Proteolytic Cleavage of Apolipoprotein E4 as the Keystone for the Heightened Risk Associated with Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to Alzheimer’s Disease
1.1. Pathology of Alzheimer’s Disease
1.2. Risk Factors Associated with Alzheimer’s Disease
2. Apolipoprotein E: Structure and Function
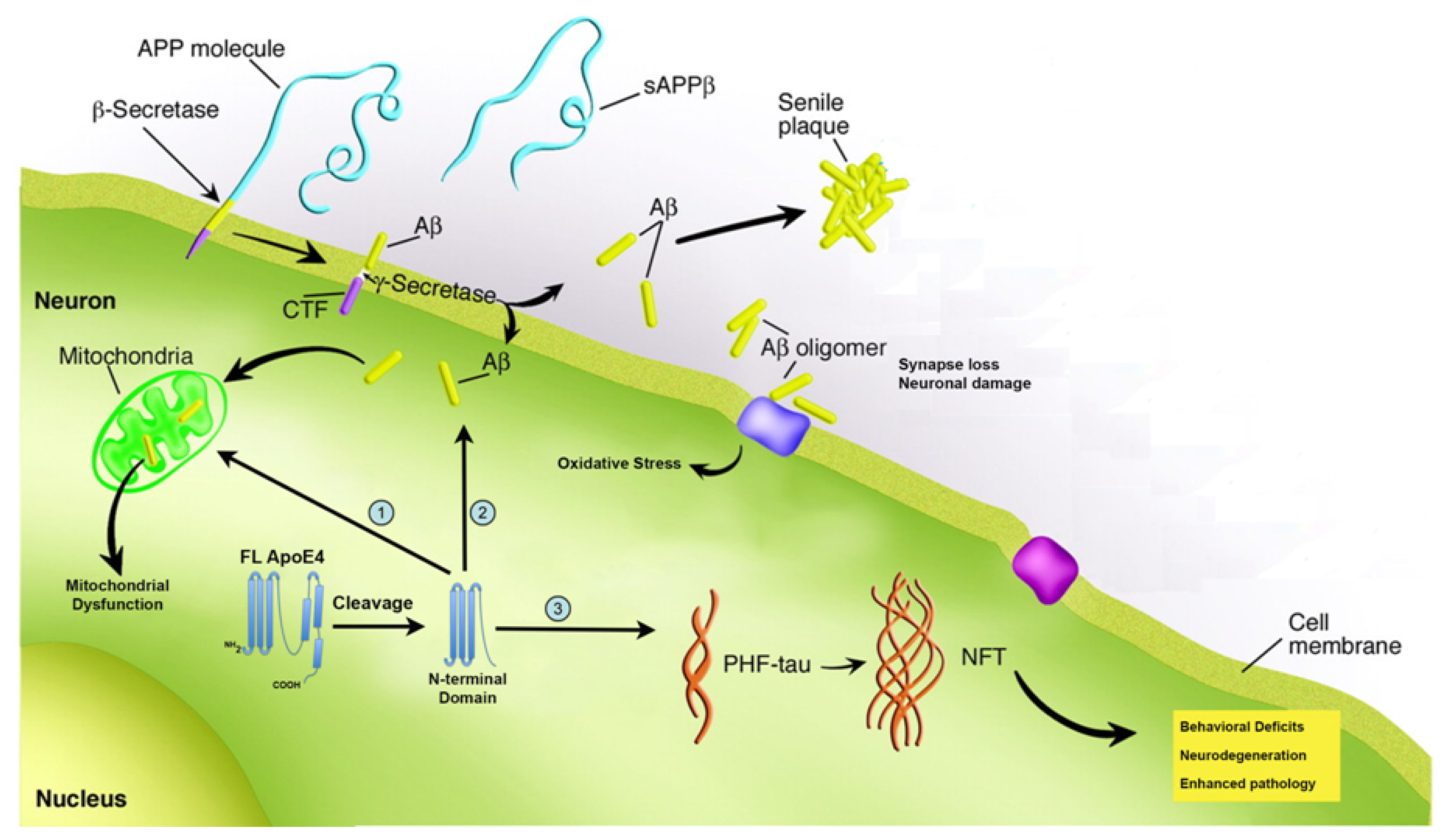
3. Proteolysis of apoE4 as a Mechanism Underlying Pathogenesis in AD
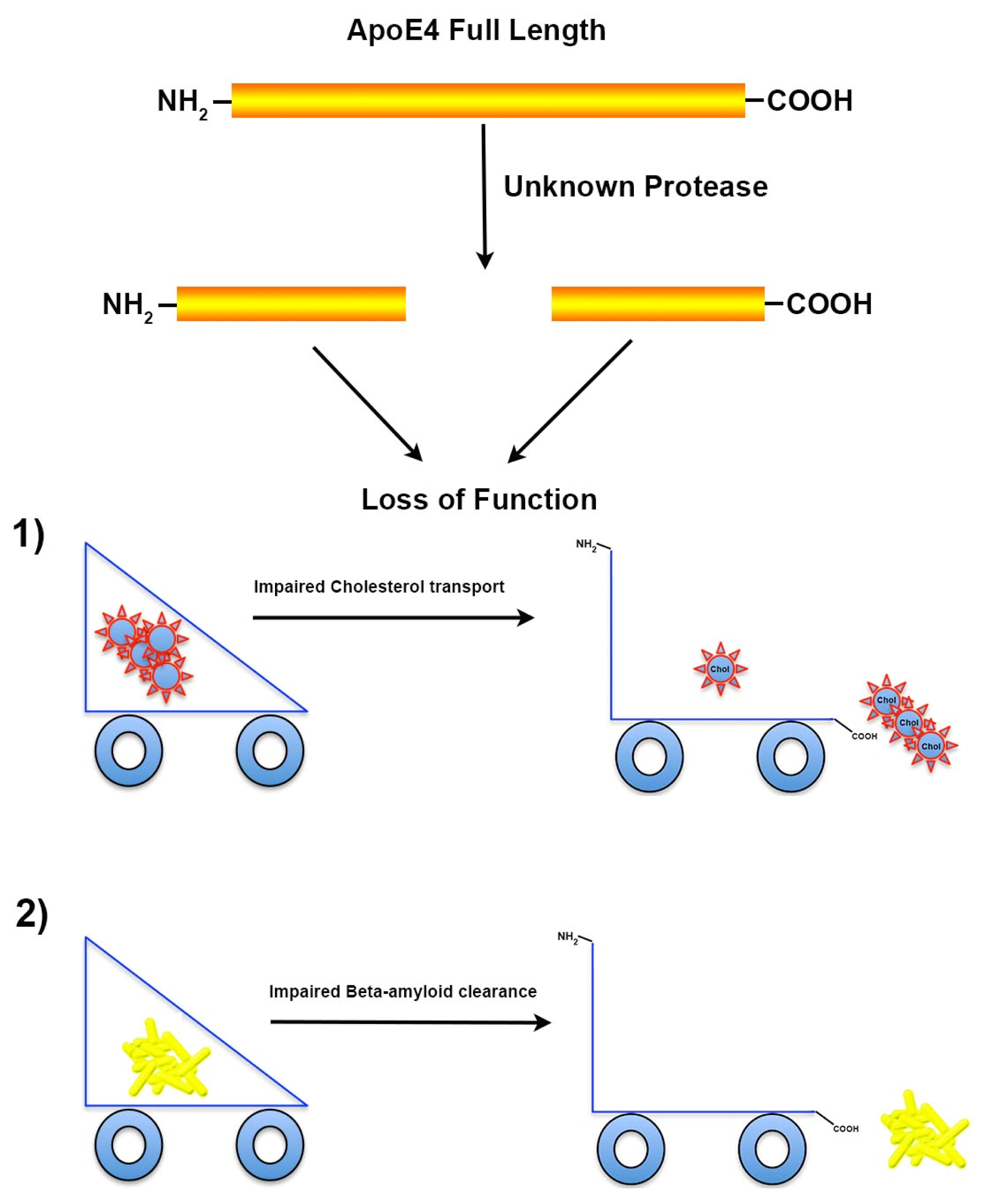
3.1. Proteolysis of apoE4 Leads to Loss of Function
3.2. Proteolysis of apoE4 Leads to a Toxicity Gain of Function
4. Targeting apoE4 Therapeutically for the Treatment of Alzheimer’s and Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar]
- Thies, W.; Bleiler, L. Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013, 9, 208–245. [Google Scholar]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 2012, 8, 1–13. [Google Scholar]
- Ferreira, S.T.; Klein, W.L. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem 2011, 96, 529–543. [Google Scholar]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol 1999, 46, 860–866. [Google Scholar]
- Broersen, K.; Rousseau, F.; Schymkowitz, J. The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer’s disease: oligomer size or conformation? Alzheimer’s Res. Ther 2010, 2, 12. [Google Scholar]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci 2004, 24, 10191–10200. [Google Scholar]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar]
- Seeman, P.; Seeman, N. Alzheimer’s disease: Beta-amyloid plaque formation in human brain. Synapse 2011, 65, 1289–1297. [Google Scholar]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar]
- Trojanowski, J.Q.; Lee, V.M. “Fatal attractions” of proteins. A comprehensive hypothetical mechanism underlying Alzheimer’s disease and other neurodegenerative disorders. Ann. N. Y. Acad. Sci 2000, 924, 62–67. [Google Scholar]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar]
- Reitz, C. Alzheimer’s disease and the amyloid cascade hypothesis: A critical review. Int. J. Alzheimer’s Dis. 2012, 2012. [Google Scholar] [CrossRef]
- Coleman, P.D.; Yao, P.J. Synaptic slaughter in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1023–1027. [Google Scholar]
- Coleman, P.; Federoff, H.; Kurlan, R. A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 2004, 63, 1155–1162. [Google Scholar]
- Bettens, K.; Sleegers, K.; van Broeckhoven, C. Genetic insights in Alzheimer’s disease. Lancet Neurol 2013, 12, 92–104. [Google Scholar]
- Schellenberg, G.D.; Bird, T.D.; Wijsman, E.M.; Orr, H.T.; Anderson, L.; Nemens, E.; White, J.A.; Bonnycastle, L.; Weber, J.L.; Alonso, M.E.; et al. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science 1992, 258, 668–671. [Google Scholar]
- Levy-Lahad, E.; Wijsman, E.M.; Nemens, E.; Anderson, L.; Goddard, K.A.; Weber, J.L.; Bird, T.D.; Schellenberg, G.D. A familial Alzheimer’s disease locus on chromosome 1. Science 1995, 269, 970–973. [Google Scholar]
- Vetrivel, K.S.; Zhang, Y.W.; Xu, H.; Thinakaran, G. Pathological and physiological functions of presenilins. Mol. Neurodegener 2006, 1, 4. [Google Scholar]
- Alonso Vilatela, M.E.; Lopez-Lopez, M.; Yescas-Gomez, P. Genetics of Alzheimer’s disease. Archives Med. Res 2012, 43, 622–631. [Google Scholar]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the risk of Alzheimer’s Disease. N. Engl. J. Med. 2012, 368, 107–116. [Google Scholar]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med 2012, 368, 117–127. [Google Scholar]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med 2005, 201, 647–657. [Google Scholar]
- Eisenstein, M. Genetics: Finding risk factors. Nature 2011, 475, S20–S22. [Google Scholar]
- Weisgraber, K.H.; Rall, S.C., Jr; Mahley, R.W. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981, 256, 9077–9083. [Google Scholar]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheime’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar]
- Wetterau, J.R.; Aggerbeck, L.P.; Rall, S.C., Jr; Weisgraber, K.H. Human apolipoprotein E3 in aqueous solution. I. Evidence for two structural domains. J. Biol. Chem. 1988, 263, 6240–6248. [Google Scholar]
- Pitas, R.E.; Boyles, J.K.; Lee, S.H.; Hui, D.; Weisgraber, K.H. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J. Biol. Chem 1987, 262, 14352–14360. [Google Scholar]
- Michikawa, M.; Fan, Q.W.; Isobe, I.; Yanagisawa, K. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J. Neurochem 2000, 74, 1008–1016. [Google Scholar]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar]
- Hayashi, H.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J. Biol. Chem 2004, 279, 14009–14015. [Google Scholar]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Tesseur, I.; Kekonius, L.; Wyss-Coray, T.; Fish, J.D.; Masliah, E.; Hopkins, P.C.; Scearce-Levie, K.; et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10966–10971. [Google Scholar]
- Zhou, W.; Scott, S.A.; Shelton, S.B.; Crutcher, K.A. Cathepsin D-mediated proteolysis of apolipoprotein E: possible role in Alzheimer’s disease. Neuroscience 2006, 143, 689–701. [Google Scholar]
- Marques, M.A.; Owens, P.A.; Crutcher, K.A. Progress toward identification of protease activity involved in proteolysis of apolipoprotein e in human brain. J. Mol. Neurosci 2004, 24, 73–80. [Google Scholar]
- Elliott, D.A.; Tsoi, K.; Holinkova, S.; Chan, S.L.; Kim, W.S.; Halliday, G.M.; Rye, K.A.; Garner, B. Isoform-specific proteolysis of apolipoprotein-E in the brain. Neurobiol. Aging 2011, 32, 257–271. [Google Scholar]
- Brecht, W.J.; Harris, F.M.; Chang, S.; Tesseur, I.; Yu, G.Q.; Xu, Q.; Fish, J.D.; Wyss-Coray, T.; Buttini, M.; Mucke, L.; et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci 2004, 24, 2527–2534. [Google Scholar]
- Bien-Ly, N.; Andrews-Zwilling, Y.; Xu, Q.; Bernardo, A.; Wang, C.; Huang, Y. C-terminal-truncated apolipoprotein (apo) E4 inefficiently clears amyloid-beta (Abeta) and acts in concert with Abeta to elicit neuronal and behavioral deficits in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 4236–4241. [Google Scholar]
- Huang, Y.; Liu, X.Q.; Wyss-Coray, T.; Brecht, W.J.; Sanan, D.A.; Mahley, R.W. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 8838–8843. [Google Scholar]
- Cho, H.S.; Hyman, B.T.; Greenberg, S.M.; Rebeck, G.W. Quantitation of apoE domains in Alzheimer disease brain suggests a role for apoE in Abeta aggregation. J. Neuropathol. Exp. Neurol 2001, 60, 342–349. [Google Scholar]
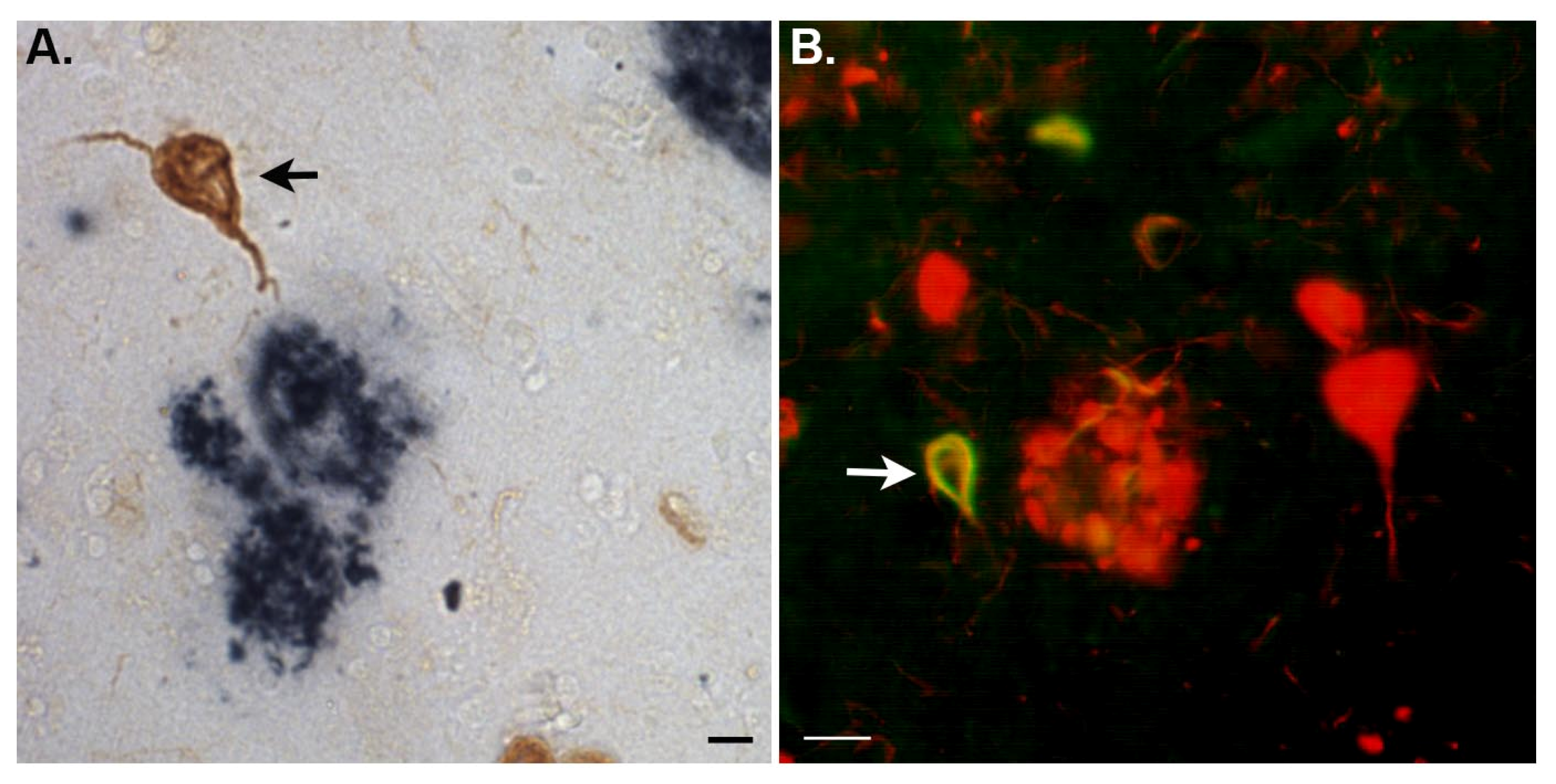
- Rohn, T.T.; Catlin, L.W.; Coonse, K.G.; Habig, J.W. Identification of an amino-terminal fragment of apolipoprotein E4 that localizes to neurofibrillary tangles of the Alzheimer’s disease brain. Brain Res 2012, 1475, 106–115. [Google Scholar]
- Raber, J.; Huang, Y.; Ashford, J.W. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 2004, 25, 641–650. [Google Scholar]
- Xu, P.T.; Gilbert, J.R.; Qiu, H.L.; Ervin, J.; Rothrock-Christian, T.R.; Hulette, C.; Schmechel, D.E. Specific regional transcription of apolipoprotein E in human brain neurons. Am. J. Pathol 1999, 154, 601–611. [Google Scholar]
- Boyles, J.K.; Pitas, R.E.; Wilson, E.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Invest 1985, 76, 1501–1513. [Google Scholar]
- Xu, P.T.; Schmechel, D.; Rothrock-Christian, T.; Burkhart, D.S.; Qiu, H.L.; Popko, B.; Sullivan, P.; Maeda, N.; Saunders, A.M.; Roses, A.D.; et al. Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: Human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol. Dis 1996, 3, 229–245. [Google Scholar]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci 2006, 26, 4985–4994. [Google Scholar]
- Dietschy, J.M.; Turley, S.D. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res 2004, 45, 1375–1397. [Google Scholar]
- Poirier, J. Apolipoprotein E represents a potent gene-based therapeutic target for the treatment of sporadic Alzheimer’s disease. Alzheimer’s Dement 2008, 4, S91–S97. [Google Scholar]
- De Chaves, E.P.; Narayanaswami, V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol 2008, 3, 505–530. [Google Scholar]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med 2004, 10, 719–726. [Google Scholar]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 2012, 335, 1503–1506. [Google Scholar]
- Fitz, N.F.; Cronican, A.A.; Lefterov, I.; Koldamova, R. Comment on “ApoE-Directed Therapeutics Rapidly Clear beta-Amyloid and Reverse Deficits in AD Mouse Models”. Science 2013, 340. [Google Scholar] [CrossRef]
- Veeraraghavalu, K.; Zhang, C.; Miller, S.; Hefendehl, J.K.; Rajapaksha, T.W.; Ulrich, J.; Jucker, M.; Holtzman, D.M.; Tanzi, R.E.; Vassar, R.; et al. Comment on “ApoE-Directed Therapeutics Rapidly Clear beta-Amyloid and Reverse Deficits in AD Mouse Models”. Science 2013, 340. [Google Scholar] [CrossRef]
- Tesseur, I.; Lo, A.C.; Roberfroid, A.; Dietvorst, S.; van Broeck, B.; Borgers, M.; Gijsen, H.; Moechars, D.; Mercken, M.; Kemp, J.; et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 2013, 340. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, e1807–e1816. [Google Scholar]
- Gylys, K.H.; Fein, J.A.; Tan, A.M.; Cole, G.M. Apolipoprotein E enhances uptake of soluble but not aggregated amyloid-beta protein into synaptic terminals. J. Neurochem 2003, 84, 1442–1451. [Google Scholar]
- Zerbinatti, C.V.; Wahrle, S.E.; Kim, H.; Cam, J.A.; Bales, K.; Paul, S.M.; Holtzman, D.M.; Bu, G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J. Biol. Chem 2006, 281, 36180–36186. [Google Scholar]
- Li, J.; Kanekiyo, T.; Shinohara, M.; Zhang, Y.; LaDu, M.J.; Xu, H.; Bu, G. Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem 2012, 287, 44593–44601. [Google Scholar]
- Chang, S.; Ma, T.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar]
- Tolar, M.; Marques, M.A.; Harmony, J.A.; Crutcher, K.A. Neurotoxicity of the 22 kDa thrombin-cleavage fragment of apolipoprotein E and related synthetic peptides is receptor-mediated. J. Neurosci 1997, 17, 5678–5686. [Google Scholar]
- Andrews-Zwilling, Y.; Bien-Ly, N.; Xu, Q.; Li, G.; Bernardo, A.; Yoon, S.Y.; Zwilling, D.; Yan, T.X.; Chen, L.; Huang, Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci 2010, 30, 13707–13717. [Google Scholar]
- Dafnis, I.; Stratikos, E.; Tzinia, A.; Tsilibary, E.C.; Zannis, V.I.; Chroni, A. An apolipoprotein E4 fragment can promote intracellular accumulation of amyloid peptide beta 42. J. Neurochem 2010, 115, 873–884. [Google Scholar]
- Christensen, D.Z.; Schneider-Axmann, T.; Lucassen, P.J.; Bayer, T.A.; Wirths, O. Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol 2010, 119, 555–566. [Google Scholar]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem 2011, 286, 5215–5221. [Google Scholar]
- Nakamura, T.; Watanabe, A.; Fujino, T.; Hosono, T.; Michikawa, M. Apolipoprotein E4 (1–272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener 2009, 4, 35. [Google Scholar]
- Valla, J.; Yaari, R.; Wolf, A.B.; Kusne, Y.; Beach, T.G.; Roher, A.E.; Corneveaux, J.J.; Huentelman, M.J.; Caselli, R.J.; Reiman, E.M. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer’s susceptibility gene. J. Alzheimers Dis 2010, 22, 307–313. [Google Scholar]
- Farol, L.T.; Hymes, K.B. Bexarotene: A clinical review. Expert Rev. Anticancer Ther 2004, 4, 180–188. [Google Scholar]
- Price, A.R.; Xu, G.; Siemienski, Z.B.; Smithson, L.A.; Borchelt, D.R.; Golde, T.E.; Felsenstein, K.M. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 2013, 340. [Google Scholar] [CrossRef]
- Mahley, R.W.; Huang, Y. Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein E4-associated neuropathology. J. Med. Chem 2012, 55, 8997–9008. [Google Scholar]
- Chen, H.K.; Liu, Z.; Meyer-Franke, A.; Brodbeck, J.; Miranda, R.D.; McGuire, J.G.; Pleiss, M.A.; Ji, Z.S.; Balestra, M.E.; Walker, D.W.; et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J. Biol. Chem 2012, 287, 5253–5266. [Google Scholar]
- Ye, S.; Huang, Y.; Mullendorff, K.; Dong, L.; Giedt, G.; Meng, E.C.; Cohen, F.E.; Kuntz, I.D.; Weisgraber, K.H.; Mahley, R.W. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc. Natl. Acad. Sci. USA 2005, 102, 18700–18705. [Google Scholar]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak, V.M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rohn, T.T. Proteolytic Cleavage of Apolipoprotein E4 as the Keystone for the Heightened Risk Associated with Alzheimer’s Disease. Int. J. Mol. Sci. 2013, 14, 14908-14922. https://doi.org/10.3390/ijms140714908
Rohn TT. Proteolytic Cleavage of Apolipoprotein E4 as the Keystone for the Heightened Risk Associated with Alzheimer’s Disease. International Journal of Molecular Sciences. 2013; 14(7):14908-14922. https://doi.org/10.3390/ijms140714908
Chicago/Turabian StyleRohn, Troy T. 2013. "Proteolytic Cleavage of Apolipoprotein E4 as the Keystone for the Heightened Risk Associated with Alzheimer’s Disease" International Journal of Molecular Sciences 14, no. 7: 14908-14922. https://doi.org/10.3390/ijms140714908
APA StyleRohn, T. T. (2013). Proteolytic Cleavage of Apolipoprotein E4 as the Keystone for the Heightened Risk Associated with Alzheimer’s Disease. International Journal of Molecular Sciences, 14(7), 14908-14922. https://doi.org/10.3390/ijms140714908