Defective Homocysteine Metabolism: Potential Implications for Skeletal Muscle Malfunction

{kind=link}
{kind=link}
{kind=link}
Abstract
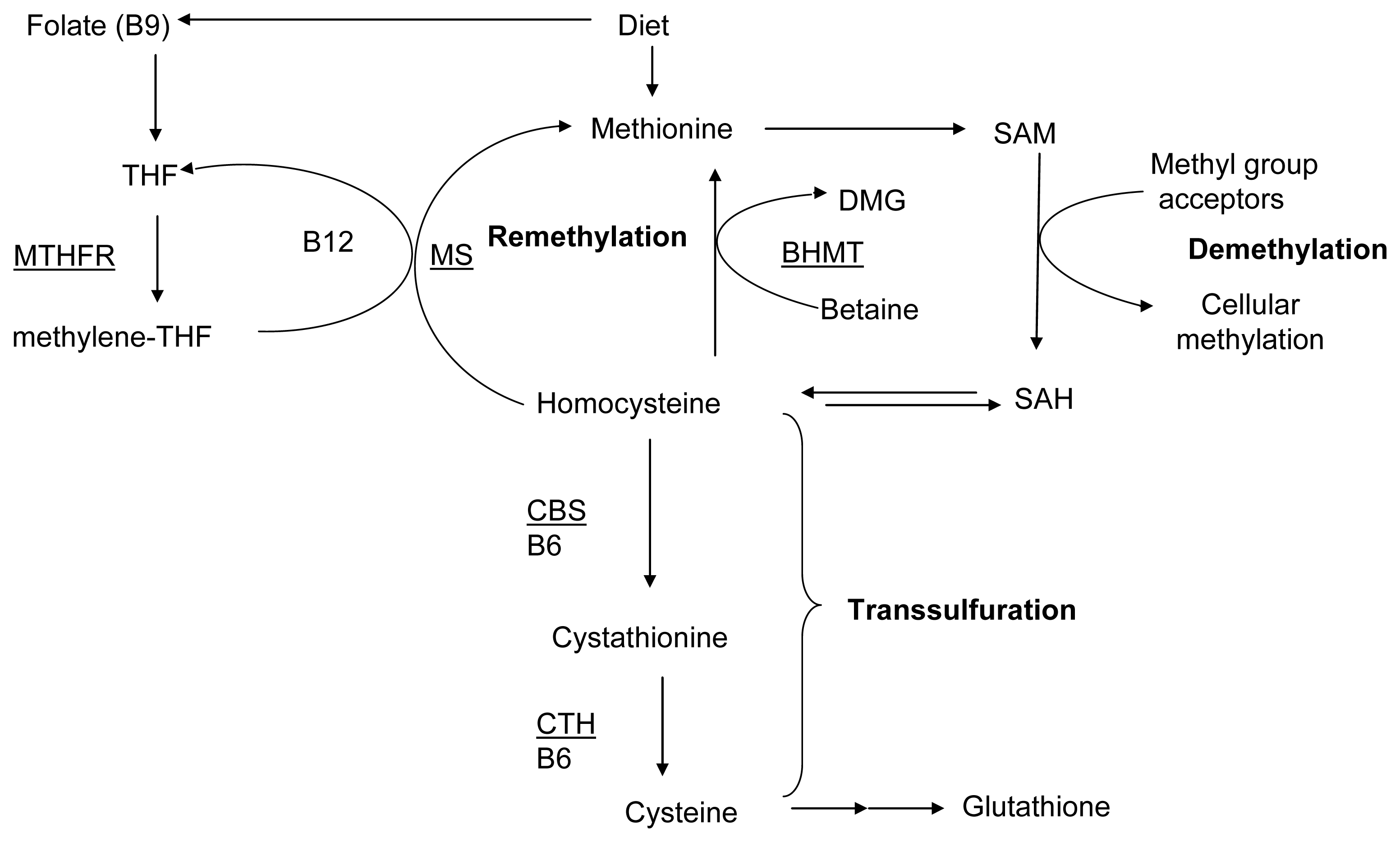
:1. Introduction
2. HHcy & Compromised Antioxidant Capability
3. HHcy & Hypomethylation
4. HHcy & Inflammation
5. HHcy & NO
6. HHcy and Endoplasmic Reticulum (ER) Stress
7. HHcy & Cell Signaling Pathways
7.1. TGF-β Signaling
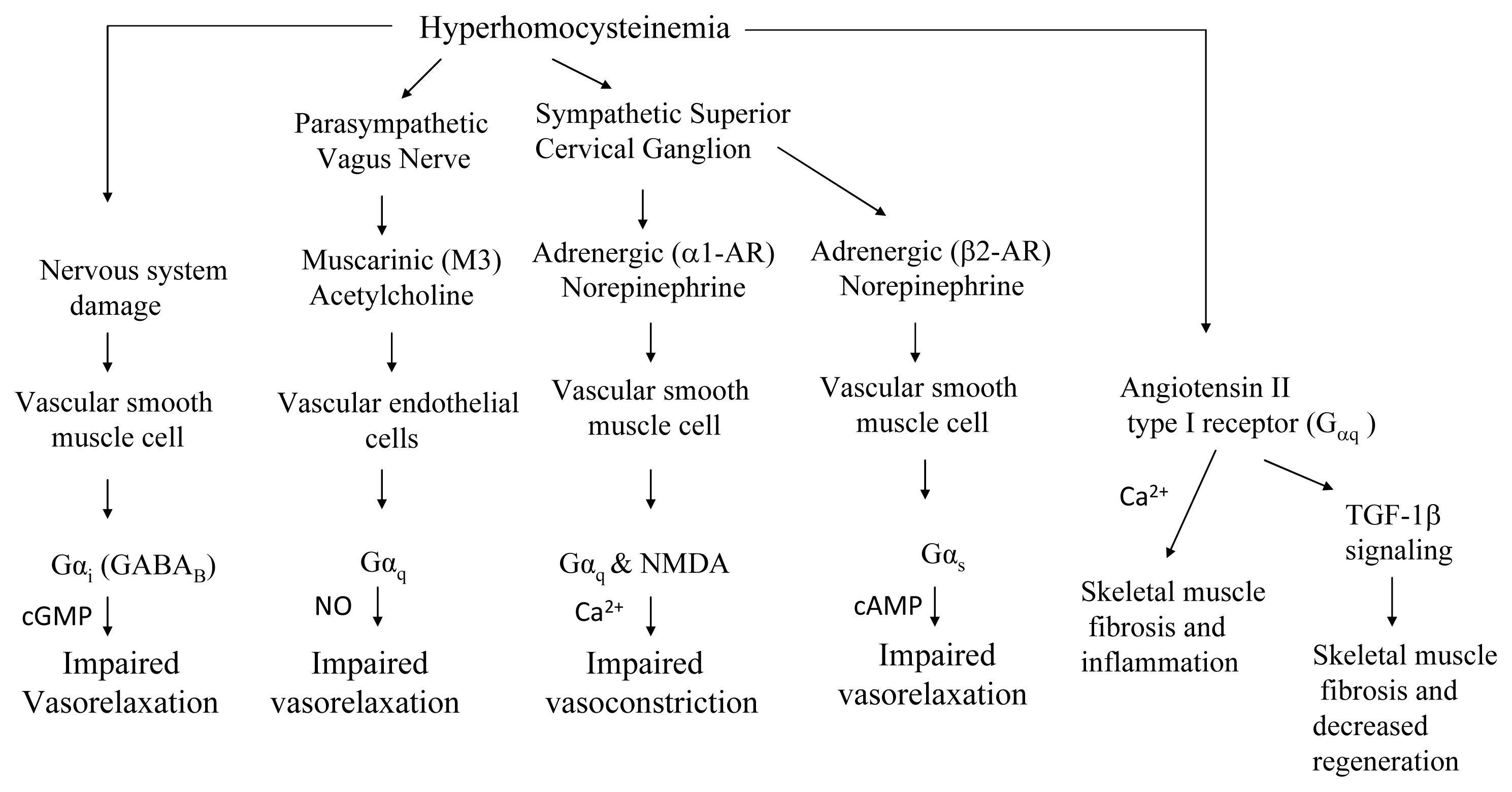
7.2. GPCR (G-protein Coupled Receptor) Signaling
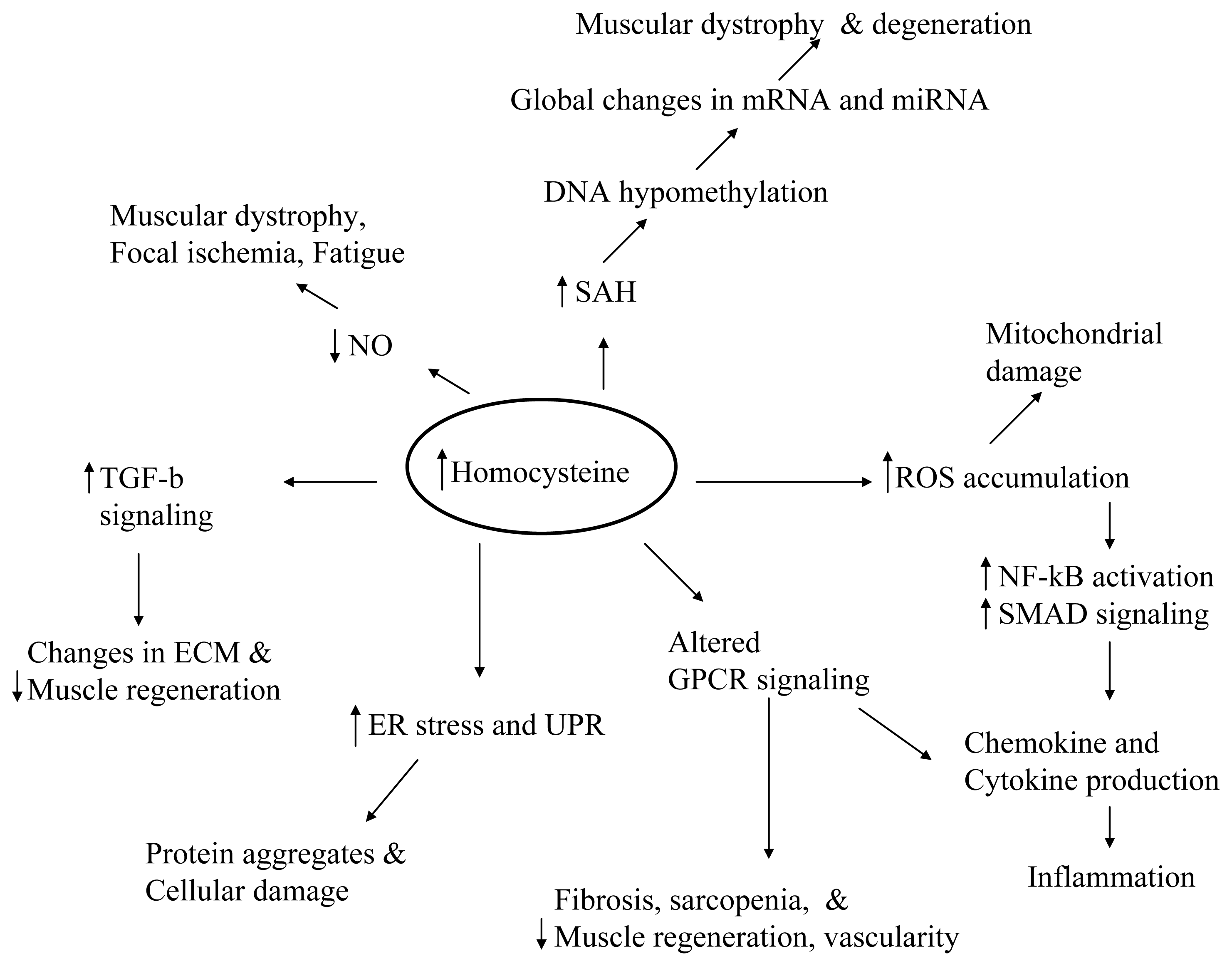
8. Conclusions and Future Perspectives
Acknowledgments
Conflict of Interest
References
- Maron, B.A.; Loscalzo, J. The treatment of hyperhomocysteinemia. Annu. Rev. Med 2009, 60, 39–54. [Google Scholar]
- Kalani, A.; Kamat, P.K.; Tyagi, S.C.; Tyagi, N. Synergy of homocysteine, microRNA, and epigenetics: A novel therapeutic approach for stroke. Mol. Neurobiol. 2013. [Google Scholar] [CrossRef]
- Schalinske, K.L.; Smazal, A.L. Homocysteine imbalance: A pathological metabolic marker. Adv. Nutr 2012, 3, 755–762. [Google Scholar]
- Steed, M.M.; Tyagi, S.C. Mechanisms of cardiovascular remodeling in hyperhomocysteinemia. Antioxid. Redox Signal 2011, 15, 1927–1943. [Google Scholar]
- Kanwar, Y.S.; Manaligod, J.R.; Wong, P.W. Morphologic studies in a patient with homocystinuria due to 5,10-methylenetetrahydrofolate reductase deficiency. Pediatr. Res 1976, 10, 598–609. [Google Scholar]
- Kolling, J.; Scherer, E.B.; Siebert, C.; Hansen, F.; Torres, F.V.; Scaini, G.; Ferreira, G.; de Andrade, R.B.; Goncalves, C.A.; Streck, E.L.; et al. Homocysteine induces energy imbalance in rat skeletal muscle: Is creatine a protector? Cell Biochem. Funct. 2012. [Google Scholar] [CrossRef]
- Valentino, F.; Bivona, G.; Butera, D.; Paladino, P.; Fazzari, M.; Piccoli, T.; Ciaccio, M.; La Bella, V. Elevated cerebrospinal fluid and plasma homocysteine levels in ALS. Eur. J. Neurol 2010, 17, 84–89. [Google Scholar]
- Zoccolella, S.; Simone, I.L.; Lamberti, P.; Samarelli, V.; Tortelli, R.; Serlenga, L.; Logroscino, G. Elevated plasma homocysteine levels in patients with amyotrophic lateral sclerosis. Neurology 2008, 70, 222–225. [Google Scholar]
- Zoccolella, S.; Tortorella, C.; Iaffaldano, P.; Direnzo, V.; D’Onghia, M.; Paolicelli, D.; Livrea, P.; Trojano, M. Elevated plasma homocysteine levels in patients with multiple sclerosis are associated with male gender. J. Neurol 2012, 259, 2105–2110. [Google Scholar]
- McDermott, M.M.; Ferrucci, L.; Guralnik, J.M.; Tian, L.; Green, D.; Liu, K.; Tan, J.; Liao, Y.; Pearce, W.H.; Schneider, J.R.; et al. Elevated levels of inflammation, d-dimer, and homocysteine are associated with adverse calf muscle characteristics and reduced calf strength in peripheral arterial disease. J. Am. Coll. Cardiol 2007, 50, 897–905. [Google Scholar]
- Kado, D.M.; Bucur, A.; Selhub, J.; Rowe, J.W.; Seeman, T. Homocysteine levels and decline in physical function: MacArthur studies of successful aging. Am. J. Med 2002, 113, 537–542. [Google Scholar]
- Swart, K.M.; Enneman, A.W.; van Wijngaarden, J.P.; van Dijk, S.C.; Brouwer-Brolsma, E.M.; Ham, A.C.; Dhonukshe-Rutten, R.A.; van der Velde, N.; Brug, J.; van Meurs, J.B.; et al. Homocysteine and the methylenetetrahydrofolate reductase 677Cshort right arrowT polymorphism in relation to muscle mass and strength, physical performance and postural sway. Eur. J. Clin. Nutr 2013, 67, 743–748. [Google Scholar]
- Van Schoor, N.M.; Swart, K.M.; Pluijm, S.M.; Visser, M.; Simsek, S.; Smulders, Y.; Lips, P. Cross-sectional and longitudinal association between homocysteine, vitamin B12 and physical performance in older persons. Eur. J. Clin. Nutr 2012, 66, 174–181. [Google Scholar]
- Ng, T.P.; Aung, K.C.; Feng, L.; Scherer, S.C.; Yap, K.B. Homocysteine, folate, vitamin B-12, and physical function in older adults: Cross-sectional findings from the Singapore Longitudinal Ageing Study. Am. J. Clin. Nutr 2012, 96, 1362–1368. [Google Scholar]
- Miller, A.; Mujumdar, V.; Shek, E.; Guillot, J.; Angelo, M.; Palmer, L.; Tyagi, S.C. Hyperhomocyst(e)inemia induces multiorgan damage. Heart Vessels 2000, 15, 135–143. [Google Scholar]
- Swart, K.M.; van Schoor, N.M.; Heymans, M.W.; Schaap, L.A.; den Heijer, M.; Lips, P. Elevated homocysteine levels are associated with low muscle strength and functional limitations in older persons. J. Nutr. Health Aging 2012, 17, 578–584. [Google Scholar]
- Hammouda, O.; Chtourou, H.; Chaouachi, A.; Chahed, H.; Ferchichi, S.; Kallel, C.; Chamari, K.; Souissi, N. Effect of short-term maximal exercise on biochemical markers of muscle damage, total antioxidant status, and homocysteine levels in football players. Asian J. Sports Med 2012, 3, 239–246. [Google Scholar]
- Deminice, R.; Vannucchi, H.; Simoes-Ambrosio, L.M.; Jordao, A.A. Creatine supplementation reduces increased homocysteine concentration induced by acute exercise in rats. Eur. J. Appl. Physiol 2011, 111, 2663–2670. [Google Scholar]
- Herrmann, M.; Wilkinson, J.; Schorr, H.; Obeid, R.; Georg, T.; Urhausen, A.; Scharhag, J.; Kindermann, W.; Herrmann, W. Comparison of the influence of volume-oriented training and high-intensity interval training on serum homocysteine and its cofactors in young, healthy swimmers. Clin. Chem. Lab. Med 2003, 41, 1525–1531. [Google Scholar]
- Gorce-Dupuy, A.M.; Vela, C.; Badiou, S.; Bargnoux, A.S.; Josse, C.; Roagna, N.; Delage, M.; Michel, F.; Vernet, M.H.; Destizons, D.; et al. Antioxidant and oligonutrient status, distribution of amino acids, muscle damage, inflammation, and evaluation of renal function in elite rugby players. Clin. Chem. Lab. Med 2012, 50, 1777–1789. [Google Scholar]
- Stead, L.M.; Au, K.P.; Jacobs, R.L.; Brosnan, M.E.; Brosnan, J.T. Methylation demand and homocysteine metabolism: Effects of dietary provision of creatine and guanidinoacetate. Am. J. Physiol. Endocrinol. Metab 2001, 281, E1095–E100. [Google Scholar]
- Cravo, M. Alcohol, methylenetetrahydrofolate 677C→T genotype, and low folate intake: Concurrent causes for hyperhomocysteinemia. Am. J. Clin. Nutr 2005, 82, 3–4. [Google Scholar]
- Chen, N.C.; Yang, F.; Capecci, L.M.; Gu, Z.; Schafer, A.I.; Durante, W.; Yang, X.F.; Wang, H. Regulation of homocysteine metabolism and methylation in human and mouse tissues. FASEB J 2010, 24, 2804–2817. [Google Scholar]
- Majors, A.K.; Pyeritz, R.E. A deficiency of cysteine impairs fibrillin-1 deposition: Implications for the pathogenesis of cystathionine beta-synthase deficiency. Mol. Genet. Metab 2000, 70, 252–260. [Google Scholar]
- Bannai, S.; Kitamura, E. Transport interaction of l-cystine and l-glutamate in human diploid fibroblasts in culture. J. Biol. Chem 1980, 255, 2372–2376. [Google Scholar]
- Budy, B.; O’Neill, R.; DiBello, P.M.; Sengupta, S.; Jacobsen, D.W. Homocysteine transport by human aortic endothelial cells: Identification and properties of import systems. Arch. Biochem. Biophys 2006, 446, 119–130. [Google Scholar]
- Jiang, X.; Yang, F.; Brailoiu, E.; Jakubowski, H.; Dun, N.J.; Schafer, A.I.; Yang, X.; Durante, W.; Wang, H. Differential regulation of homocysteine transport in vascular endothelial and smooth muscle cells. Arterioscler. Thromb. Vasc. Biol 2007, 27, 1976–1983. [Google Scholar]
- Ishii, I.; Akahoshi, N.; Yamada, H.; Nakano, S.; Izumi, T.; Suematsu, M. Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem 2010, 285, 26358–26368. [Google Scholar]
- Martensson, J.; Meister, A. Mitochondrial damage in muscle occurs after marked depletion of glutathione and is prevented by giving glutathione monoester. Proc. Natl. Acad. Sci. USA 1989, 86, 471–475. [Google Scholar]
- Lang, D.; Kredan, M.B.; Moat, S.J.; Hussain, S.A.; Powell, C.A.; Bellamy, M.F.; Powers, H.J.; Lewis, M.J. Homocysteine-induced inhibition of endothelium-dependent relaxation in rabbit aorta: Role for superoxide anions. Arterioscler. Thromb. Vasc. Biol 2000, 20, 422–427. [Google Scholar]
- Tyagi, N.; Moshal, K.S.; Sen, U.; Lominadze, D.; Ovechkin, A.V.; Tyagi, S.C. Ciglitazone ameliorates homocysteine-mediated mitochondrial translocation and matrix metalloproteinase-9 activation in endothelial cells by inducing peroxisome proliferator activated receptor-gamma activity. Cell. Mol. Biol 2006, 52, 21–27. [Google Scholar]
- Liu, Z.; Luo, H.; Zhang, L.; Huang, Y.; Liu, B.; Ma, K.; Feng, J.; Xie, J.; Zheng, J.; Hu, J.; et al. Hyperhomocysteinemia exaggerates adventitial inflammation and angiotensin II-induced abdominal aortic aneurysm in mice. Circ. Res 2012, 111, 1261–1273. [Google Scholar]
- Jamaluddin, M.S.; Yang, X.; Wang, H. Hyperhomocysteinemia, DNA methylation and vascular disease. Clin. Chem. Lab. Med 2007, 45, 1660–1666. [Google Scholar]
- Caudill, M.A.; Wang, J.C.; Melnyk, S.; Pogribny, I.P.; Jernigan, S.; Collins, M.D.; Santos-Guzman, J.; Swendseid, M.E.; Cogger, E.A.; James, S.J. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. J. Nutr 2001, 131, 2811–2818. [Google Scholar]
- Tsai, J.C.; Wang, H.; Perrella, M.A.; Yoshizumi, M.; Sibinga, N.E.; Tan, L.C.; Haber, E.; Chang, T.H.; Schlegel, R.; Lee, M.E. Induction of cyclin A gene expression by homocysteine in vascular smooth muscle cells. J. Clin. Invest 1996, 97, 146–153. [Google Scholar]
- Niesen, M.I.; Osborne, A.R.; Yang, H.; Rastogi, S.; Chellappan, S.; Cheng, J.Q.; Boss, J.M.; Blanck, G. Activation of a methylated promoter mediated by a sequence-specific DNA-binding protein, RFX. J. Biol. Chem 2005, 280, 38914–38922. [Google Scholar]
- Kangaspeska, S.; Stride, B.; Metivier, R.; Polycarpou-Schwarz, M.; Ibberson, D.; Carmouche, R.P.; Benes, V.; Gannon, F.; Reid, G. Transient cyclical methylation of promoter DNA. Nature 2008, 452, 112–115. [Google Scholar]
- Metivier, R.; Gallais, R.; Tiffoche, C.; Le Peron, C.; Jurkowska, R.Z.; Carmouche, R.P.; Ibberson, D.; Barath, P.; Demay, F.; Reid, G.; et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008, 452, 45–50. [Google Scholar]
- Acharyya, S.; Sharma, S.M.; Cheng, A.S.; Ladner, K.J.; He, W.; Kline, W.; Wang, H.; Ostrowski, M.C.; Huang, T.H.; Guttridge, D.C. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: Implications in duchenne muscular dystrophy. PLoS One 2010, 5, e12479. [Google Scholar]
- Chen, J.F.; Callis, T.E.; Wang, D.Z. microRNAs and muscle disorders. J. Cell Sci 2009, 122, 13–20. [Google Scholar]
- Eisenberg, I.; Eran, A.; Nishino, I.; Moggio, M.; Lamperti, C.; Amato, A.A.; Lidov, H.G.; Kang, P.B.; North, K.N.; Mitrani-Rosenbaum, S.; et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 17016–17021. [Google Scholar]
- Liu, N.; Bezprozvannaya, S.; Shelton, J.M.; Frisard, M.I.; Hulver, M.W.; McMillan, R.P.; Wu, Y.; Voelker, K.A.; Grange, R.W.; Richardson, J.A.; et al. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J. Clin. Invest 2011, 121, 3258–3268. [Google Scholar]
- Kochegarov, A.; Moses, A.; Lian, W.; Meyer, J.; Hanna, M.C.; Lemanski, L.F. A new unique form of microRNA from human heart, microRNA-499c, promotes myofibril formation and rescues cardiac development in mutant axolotl embryos. J. Biomed. Sci 2013, 20, 20. [Google Scholar]
- Cacchiarelli, D.; Martone, J.; Girardi, E.; Cesana, M.; Incitti, T.; Morlando, M.; Nicoletti, C.; Santini, T.; Sthandier, O.; Barberi, L.; et al. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab 2010, 12, 341–351. [Google Scholar]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and creatinine metabolism. Physiol. Rev 2000, 80, 1107–1213. [Google Scholar]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar]
- Brock, T.A.; Alexander, R.W.; Ekstein, L.S.; Atkinson, W.J.; Gimbrone, M.A., Jr. Angiotensin increases cytosolic free calcium in cultured vascular smooth muscle cells. Hypertension 1985, 7, I105–I109. [Google Scholar]
- Dalakas, M.C. Review: An update on inflammatory and autoimmune myopathies. Neuropathol. Appl. Neurobiol 2011, 37, 226–242. [Google Scholar]
- Zhang, C.; Boini, K.M.; Xia, M.; Abais, J.M.; Li, X.; Liu, Q.; Li, P.L. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension 2012, 60, 154–162. [Google Scholar]
- Da Cunha, A.A.; Ferreira, A.G.; Loureiro, S.O.; da Cunha, M.J.; Schmitz, F.; Netto, C.A.; Wyse, A.T. Chronic hyperhomocysteinemia increases inflammatory markers in hippocampus and serum of rats. Neurochem. Res 2012, 37, 1660–1669. [Google Scholar]
- Da Cunha, A.A.; Ferreira, A.G.; Wyse, A.T. Increased inflammatory markers in brain and blood of rats subjected to acute homocysteine administration. Metab. Brain Dis 2010, 25, 199–206. [Google Scholar]
- Semmler, A.; Prost, J.C.; Smulders, Y.; Smith, D.; Blom, H.; Bigler, L.; Linnebank, M. Methylation metabolism in sepsis and systemic inflammatory response syndrome. Scand. J. Clin. Lab. Invest. 2013. [Google Scholar] [CrossRef]
- Zeng, X.; Dai, J.; Remick, D.G.; Wang, X. Homocysteine mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Circ. Res 2003, 93, 311–320. [Google Scholar]
- Villanueva, C.; Giulivi, C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic. Biol. Med 2010, 49, 307–316. [Google Scholar]
- Baum, O.; Vieregge, M.; Koch, P.; Gul, S.; Hahn, S.; Huber-Abel, F.A.; Pries, A.R.; Hoppeler, H.H. Phenotype of capillaries in skeletal muscle of nNOS-knockout mice. Am. J. Physiol. Regul. Integr. Comp. Physiol 2013, 304, R1175–R1182. [Google Scholar]
- Dankner, R.; Chetrit, A.; Dror, G.K.; Sela, B.A. Physical activity is inversely associated with total homocysteine levels, independent of C677T MTHFR genotype and plasma B vitamins. Age 2007, 29, 219–227. [Google Scholar]
- Dominguez, L.J.; Galioto, A.; Pineo, A.; Ferlisi, A.; Ciaccio, M.; Putignano, E.; Belvedere, M.; Costanza, G.; Barbagallo, M. Age, homocysteine, and oxidative stress: Relation to hypertension and type 2 diabetes mellitus. J. Am. Coll. Nutr 2010, 29, 1–6. [Google Scholar]
- Looft-Wilson, R.C.; Payne, G.W.; Segal, S.S. Connexin expression and conducted vasodilation along arteriolar endothelium in mouse skeletal muscle. J. Appl. Physiol 2004, 97, 1152–1158. [Google Scholar]
- De Wit, C.; Roos, F.; Bolz, S.S.; Kirchhoff, S.; Kruger, O.; Willecke, K.; Pohl, U. Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ. Res 2000, 86, 649–655. [Google Scholar]
- Givvimani, S.; Narayanan, N.; Armaghan, F.; Pushpakumar, S.; Tyagi, S.C. Attenuation of conducted vasodilation in skeletal muscle arterioles during hyperhomocysteinemia. Pharmacology 2013, 91, 287–296. [Google Scholar]
- Deldicque, L.; Hespel, P.; Francaux, M. Endoplasmic reticulum stress in skeletal muscle: Origin and metabolic consequences. Exerc. Sport Sci. Rev 2012, 40, 43–49. [Google Scholar]
- Rayavarapu, S.; Coley, W.; Nagaraju, K. Endoplasmic reticulum stress in skeletal muscle homeostasis and disease. Curr. Rheumatol. Rep 2012, 14, 238–243. [Google Scholar]
- Askanas, V.; Engel, W.K. Sporadic inclusion-body myositis: Conformational multifactorial ageing-related degenerative muscle disease associated with proteasomal and lysosomal inhibition, endoplasmic reticulum stress, and accumulation of amyloid-β42 oligomers and phosphorylated tau. Presse Med 2011, 40, e219–e235. [Google Scholar]
- Outinen, P.A.; Sood, S.K.; Liaw, P.C.; Sarge, K.D.; Maeda, N.; Hirsh, J.; Ribau, J.; Podor, T.J.; Weitz, J.I.; Austin, R.C. Characterization of the stress-inducing effects of homocysteine. Biochem. J 1998, 332, 213–221. [Google Scholar]
- Kokame, K.; Kato, H.; Miyata, T. Homocysteine-respondent genes in vascular endothelial cells identified by differential display analysis. GRP78/BiP and novel genes. J. Biol. Chem 1996, 271, 29659–29665. [Google Scholar]
- Miyata, T.; Kokame, K.; Agarwala, K.L.; Kato, H. Analysis of gene expression in homocysteine-injured vascular endothelial cells: Demonstration of GRP78/BiP expression, cloning and characterization of a novel reducing agent-tunicamycin regulated gene. Semin. Thromb. Hemost 1998, 24, 285–291. [Google Scholar]
- Lentz, S.R.; Sadler, J.E. Homocysteine inhibits von Willebrand factor processing and secretion by preventing transport from the endoplasmic reticulum. Blood 1993, 81, 683–689. [Google Scholar]
- Outinen, P.A.; Sood, S.K.; Pfeifer, S.I.; Pamidi, S.; Podor, T.J.; Li, J.; Weitz, J.I.; Austin, R.C. Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood 1999, 94, 959–967. [Google Scholar]
- Glushchenko, A.V.; Jacobsen, D.W. Molecular targeting of proteins by l-homocysteine: Mechanistic implications for vascular disease. Antioxid. Redox. Signal 2007, 9, 1883–1898. [Google Scholar]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med 2007, 13, 204–210. [Google Scholar]
- Raaf, L.; Noll, C.; Cherifi Mel, H.; Samuel, J.L.; Delcayre, C.; Delabar, J.M.; Benazzoug, Y.; Janel, N. Myocardial fibrosis and TGFB expression in hyperhomocysteinemic rats. Mol. Cell. Biochem 2011, 347, 63–70. [Google Scholar]
- Liu, X.; Luo, F.; Li, J.; Wu, W.; Li, L.; Chen, H. Homocysteine induces connective tissue growth factor expression in vascular smooth muscle cells. J. Thromb. Haemost 2008, 6, 184–192. [Google Scholar]
- Mujumdar, V.S.; Hayden, M.R.; Tyagi, S.C. Homocyst(e)ine induces calcium second messenger in vascular smooth muscle cells. J. Cell. Physiol 2000, 183, 28–36. [Google Scholar]
- Moshal, K.S.; Sen, U.; Tyagi, N.; Henderson, B.; Steed, M.; Ovechkin, A.V.; Tyagi, S.C. Regulation of homocysteine-induced MMP-9 by ERK1/2 pathway. Am. J. Physiol. Cell Physiol 2006, 290, C883–C891. [Google Scholar]
- Vacek, T.P.; Sen, U.; Tyagi, N.; Kumar, M.; Moshal, K.S.; Passmore, J.C.; Tyagi, S.C. Homocysteine effects classical pathway of GPCR down regulation: Galpha(q/11), Galpha(12/13), G(i/o). Mol. Cell. Biochem 2009, 321, 1–8. [Google Scholar]
- Morales, M.G.; Vazquez, Y.; Acuna, M.J.; Rivera, J.C.; Simon, F.; Salas, J.D.; Alvarez Ruf, J.; Brandan, E.; Cabello-Verrugio, C. Angiotensin II-induced pro-fibrotic effects require p38MAPK activity and transforming growth factor beta 1 expression in skeletal muscle cells. Int. J. Biochem. Cell Biol 2012, 44, 1993–2002. [Google Scholar]
- Cabello-Verrugio, C.; Acuna, M.J.; Morales, M.G.; Becerra, A.; Simon, F.; Brandan, E. Fibrotic response induced by angiotensin-II requires NAD(P)H oxidase-induced reactive oxygen species (ROS) in skeletal muscle cells. Biochem. Biophys. Res. Commun 2011, 410, 665–670. [Google Scholar]
- Limas, C.J. Effect of phospholipid methylation on beta-adrenergic receptors in the normal and hypertrophied rat myocardium. Circ. Res 1980, 47, 536–541. [Google Scholar]
- Mishra, P.K.; Awe, O.; Metreveli, N.; Qipshidze, N.; Joshua, I.G.; Tyagi, S.C. Exercise mitigates homocysteine—beta2-adrenergic receptor interactions to ameliorate contractile dysfunction in diabetes. Int. J. Physiol. Pathophysiol. Pharmacol 2011, 3, 97–106. [Google Scholar]
- Santulli, G.; Iaccarino, G. Pinpointing beta adrenergic receptor in ageing pathophysiology: Victim or executioner? Evidence from crime scenes. Immun. Ageing 2013, 10, 10. [Google Scholar]
- French, J.F.; Rapoport, R.M.; Matlib, M.A. Possible mechanism of benzodiazepine-induced relaxation of vascular smooth muscle. J. Cardiovasc. Pharmacol 1989, 14, 405–411. [Google Scholar]
- Eckly-Michel, A.; Martin, V.; Lugnier, C. Involvement of cyclic nucleotide-dependent protein kinases in cyclic AMP-mediated vasorelaxation. Br. J. Pharmacol 1997, 122, 158–164. [Google Scholar]
- Webb, R.C. Smooth muscle contraction and relaxation. Adv. Physiol. Educ 2003, 27, 201–206. [Google Scholar]
- Fujiwara, M.; Muramatsu, I. Gamma-aminobutyric acid receptor on vascular smooth muscle of dog cerebral arteries. Br. J. Pharmacol 1975, 55, 561–562. [Google Scholar]
- Bauer, M.B.; Murphy, S.; Gebhart, G.F. Muscarinic cholinergic stimulation of the nitric oxide-cyclic GMP signaling system in cultured rat sensory neurons. Neuroscience 1994, 62, 351–359. [Google Scholar]
- Grange, R.W.; Isotani, E.; Lau, K.S.; Kamm, K.E.; Huang, P.L.; Stull, J.T. Nitric oxide contributes to vascular smooth muscle relaxation in contracting fast-twitch muscles. Physiol. Genomics 2001, 5, 35–44. [Google Scholar]
- Phillippe, M.; Bangalore, S. Adrenergic stimulation of inositol-phosphate production in a genital tract smooth muscle cell line. Biol. Reprod 1989, 41, 49–53. [Google Scholar]
- Lu, W.Y.; Xiong, Z.G.; Lei, S.; Orser, B.A.; Dudek, E.; Browning, M.D.; MacDonald, J.F. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat. Neurosci 1999, 2, 331–338. [Google Scholar]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev 2003, 83, 1325–1358. [Google Scholar]
- Mathes, C.; Thompson, S.H. The nitric oxide/cGMP pathway couples muscarinic receptors to the activation of Ca2+ influx. J. Neurosci 1996, 16, 1702–1709. [Google Scholar]
- Morales, M.G.; Cabrera, D.; Cespedes, C.; Vio, C.P.; Vazquez, Y.; Brandan, E.; Cabello-Verrugio, C. Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2). Cell Tissue Res 2013, 353, 173–187. [Google Scholar]
- Sen, U.; Herrmann, M.; Herrmann, W.; Tyagi, S.C. Synergism between AT1 receptor and hyperhomocysteinemia during vascular remodeling. Clin. Chem. Lab. Med 2007, 45, 1771–1776. [Google Scholar]



© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Veeranki, S.; Tyagi, S.C. Defective Homocysteine Metabolism: Potential Implications for Skeletal Muscle Malfunction. Int. J. Mol. Sci. 2013, 14, 15074-15091. https://doi.org/10.3390/ijms140715074
Veeranki S, Tyagi SC. Defective Homocysteine Metabolism: Potential Implications for Skeletal Muscle Malfunction. International Journal of Molecular Sciences. 2013; 14(7):15074-15091. https://doi.org/10.3390/ijms140715074
Chicago/Turabian StyleVeeranki, Sudhakar, and Suresh C. Tyagi. 2013. "Defective Homocysteine Metabolism: Potential Implications for Skeletal Muscle Malfunction" International Journal of Molecular Sciences 14, no. 7: 15074-15091. https://doi.org/10.3390/ijms140715074
APA StyleVeeranki, S., & Tyagi, S. C. (2013). Defective Homocysteine Metabolism: Potential Implications for Skeletal Muscle Malfunction. International Journal of Molecular Sciences, 14(7), 15074-15091. https://doi.org/10.3390/ijms140715074