral sclerosis. Environ. Health Perspect 2008, 116, 943–947. [Google Scholar]
- Daniele, L.; Garzillo, E.; Trojsi, F.; Monsurrò, M.; Tedeschi, G. Role of Occupational and Environmental Exposure to Heavy Metals in Amyotrophic Lateral Sclerosis: A Case-control Study in Campania Region. In Neurol. Sciences, Proceedings of the 43rd Congress of the Italian Neurological Society, Rimini, Italy, 6–9 October 2012; Springer-Verlag Italia: Milan, Italy, 2012. [Google Scholar]
- Albalak, R.; McElroy, R.H.; Noonan, G.; Buchanan, S.; Jones, R.L.; Flanders, W.D.; Gotway-Crawford, C.; Kim, D.; Dignam, T.; Daley, W.R.; et al. Blood lead levels and risk factors for lead poisoning among children in a Mexican smelting community. Arch. Environ. Health 2003, 58, 172–183. [Google Scholar]
- Oh, S.S.; Kim, E.A.; Lee, S.W.; Kim, M.K.; Kang, S.K. A case of amyotrophic lateral sclerosis in electronic parts manufacturing worker exposed to lead. Neurotoxicology 2007, 28, 324–327. [Google Scholar]
- Yan, D.; Xiao, C.; Ma, F.L.; Wang, L.; Luo, Y.Y.; Liu, J.; Wang, H.L.; Chen, J.T.; Ruan, D.Y. Excitatory effects of low-level lead exposure on action potential firing of pyramidal neurons in CA1 region of rat hippocampal slices. J. Neurosci. Res 2008, 86, 3665–3673. [Google Scholar]
- Fang, F.; Kwee, L.C.; Allen, K.D.; Umbach, D.M.; Ye, W.; Watson, M.; Keller, J.; Oddone, E.Z.; Sandler, D.P.; Schmidt, S.; et al. Association between blood lead and the risk of amyotrophic lateral sclerosis. Am. J. Epidemiol 2010, 171, 1126–1133. [Google Scholar]
- Luo, W.; Ruan, D.; Yan, C.; Yin, S.; Chen, J. Effects of chronic lead exposure on functions of nervous system in Chinese children and developmental rats. Neurotoxicology 2012, 33, 862–871. [Google Scholar]
- Conradi, S.; Ronnevi, L.O.; Vesterberg, O. Abnormal tissue distribution of lead in amyo-trophic lateral sclerosis. J. Neurol. Sci 1976, 29, 259–265. [Google Scholar]
- Barbeito, A.G.; Cassina, P. Lead exposure stimulates VEGF expression in the spinal cord and extends survival in a mouse model of ALS. Neurobiol. Dis 2010, 37, 574–580. [Google Scholar]
- Kamel, F.; Umbach, D.M.; Lehman, T.A.; Park, L.P.; Munsat, T.L.; Shefner, J.M.; Sandler, D.P.; Hu, H.; Taylor, J.A. Amyotrophic lateral sclerosis, lead, and genetic susceptibility: Polymorphisms in the delta-aminolevulinic acid dehydratase and vitamin D receptor genes. Environ. Health Perspect 2003, 111, 1335–1339. [Google Scholar]
- Pilsner, J.R.; Hu, H.; Ettinger, A.; Sánchez, B.N.; Wright, R.O.; Cantonwine, D.; Lazarus, A.; Lamadrid-Figueroa, H.; Mercado-García, A.; Téllez-Rojo, M.M.; et al. Influence of prenatal lead exposure on genomic methylation of cord blood DNA. Environ. Health Perspect 2009, 117, 1466–1471. [Google Scholar]
- Díez, S. Human health effects of methylmercury exposure. Rev. Environ. Contam. Toxicol 2009, 198, 111–132. [Google Scholar]
- Adams, C.R.; Ziegler, D.K.; Lin, J.T. Mercury intoxication simulating amyotrophic lateral sclerosis. JAMA 1983, 250, 642–643. [Google Scholar]
- Provinciali, L.; Giovagnoli, A.R. Antecedent events in amyotrophic lateral sclerosis: Do they influence clinical onset and progression? Neuroepidemiology 1990, 9, 255–262. [Google Scholar]
- Praline, J.; Guennoc, A.; Limousin, N.; Hallak, H.; de Toffol, B.; Corcia, P. Case report; ALS and mercury intoxication: A relationship? Clin. Neurol. Neurosurg 2007, 109, 880–883. [Google Scholar]
- Schwarz, S.; Husstedt, I.; Bertram, H.P.; Kuchelmeister, K. Amyotrophic lateral sclerosis after accidental injection of mercury. J. Neurol. Neurosurg. Psychiatry 1996, 60, 698. [Google Scholar]
- Barber, T.E. Inorganic mercury intoxication reminiscent of amyotrophic lateral sclerosis. J. Occup. Med 1978, 20, 667–669. [Google Scholar]
- Pamphlett, R.; Waley, P. Mercury in human spinal motor neurons. Acta Neuropathol 1998, 96, 515–519. [Google Scholar]
- Gresham, L.S.; Molgaard, C.A.; Golbeck, A.L.; Smith, R. Amyotrophic lateral sclerosis and occupational heavy metal exposure: A case-control study. Neuroepidemiology 1986, 5, 29–38. [Google Scholar]
- Moriwaka, F.; Tashiro, K.; Doi, R.; Satoh, H.; Fukuchi, Y. A clinical evaluation of the inorganic mercurialism—Its pathogenic relation to amyotrophic lateral sclerosis. Rinsho Shinkeigaku 1991, 31, 885–887. [Google Scholar]
- Arvidson, B. Inorganic mercury is transported from muscular nerve terminals to spinal and brainstem motoneurons. Muscle Nerve 1992, 15, 1089–1094. [Google Scholar]
- Chuu, J.J.; Liu, S.H.; Lin-Shiau, S.Y. Differential neurotoxic effects of methylmercury and mercuric sulfide in rats. Toxicol. Lett 2007, 169, 109–120. [Google Scholar]
- Johnson, F.; Atchison, W. Postnatal Exposure to Methylmercury Enhances Development of Paralytic Phenotype in SOD1-G93A Female Mice. In The toxicologist, Proceedings of the 48th Annual Meeting and ToxExpo™, Baltimore, MD, USA, 15–19 March 2009; Oxford University Press Society of Toxicology: Reston, VA, USA, 2009. [Google Scholar]
- Bassett, T.; Bach, P.; Chan, H.M. Effects of methylmercury on the secretion of pro-inflammatory cytokines from primary microglial cells and astrocytes. Neurotoxicology 2012, 33, 229–234. [Google Scholar]
- Choonara, Y.A.; Pillay, V.; du Toit, L.C.; Modi, G.; Naidoo, D.; Ndesendo, V.M.K.; Sibambo, S.R. Trends in the molecular pathogenesis and clinical therapeutics of common neurodegenerative disorders. Int. J. Mol. Sci 2009, 10, 2510–2557. [Google Scholar]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Marchetti, B. Glia as a turning point in the therapeutic strategy of Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 349–372. [Google Scholar]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol 2011, 10, 253–263. [Google Scholar]
- Bridges, C.; Zalups, R.K. Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol 2005, 204, 274–308. [Google Scholar]
- Rooney, J. Further thoughts on mercury, epigenetics, genetics and amyotrophic lateral sclerosis. Neurodegener. Dis 2011, 8, 523–524. [Google Scholar]
- Kilness, A.W.; Hochberg, F.H. Amyotrophic lateral sclerosis in a high selenium environment. JAMA 1977, 237, 2843–2844. [Google Scholar]
- Vinceti, M.; Guidetti, D.; Pinotti, M.; Rovesti, S.; Merlin, M.; Vescovi, L.; Bergomi, M.; Vivoli, G. Amyotrophic lateral sclerosis after long-term exposure to drinking water with high selenium content. Epidemiology 1996, 7, 529–532. [Google Scholar]
- Yang, G.Q.; Wang, S.Z.; Zhou, R.H.; Sun, S.Z. Endemic selenium intoxication of humans in China. Am. J. Clin. Nutr 1983, 37, 872–881. [Google Scholar]
- Vinceti, M.; Maraldi, T.; Bergomi, M.; Malagoli, C. Risk of chronic low-dose selenium overexposure in humans: Insights from epidemiology and biochemistry. Rev. Environ. Health 2009, 24, 231–248. [Google Scholar]
- Matsumura, H.; Takahata, R.; Hayaishi, O. Inhibition of sleep in rats by inorganic selenium compounds, inhibitors of prostaglandin D synthase. Proc. Natl. Acad. Sci. USA 1991, 88, 9046–9050. [Google Scholar]
- Nehru, B.; Iyer, A. Effect of selenium on lead-induced neurotoxicity in different brain regions of adult rats. J. Environ. Pathol. Toxicol. Oncol 1994, 13, 265–268. [Google Scholar]
- Rasekh, H.R.; Soliman, K.F.A.; Davis, M.D. Effect of selenium on central nervous system of male S-D rats: Evidences for neurotoxicity of selenium. Toxicol. Lett 1998, 95, 64. [Google Scholar]
- Tsunoda, M.; Johnson, V.J.; Sharma, R.P. Increase in dopamine metabolites in murine striatum after oral exposure to inorganic but not organic form of selenium. Arch. Environ. Contam. Toxicol 2000, 39, 32–37. [Google Scholar]
- Nogueira, C.W.; Meotti, F.C.; Curte, E.; Pilissao, C.; Zeni, G.; Rocha, J.B. Investigations into the potential neurotoxicity induced by diselenides in mice and rats. Toxicology 2003, 183, 29–37. [Google Scholar]
- Liu, S.H.; Fu, W.M.; Lin-Shiau, S.Y. Effects of sodium selenite on neuromuscular junction of the mouse phrenic nerve-diaphragm preparation. Neuropharmacology 1989, 28, 733–739. [Google Scholar]
- Lin-Shiau, S.Y.; Liu, S.H.; Fu, W.M. Neuromuscular actions of sodium selenite on chick biventer cervicis nerve-muscle preparation. Neuropharmacology 1990, 29, 493–501. [Google Scholar]
- Togna, G.; Russo, P.; Pierconti, F.; Caprino, L. Effects of sodium selenite on vascular smooth muscle reactivity. Pharmacol. Res 2000, 41, 195–199. [Google Scholar]
- Ayaz, M.; Dalkilic, N.; Bariskaner, H.; Tuncer, S.; Demirel, I. Gender-dependent effects of selenite on the perfused rat heart: A toxicological study. Biol. Trace Elem. Res 2007, 116, 301–310. [Google Scholar]
- Davidson-York, D.; Galey, F.D.; Blanchard, P.; Gardner, I.A. Selenium elimination in pigs after an outbreak of selenium toxicosis. J. Vet. Diagn. Invest 1999, 11, 352–357. [Google Scholar]
- Casteignau, A.; Fontán, A.; Morillo, A.; Oliveros, J.A.; Segalés, J. Clinical, pathological and toxicological findings of a iatrogenic selenium toxicosis case in feeder pigs. J. Vet. Med. A 2006, 53, 323–326. [Google Scholar]
- Vinceti, M.; Wei, E.T.; Malagoli, C.; Bergomi, M.; Vivoli, G. Adverse health effects of selenium in humans. Rev. Environ. Health 2001, 16, 233–251. [Google Scholar]
- Maag, D.D.; Orsborn, J.S.; Clopton, J.R. The effect of sodium selenite on cattle. Am. J. Vet. Res 1960, 21, 1049–1053. [Google Scholar]
- Estevez, A.O.; Mueller, C.L.; Morgan, K.L.; Szewczyk, N.J.; Teece, L.; Miranda-Vizuete, A.; Estevez, M. Selenium induces cholinergic motor neuron degeneration in Caenorhabditis elegans. Neurotoxicology 2012, 33, 1021–1032. [Google Scholar]
- Maraldi, T.; Riccio, M.; Zambonin, L.; Vinceti, M.; de Pol, A.; Hakim, G. Low levels of selenium compounds are selectively toxic for a human neuron cell line through ROS/RNS increase and apoptotic process activation. Neurotoxicology 2011, 32, 180–187. [Google Scholar]
- Bendotti, C.; Carrì, M.T. Amyotrophic lateral sclerosis: Mechanisms and countermeasures. Antioxid. Redox Signal 2009, 11, 1519–1522. [Google Scholar]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci 2005, 6, 449–462. [Google Scholar]
- Sensi, S.L.; Jeng, J.M. Rethinking the excitotoxic ionic milieu: The emerging role of Zn2+ in ischemic neuronal injury. Curr. Mol. Med 2004, 4, 87–111. [Google Scholar]
- Yao, X. Effect of zinc exposure on HNE and GLT-1 in spinal cord culture. Neurotoxicology 2009, 30, 121–126. [Google Scholar]
- Nutini, M.; Frazzini, V.; Marini, C.; Spalloni, A.; Sensi, S.L.; Longone, P. Zinc pre-treatment enhances NMDAR-mediated excitotoxicity in cultured cortical neurons from SOD1G93A mouse, a model of amyotrophic lateral sclerosis. Neuropharmacology 2011, 60, 1200–1208. [Google Scholar]
- Groeneveld, G.J.; de Leeuw van Weenen, J.; van Muiswinkel, F.L.; Veldman, H.; Veldink, J.H.; Wokke, J.H.; Bär, P.R.; van den Berg, L.H. Zinc amplifies mSOD1-mediated toxicity in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci. Lett 2003, 352, 175–178. [Google Scholar]
- Ermilova, P.I.; Ermilov, B.V.; Levy, M.; Hob, E.; Pereira, C.; Beckman, S.B. Protection by dietary zinc in ALS mutant G93A SOD transgenic mice. Neurosci. Lett 2005, 379, 42–46. [Google Scholar]
- Ahtoniemi, T.; Goldsteins, G.; Keksa-Goldsteine, V.; Malm, T.; Kanninen, K.; Salminen, A.; Koistinaho, J. Pyrrolidine dithiocarbamate inhibits induction of immunoproteasome and decreases survival in a rat model of amyotrophic lateral sclerosis. Mol. Pharmacol. 2007, 71, 30–37. [Google Scholar]
- Li, Q.X.; Mok, S.S.; Laughton, K.M.; McLean, C.A.; Volitakis, I.; Cherny, R.A.; Cheung, N.S.; White, A.R.; Masters, C.L. Overexpression of Abeta is associated with acceleration of onset of motor impairment and superoxide dismutase 1 aggregation in an amyotrophic lateral sclerosis mouse model. Aging Cell 2006, 5, 153–165. [Google Scholar]
- Tokuda, E.; Ono, S.; Ishige, K.; Naganuma, A.; Ito, Y.; Suzuki, T. Metallothionein proteins expression, copper and zinc concentrations, and lipid peroxidation level in a rodent model for amyotrophic lateral sclerosis. Toxicology 2007, 229, 33–41. [Google Scholar]
- Hayward, L.J.; Rodriguez, J.A.; Kim, J.W.; Tiwari, A.; Goto, J.J.; Cabelli, D.E.; Valentine, J.S.; Brown, R.H., Jr. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 15923–159231. [Google Scholar]
- Valentine, J.S.; Hart, P.J. Misfolded, CuZn SOD and amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 3617–3622. [Google Scholar]
- Hart, P.J. Pathogenic superoxide dismutase structure, folding, aggregation and turnover. Curr. Opin. Chem. Biol 2006, 10, 131–138. [Google Scholar]
- Martyshkin, D.V.; Mirov, S.B.; Zhuang, Y.X.; Crow, J.P.; Ermilov, V.; Beckman, J.S. Fluorescence assay for monitoring Zn-deficient superoxide dismutase in vitro. Spectrochim. Acta A 2003, 59, 3165–3175. [Google Scholar]
- Estévez, A.G.; Crow, J.P.; Sampson, J.B.; Reiter, C.; Zhuang, Y.; Richardson, G.J.; Tarpey, M.M.; Barbeito, L.; Beckman, J.S. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 1999, 286, 2498–2500. [Google Scholar]
- Goto, J.J.; Gralla, E.B.; Valentine, J.S.; Cabelli, D.E. Reactions of hydrogen peroxide with familial amyotrophic lateral sclerosis mutant human copper-zinc superoxide dismutases studied by pulse radiolysis. J. Biol. Chem 1998, 273, 30104–30109. [Google Scholar]
- Rivera-Mancía, S.; Pérez-Neria, I.; Ríos, C.; Tristán-López, L.; Rivera-Espinosa, L.; Montes, S. The transition metals copper and iron in neurodegenerative diseases. Chem. Biol. Interact 2010, 186, 184–199. [Google Scholar]
- Ji, H.F.; Shen, L.; Zhang, H.Y. Beta-lactam antibiotics are multipotent agents to combat neurological diseases. Biochem. Biophys. Res. Commun 2005, 333, 661–663. [Google Scholar]
- Hottinger, A.F.; Fine, E.G.; Gurney, M.E.; Zurn, A.D.; Aebischer, P. The copper chelator d-penicillamine delays onset of disease and extends survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur. J. Neurosci 1997, 9, 1548–1551. [Google Scholar]
- Andreassen, O.A.; Dedeoglu, A.; Klivenyi, P.; Beal, M.F.; Bush, A.I. N-acetyl-l-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. Neuroreport 2000, 11, 2491–2493. [Google Scholar]
- Ho, Y.S.; Yang, X.; Yeung, S.C.; Chiu, K.; Lau, C.F.; Tsang, A.W.; Mak, J.C.; Chang, R.C. Cigarette smoking accelerated brain aging and induced pre-Alzheimer-like neuropathology in rats. PLoS One 2012, 7, e36752. [Google Scholar]
- Weisskopf, M.G.; Grodstein, F.; Ascherio, A. Smoking and cognitive function in Parkinson’s disease. Mov. Disord 2007, 22, 660–665. [Google Scholar]
- Alonso, A.; Logroscino, G.; Hernán, M.A. Smoking and the risk of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1249–1252. [Google Scholar]
- Wang, H.; O’Reilly, E.J.; Weisskopf, M.G.; Logroscino, G.; McCullough, M.L.; Thun, M.J.; Schatzkin, A.; Kolonel, L.N.; Ascherio, A. Smoking and risk of amyotrophic lateral sclerosis: A pooled analysis of 5 prospective cohorts. Arch. Neurol 2011, 68, 207–213. [Google Scholar]
- De Jong, S.W.; Huisman, M.H.B.; Sutedja, N.A.; van der Kooi, A.J.; de Visser, M.; Schelhaas, H.J.; Fischer, K.; Veldink, J.H.; van den Berg, L.H. Smoking, alcohol consumption, and the risk of amyotrophic lateral sclerosis: A population-based study. Am. J. Epidemiol 2012, 176, 233–239. [Google Scholar]
- Fang, F.; Bellocco, R.; Hernán, M.A.; Ye, W. Smoking, snuff dipping and the risk of amyotrophic lateral sclerosis-a prospective cohort study. Neuroepidemiology 2006, 27, 217–221. [Google Scholar]
- Djordjevic, M.V.; Fan, J.; Hoffmann, D. Assessment of chlorinated pesticide residues in cigarette tobacco based on supercritical fluid extraction and GC-ECD. Carcinogenesis 1995, 16, 2627–2632. [Google Scholar]
- Hoffmann, D.; Hoffmann, I.; El-Bayoumy, K. The less harmful cigarette: A controversial issue. A tribute to Ernst L. Wynder. Chem. Res. Toxicol 2001, 14, 767–790. [Google Scholar]
- Morselt, A.F. Environmental pollutants and diseases. A cell biological approach using chronic cadmium exposure in the animal model as a paradigm case. Toxicology 1991, 70, 1–132. [Google Scholar]
- Huang, Y.H.; Shih, C.M.; Huang, C.J.; Lin, C.M.; Chou, C.M.; Tsai, M.L.; Liu, T.P.; Chiu, J.F.; Chen, C.T. Effects of cadmium on structure and enzymatic activity of Cu,Zn-SOD and oxidative status in neural cells. J. Cell. Biochem 2006, 98, 577–589. [Google Scholar]
- Bauer, R.; Demeter, I.; Hasemann, V.; Johansen, J.T. Structural properties of the zinc site in Cu,Zn-superoxide dismutase; perturbed angular correlation of gamma ray spectroscopy on the Cu, 111Cd-superoxide dismutase derivative. Biochem. Biophys. Res. Commun 1980, 94, 1296–1302. [Google Scholar]
- Kofod, P.; Bauer, R.; Danielsen, E.; Larsen, E.; Bjerrem, M.J. 113Cd-NMR investigation of a cadmium-substituted copper, zinc-containing superoxide dismutase from yeast. Eur. J. Biochem 1991, 198, 607–611. [Google Scholar]
- Bar-Sela, S.; Reingold, S.; Richter, E.D. Amyotrophic lateral sclerosis in a battery-factory worker exposed to cadmium. Int. J. Occup. Environ. Health 2001, 7, 109–112. [Google Scholar]
- Gurel, A.; Coskun, O.; Armutcu, F.; Kanter, M.; Ozen, O.A. Vitamin E against oxidative damage caused by formaldehyde in frontal cortex and hippocampus: Biochemical and histological studies. J. Chem. Neuroanat 2005, 29, 173–178. [Google Scholar]
- Nie, C.L.; Wang, X.S.; Liu, Y.; Perrett, S.; He, R.Q. Amyloid-like aggregates of neuronal tau induced by formaldehyde promote apoptosis of neuronal cells. BMC Neurosci 2007, 8, 9. [Google Scholar]
- Strubelt, O.; Younes, M.; Pentz, R.; Kühnel, W. Mechanistic study on formaldehyde-induced hepatotoxicity. J. Toxicol. Environ. Health 1989, 27, 351–366. [Google Scholar]
- Ticozzi, N.; LeClerc, A.L.; Keagle, P.; Glass, J.D.; Wills, A.M.; van Blitterswijk, M.; Bosco, D.A.; Rodriguez-Leyva, I.; Gellera, C.; Ratti, A.; et al. Paraoxonase gene mutations in amyotrophic lateral sclerosis. Ann. Neurol 2010, 68, 102–107. [Google Scholar]
- Imaizumi, T.; Higaki, Y.; Hara, M.; Sakamoto, T.; Horita, M.; Mizuta, T.; Eguchi, Y.; Yasutake, T.; Ozaki, I.; Yamamoto, K.; et al. Interaction between cytochrome P450 1A2 genetic polymorphism and cigarette smoking on the risk of hepatocellular carcinoma in a Japanese population. Carcinogenesis 2009, 30, 1729–1734. [Google Scholar]
- Boccia, S.; Sayed-Tabatabaei, F.A.; Persiani, R.; Gianfagna, F.; Rausei, S.; Arzani, D.; La Greca, A.; D’Ugo, D.; La Torre, G.; van Duijn, C.M.; et al. Polymorphisms in metabolic genes, their combination and interaction with tobacco smoke and alcohol consumption and risk of gastric cancer: A case-control study in an Italian population. BMC Cancer 2007, 7, 206. [Google Scholar]
- Diekstra, F.P.; Saris, C.G.J.; van Rheenen, W.; Franke, L.; Jansen, R.C.; van Es, M.A.; van Vught, P.W.J.; Blauw, H.M.; Groen, E.J.N.; Horvath, S.; et al. Mapping of gene expression reveals CYP27A1 as a susceptibility gene for sporadic ALS. PLoS One 2012, 7, e35333. [Google Scholar]
- Zhou, H.; Chen, G.; Chen, C.; Yu, Y.; Xu, Z. Association between extremely low-frequency electromagnetic fields occupations and amyotrophic lateral sclerosis: A meta-analysis. PLoS One 2012, 7, e48354. [Google Scholar]
- Abhinav, K.; Al-Chalabi, A.; Hortobagyi, T.; Leigh, P.N. Electrical injury and amyotrophic lateral sclerosis: A systematic review of the literature. J. Neurol. Neurosurg. Psychiatry 2007, 78, 450–453. [Google Scholar]
- Falone, S.; Mirabilio, A.; Carbone, M.C.; Zimmitti, V.; di Loreto, S.; Mariggiò, M.A.; Mancinelli, R.; di Ilio, C.; Amicarelli, F. Chronic exposure to 50 Hz magnetic fields causes a significant weakening of antioxidant defence systems in aged rat brain. Int. J. Biochem. Cell. Biol 2008, 40, 2762–2770. [Google Scholar]
- Martinez-Samano, J.; Torres-Duran, P.V.; Juarez-Oropeza, M.A.; Verdugo-Diaz, L. Effect of acute extremely low frequency electromagnetic field exposure on the antioxidant status and lipid levels in rat brain. Arch. Med. Res 2012, 43, 183–189. [Google Scholar]
- Poulletier de Gannes, F.; Ruffié, G.; Taxile, M.; Ladevèze, E.; Hurtier, A.; Haro, E.; Duleu, S.; Charlet de Sauvage, R.; Billaudel, B.; Geffard, M.; et al. Amyotrophic lateral sclerosis (ALS) and extremely-low frequency (ELF) magnetic fields: A study in the SOD-1 transgenic mouse model. Amyotroph. Lateral. Scler 2009, 10, 370–373. [Google Scholar]
- Elbaz, A.; Dufouil, C.; Alpérovitch, A. Interaction between genes and environment in neurodegenerative diseases. C. R. Biol 2007, 330, 318–328. [Google Scholar]
- Stozická, Z.; Zilka, N.; Novák, M. Risk and protective factors for sporadic Alzheimer’s disease. Acta Virol 2007, 51, 205–222. [Google Scholar]
- Savettieri, G.; Salemi, G.; Arcara, A.; Cassata, M.; Castiglione, M.G.; Fierro, B. A case-control study of amyotrophic lateral sclerosis. Neuroepidemiology 1991, 10, 242–245. [Google Scholar]
- Gunnarsson, L.G.; Bodin, L.; Soderfeldt, B.; Axelson, O. A case-control study of motor neurone disease—Its relation to heritability, and occupational exposures, particularly to solvents. Br. J. Ind. Med 1992, 49, 791–798. [Google Scholar]
- Chancellor, A.M.; Slattery, J.M.; Fraser, H.; Warlow, C.P. Risk factors for motor neuron disease—A case-control study based on patients from the Scottish motor neuron disease register. J. Neurol. Neurosurg. Psychiatry 1993, 56, 1200–1206. [Google Scholar]
- McGuire, V.; Longstreth, W.T., Jr; Nelson, L.M.; Koepsell, T.D.; Checkoway, H.; Morgan, M.S.; van Belle, G. Occupational exposures and amyotrophic lateral sclerosis: A population-based case-control study. Am. J. Epidemiol. 1997, 145, 1076–1088. [Google Scholar]
- Bonvicini, F.; Marcello, N.; Mandrioli, J.; Pietrini, V.; Vinceti, M. Exposure to pesticides and risk of amyotrophic lateral sclerosis: A population-based case-control study. Ann. Ist. Super. Sanità 2010, 46, 284–287. [Google Scholar]
- Kamel, F.; Umbach, D.M.; Bedlack, R.S.; Richards, M.; Watson, M.; Alavanja, M.C.R.; Blair, A.; Hoppin, J.A.; Schmidt, S.; Sandler, D.P. Pesticide exposure and amyotrophic lateral sclerosis. Neurotoxicology 2012, 33, 457–462. [Google Scholar]
- Burns, C.J.; Beard, K.K.; Cartmill, J.B. Mortality in chemical workers potentially exposed to 2,4-dichlorophenoxyacetic acid (2,4-D) 1945–94: An update. Occup. Environ. Med 2001, 58, 24–30. [Google Scholar]
- Granieri, E.; Carreras, M.; Tola, R.; Paolino, E.; Tralli, G.; Eleopra, R.; Serra, G. Motor neuron disease in the province of Ferrara, Italy, in 1964–1982. Neurology 1988, 38, 1604–1608. [Google Scholar]
- Malek, A.M.; Barchowsky, A.; Bowser, R.; Youk, A.; Talbott, E.O. Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: A meta-analysis of epidemiological studies: Pesticide exposure as a risk factor for ALS. Environ. Res 2012, 117, 112–119. [Google Scholar]
- Weiss, B.; Amler, S.; Amler, W.R. Pesticides. Pediatrics 2004, 113, 1030–1036. [Google Scholar]
- Chen, Y. Organophosphate-induced brain damage: Mechanisms, neuropsychiatric and neurological consequences, and potential therapeutic strategies. Neurotoxicology 2012, 33, 391–400. [Google Scholar]
- Morahan, J.M.; Yu, B.; Trent, R.J.; Pamphlett, R. Genetic susceptibility to environmental toxicants in ALS. Am. J. Med. Genet. B. Neuropsychiatr. Genet 2007, 144B, 885–890. [Google Scholar]
- Wills, A.M.; Landers, J.E.; Zhang, H.; Richter, R.J.; Caraganis, A.J.; Cudkowicz, M.E.; Furlong, C.E.; Brown, R.H., Jr. Paraoxonase 1 (PON1) organophosphate hydrolysis is not reduced in ALS. Neurology 2008, 70, 929–934. [Google Scholar]
- Costa, L.G.; Giordano, G.; Cole, T.B.; Marsillach, J.; Furlong, C.E. Paraoxonase 1 (PON1) as a genetic determinant of susceptibility to organophosphate toxicity. Toxicology 2013, 307, 115–122. [Google Scholar]
- Landers, J.E.; Shi, L.; Cho, T.J.; Glass, J.D.; Shaw, C.E.; Leigh, P.N.; Diekstra, F.; Polak, M.; Rodriguez-Leyva, I.; Niemann, S.; et al. A common haplotype within the PON1 promoter region is associated with sporadic ALS. Amyotroph. Lateral Scler 2008, 9, 306–314. [Google Scholar]
- Richter, R.J.; Jarvik, G.P.; Furlong, C.E. Paraoxonase 1 (PON1) status and substrate hydrolysis. Toxicol. Appl. Pharmacol 2009, 235, 1–9. [Google Scholar]
- Saeed, M.; Siddique, N.; Hung, W.Y.; Usacheva, E.; Liu, E.; Sufit, R.L.; Heller, S.L.; Haines, J.L.; Pericak-Vance, M.; Siddique, T. Paraoxonase cluster polymorphisms are associated with sporadic ALS. Neurology 2006, 67, 771–776. [Google Scholar]
- Slowik, A.; Tomik, B.; Wolkow, P.P.; Partyka, D.; Turaj, W.; Malecki, M.T.; Pera, J.; Dziedzic, T.; Szczudlik, A.; Figlewicz, D.A. Paraoxonase gene polymorphisms and sporadic ALS. Neurology 2006, 67, 766–770. [Google Scholar]
- Cronin, S.; Greenway, M.J.; Prehn, J.H.; Hardiman, O. Paraoxonase promoter and intronic variants modify risk of sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2007, 78, 984–986. [Google Scholar]
- Valdmanis, P.N.; Kabashi, E.; Dyck, A.; Hince, P.; Lee, J.; Dion, P.; D’Amour, M.; Souchon, F.; Bouchard, J.P.; Salachas, F.; et al. Association of paraoxonase gene cluster polymorphisms with ALS in France, Quebec, and Sweden. Neurology 2008, 71, 514–520. [Google Scholar]
- Simpson, C.L.; Al-Chalabi, A. Amyotrophic lateral sclerosis as a complex genetic disease. Biochim. Biophys. Acta 2006, 1762, 973–985. [Google Scholar]
- Van Blitterswijk, M.; Blokhuis, A.; van Es, M.A.; van Vught, P.W.; Rowicka, P.A.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. Rare and common paraoxonase gene variants in amyotrophic lateral sclerosis patients. Neurobiol. Aging 2012, 33, 1845, e1–1845.e3.. [Google Scholar]
- Vinceti, M.; Fiore, M.; Signorelli, C.; Odone, A.; Tesauro, M.; Consonni, M.; Arcolin, E.; Malagoli, C.; Mandrioli, J.; Marmiroli, S.; et al. Environmental risk factors for amyotrophic lateral sclerosis: Methodological issues in epidemiologic studies. Ann. Ig 2012, 24, 407–415. [Google Scholar]
- Qureshi, I.A.; Mehler, M.F. Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr. Neurol. Neurosci. Rep 2011, 11, 464–473. [Google Scholar]
- Marques, S.C.F.; Oliveira, C.R.; Pereira, C.M.F.; Outeiro, T.F. Epigenetics in neurodegeneration: A new layer of complexity. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 348–355. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Trojsi, F.; Monsurrò, M.R.; Tedeschi, G. Exposure to Environmental Toxicants and Pathogenesis of Amyotrophic Lateral Sclerosis: State of the Art and Research Perspectives. Int. J. Mol. Sci. 2013, 14, 15286-15311. https://doi.org/10.3390/ijms140815286
Trojsi F, Monsurrò MR, Tedeschi G. Exposure to Environmental Toxicants and Pathogenesis of Amyotrophic Lateral Sclerosis: State of the Art and Research Perspectives. International Journal of Molecular Sciences. 2013; 14(8):15286-15311. https://doi.org/10.3390/ijms140815286
Chicago/Turabian StyleTrojsi, Francesca, Maria Rosaria Monsurrò, and Gioacchino Tedeschi. 2013. "Exposure to Environmental Toxicants and Pathogenesis of Amyotrophic Lateral Sclerosis: State of the Art and Research Perspectives" International Journal of Molecular Sciences 14, no. 8: 15286-15311. https://doi.org/10.3390/ijms140815286