Micromanaging Abdominal Aortic Aneurysms

Abstract
:1. Abdominal Aortic Aneurysm Disease
2. Pathology and Cellular Mechanisms
2.1. Impaired Homeostasis of Vascular Smooth Muscle Cells and Extracellular Matrix
2.2. Inflammation
3. MicroRNA Biogenesis and Function
4. miRs in AAA Disease
4.1. miR-21
4.2. miR-26a
4.3. miR-29b
5. miR-143/145
6. Other miRs
6.1. miR-126
6.2. miR-146a
6.3. miR-155
7. Therapeutic Approaches Using miR Modulators
8. miRs as Biomarkers in AAA Disease
9. Summary and Perspectives
Acknowledgments
Conflict of Interest
References
- Golledge, J.; Muller, J.; Daugherty, A.; Norman, P. Abdominal aortic aneurysm: Pathogenesis and implications for management. Arterioscler. Thromb. Vasc. Biol 2006, 26, 2605–2613. [Google Scholar]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Executive summary: Heart disease and stroke statistics—2013 update: A report from the American Heart Association. Circulation 2013, 127, 143–152. [Google Scholar]
- Svensjo, S.; Martin Björck, M.; Gürtelschmid, M.; Gidlund, K.D.; Hellberg, A.; Wanhainen, A. Low prevalence of abdominal aortic aneurysm among 65-year-old Swedish men indicates a change in the epidemiology of the disease. Circulation 2011, 124, 1118–1123. [Google Scholar]
- Golledge, J.; Norman, P.E. Current status of medical management for abdominal aortic aneurysm. Atherosclerosis 2011, 217, 57–63. [Google Scholar]
- Thom, T.; Haase, N.; Rosamond, W.; Howard, V.J.; Rumsfeld, J.; Manolio, T.; Zheng, Z.J.; Flegal, K.; O’Donnell, C.; Kittner, S.; et al. Heart disease and stroke statistics—2006 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006, 113, e85–e151. [Google Scholar]
- Golledge, J.; Tsao, P.S.; Dalman, R.L.; Norman, P.E. Circulating markers of abdominal aortic aneurysm presence and progression. Circulation 2008, 118, 2382–2392. [Google Scholar]
- Jones, D.W.; Easton, J.D.; Halperin, J.L.; Hirsch, A.T.; Matsumoto, A.H.; O’Gara, P.T.; Safian, R.D.; Schwartz, G.L.; Spittell, J.A. American Heart Association. Atherosclerotic Vascular Disease Conference: Writing Group V: Medical decision making and therapy. Circulation 2004, 109, 2634–2642. [Google Scholar]
- Weintraub, N.L. Understanding abdominal aortic aneurysm. N. Engl. J. Med 2009, 361, 1114–1116. [Google Scholar]
- Franks, P.J.; Edwards, R.J.; Greenhalgh, R.M.; Powell, J.T. Smoking as a risk factor for abdominal aortic aneurysm. Ann. N. Y. Acad. Sci 1996, 800, 246–248. [Google Scholar]
- Norman, P.E.; Powell, J.T. Abdominal aortic aneurysm: The prognosis in women is worse than in men. Circulation 2007, 115, 2865–2869. [Google Scholar]
- Powell, J.T.; Greenhalgh, R.M. Clinical practice. Small abdominal aortic aneurysms. N. Engl. J. Med 2003, 348, 1895–1901. [Google Scholar]
- Lu, H.; Rateri, D.L.; Bruemmer, D.; Cassis, L.A.; Daugherty, A. Novel mechanisms of abdominal aortic aneurysms. Curr. Atheroscler. Rep 2012, 14, 402–412. [Google Scholar]
- Lu, H.; Rateri, D.L.; Bruemmer, D.; Cassis, L.A.; Daugherty, A. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clin. Sci 2012, 123, 531–543. [Google Scholar]
- Daugherty, A.; Cassis, L.A. Mouse models of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol 2004, 24, 429–434. [Google Scholar]
- Milewicz, D.M. MicroRNAs, fibrotic remodeling, and aortic aneurysms. J. Clin. Invest 2012, 122, 490–493. [Google Scholar]
- Lindsay, M.E.; Dietz, H.C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011, 473, 308–316. [Google Scholar]
- Guo, D.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet 2007, 39, 1488–1493. [Google Scholar]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; de Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med 2006, 355, 788–798. [Google Scholar]
- Regalado, E.S.; Guo, D.; Villamizar, C.; Avidan, N.; Gilchrist, D.; McGillivray, B.; Clarke, L.; Bernier, F.; Santos-Cortez, R.L.; Leal, S.M. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ. Res 2011, 109, 680–686. [Google Scholar]
- Van de Laar, I.M.B.H.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.A.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.; Venselaar, H. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet 2011, 43, 121–126. [Google Scholar]
- Dai, J.; Losy, F.; Guinault, A.M.; Pages, C.; Anegon, I.; Desgranges, P.; Becquemin, J.P.; Allaire, E. Overexpression of transforming growth factor-beta1 stabilizes already-formed aortic aneurysms: A first approach to induction of functional healing by endovascular gene therapy. Circulation 2005, 112, 1008–1015. [Google Scholar]
- Biros, E.; Walker, P.J.; Nataatmadja, M.; West, M.; Golledge, J. Downregulation of transforming growth factor, beta receptor 2 and Notch signaling pathway in human abdominal aortic aneurysm. Atherosclerosis 2012, 221, 383–386. [Google Scholar]
- Golledge, J.; Clancy, P.; Jones, G.T.; Cooper, M.; Palmer, L.J.; van Rij, A.M.; Norman, P.E. Possible association between genetic polymorphisms in transforming growth factor beta receptors, serum transforming growth factor beta1 concentration and abdominal aortic aneurysm. Br. J. Surg 2009, 96, 628–632. [Google Scholar]
- Wang, L.; Guo, D.; Cao, J.; Gong, L.; Kamm, K.E.; Regalado, E.; Li, L.; Shete, S.; He, W.; Zhu, M.; et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet 2010, 87, 701–707. [Google Scholar]
- Zhu, L.; Vranckx, R.; van Kien, P.K.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet 2006, 38, 343–349. [Google Scholar]
- Takagi, H.; Manabe, H.; Kawai, N.; Goto, S.; Umemoto, T. Circulating matrix metalloproteinase-9 concentrations and abdominal aortic aneurysm presence: A meta-analysis. Interact. Cardiovasc. Thorac. Surg 2009, 9, 437–440. [Google Scholar]
- Freestone, T.; Turner, R.J.; Higman, D.J.; Lever, M.J.; Powell, J.T. Influence of hypercholesterolemia and adventitial inflammation on the development of aortic aneurysm in rabbits. Arterioscler. Thromb. Vasc. Biol 1997, 17, 10–17. [Google Scholar]
- Anidjar, S.; Salzmann, J.L.; Gentric, D.; Lagneau, P.; Camilleri, J.P.; Michel, J.B. Elastase-induced experimental aneurysms in rats. Circulation 1990, 82, 973–981. [Google Scholar]
- Saraff, K.; Babamusta, F.; Cassis, L.A.; Daugherty, A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol 2003, 23, 1621–1626. [Google Scholar]
- Rateri, D.L.; Howatt, D.A.; Moorleghen, J.J.; Charnigo, R.; Cassis, L.A.; Daugherty, A. Prolonged infusion of angiotensin II in apoE(−/−) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am. J. Pathol 2011, 179, 1542–1548. [Google Scholar]
- Daugherty, A.; Rateri, D.L.; Charo, I.F.; Phillip Owens, A., III; Howatt, D.A.; Cassis, L.A. Angiotensin II infusion promotes ascending aortic aneurysms: Attenuation by CCR2 deficiency in apoE−/− mice. Clin. Sci. 2010, 118, 681–689. [Google Scholar]
- MacTaggart, J.N.; Xiong, W.; Knispel, R.; Baxter, B.T. Deletion of CCR2 but not CCR5 or CXCR3 inhibits aortic aneurysm formation. Surgery 2007, 142, 284–288. [Google Scholar]
- Phillip Owens, A., III; Rateri, D.L.; Howatt, D.A.; Moore, K.J.; Tobias, P.S.; Curtiss, L.K.; Lu, H.; Cassis, L.A.; Daugherty, A. MyD88 deficiency attenuates angiotensin II-induced abdominal aortic aneurysm formation independent of signaling through Toll-like receptors 2,4. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2813–2819. [Google Scholar]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Invest 2000, 105, 1605–1612. [Google Scholar]
- Yin, M.; Zhang, J.; Wang, Y.; Wang, S.; Böckler, D.; Duan, Z.; Xin, S. Deficient CD4+CD25+ T regulatory cell function in patients with abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol 2010, 30, 1825–1831. [Google Scholar]
- Eliason, J.L.; Hannawa, K.K.; Ailawadi, G.; Sinha, I.; Ford, J.W.; Deogracias, M.P.; Roelofs, K.J.; Woodrum, D.T.; Ennis, T.L.; Henke, P.K.; et al. Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation 2005, 112, 232–240. [Google Scholar]
- Hannawa, K.K.; Eliason, J.L.; Woodrum, D.T.; Pearce, C.G.; Roelofs, K.J.; Grigoryants, V.; Eagleton, M.J.; Henke, P.K.; Wakefield, T.W.; Myers, D.D.; et al. L-selectin-mediated neutrophil recruitment in experimental rodent aneurysm formation. Circulation 2005, 112, 241–247. [Google Scholar]
- Middleton, R.K.; Lloyd, G.M.; Bown, M.J.; Cooper, N.J.; London, N.J.; Sayers, R.D. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: A protein array study. J. Vasc. Surg 2007, 45, 574–580. [Google Scholar]
- Kaneko, H.; Anzai, T.; Horiuchi, K.; Kohno, T.; Nagai, T.; Anzai, A.; Takahashi, T.; Sasaki, A.; Shimoda, M.; Maekawa, Y.; et al. Tumor necrosis factor-alpha converting enzyme is a key mediator of abdominal aortic aneurysm development. Atherosclerosis 2011, 218, 470–478. [Google Scholar]
- Satoh, H.; Nakamura, M.; Satoh, M.; Nakajima, T.; Izumoto, H.; Maesawa, C.; Kawazoe, K.; Masuda, T.; Hiramori, K. Expression and localization of tumour necrosis factor-alpha and its converting enzyme in human abdominal aortic aneurysm. Clin. Sci 2004, 106, 301–306. [Google Scholar]
- Juvonen, J.; Surcel, H.; Satta, J.; Teppo, A.; Bloigu, A.; Syrjälä, H.; Airaksinen, J.; Leinonen, M.; Saikku, P.; Juvonen, T. Elevated circulating levels of inflammatory cytokines in patients with abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol 1997, 17, 2843–2847. [Google Scholar]
- Koole, D.; Hurks, R.; Schoneveld, A.; Vink, A.; Golledge, J.; Moran, C.S.; de Kleijn, D.P.; van Herwaarden, J.A.; de Vries, J.; Laman, J.D.; et al. Osteoprotegerin is associated with aneurysm diameter and proteolysis in abdominal aortic aneurysm disease. Arterioscler. Thromb. Vasc. Biol 2012, 32, 1497–1504. [Google Scholar]
- Xiong, W.; MacTaggart, J.; Knispel, R.; Worth, J.; Persidsky, Y.; Baxter, B.T. Blocking TNF-α attenuates aneurysm formation in a murine model. J. Immunol 2009, 183, 2741–2746. [Google Scholar]
- Kloosterman, W.P.; Plasterk, R.H. The diverse functions of microRNAs in animal development and disease. Dev. Cell 2006, 11, 441–450. [Google Scholar]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar]
- Yeom, K.; Lee, Y.; Han, J.; Suh, M.R.; Kim, V.N. Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing. Nucleic. Acids Res 2006, 34, 4622–4629. [Google Scholar]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet 2010, 11, 597–610. [Google Scholar]
- Kim, V.N. MicroRNA precursors in motion: Exportin-5 mediates their nuclear export. Trends Cell Biol 2004, 14, 156–159. [Google Scholar]
- Hutvagner, G. Small RNA asymmetry in RNAi: Function in RISC assembly and gene Regulation. FEBS Lett 2005, 579, 5850–5857. [Google Scholar]
- Okamura, K.; Phillips, M.D.; Tyler, D.M.; Duan, H.; Chou, Y.T.; Lai, E.C. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat. Struct. Mol. Biol 2008, 15, 354–363. [Google Scholar]
- Guo, L.; Lu, Z. The fate of miRNA* strand through evolutionary analysis: Implication for degradation as merely carrier strand or potential regulatory molecule? PLoS One 2010, 5, e11387. [Google Scholar]
- Chang, K.W.; Kao, S.Y.; Wu, Y.H.; Tsai, M.M.; Tu, H.F.; Liu, C.J.; Lui, M.T.; Lin, S.C. Passenger strand miRNA miR-31* regulates the phenotypes of oral cancer cells by targeting RhoA. Oral. Oncol 2013, 49, 27–33. [Google Scholar]
- Yang, J.; Phillips, M.D.; Betel, D.; Mu, P.; Ventura, A.; Siepel, A.C.; Chen, K.C.; Lai, E.C. Widespread regulatory activity of vertebrate microRNA* species. RNA 2011, 17, 312–326. [Google Scholar]
- Zhou, H.; Huang, X.; Cui, H.; Luo, X.; Tang, Y.; Chen, S.; Wu, L.; Shen, N. miR-155 and its star-form partner miR-155* cooperatively regulate type I interferon production by human plasmacytoid dendritic cells. Blood 2010, 116, 5885–5894. [Google Scholar]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev 2004, 18, 504–511. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet 2011, 12, 99–110. [Google Scholar]
- Pillai, R.S.; Bhattacharyya, S.N.; Artus, C.G.; Zoller, T.; Cougot, N.; Basyuk, E.; Bertrand, E.; Filipowicz, W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 2005, 309, 1573–1576. [Google Scholar]
- Maegdefessel, L.; Azuma, J.; Toh, R.; Deng, A.; Merk, D.R.; Raiesdana, A.; Leeper, N.J.; Raaz, U.; Schoelmerich, A.M.; McConnell, M.V.; et al. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci. Transl. Med 2012, 4, 122r, a22.. [Google Scholar]
- Leeper, N.J.; Raiesdana, A.; Kojima, Y.; Chun, H.J.; Azuma, J.; Maegdefessel, L.; Kundu, R.K.; Quertermous, T.; Tsao, P.S.; Spin, J.M. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J. Cell Physiol 2011, 226, 1035–1043. [Google Scholar]
- Boon, R.A.; Seeger, T.; Heydt, S.; Fischer, A.; Hergenreider, E.; Horrevoets, A.J.G.; Vinciguerra, M.; Rosenthal, N.; Sciacca, S.; Pilato, M.; et al. MicroRNA-29 in aortic dilation: Implications for aneurysm formation. Circ. Res 2011, 109, 1115–1119. [Google Scholar]
- Maegdefessel, L.; Azuma, J.; Toh, R.; Merk, D.R.; Deng, A.; Chin, J.T.; Raaz, U.; Schoelmerich, A.M.; Raiesdana, A.; Leeper, N.J.; et al. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J. Clin. Invest 2012, 122, 497–506. [Google Scholar]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.G.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ 2009, 16, 1590–1598. [Google Scholar]
- Lee, Y.S.; Dutta, A. MicroRNAs in cancer. Annu. Rev. Pathol 2009, 4, 199–227. [Google Scholar]
- Jazbutyte, V.; Thum, T. MicroRNA-21: From cancer to cardiovascular disease. Curr. Drug Targets 2010, 11, 926–35. [Google Scholar]
- Cheng, Y.; Zhang, C. MicroRNA-21 in cardiovascular disease. J. Cardiovasc. Transl. Res 2010, 3, 251–255. [Google Scholar]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61. [Google Scholar]
- Ji, R.; Cheng, Y.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ. Res 2007, 100, 1579–1588. [Google Scholar]
- Kang, H.; Hata, A. MicroRNA regulation of smooth muscle gene expression and phenotype. Curr. Opin. Hematol 2012, 19, 224–231. [Google Scholar]
- Sarkar, J.; Gou, D.; Turaka, P.; Viktorova, E.; Ramchandran, R.; Usha Raj, J. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am. J. Physiol. Lung Cell Mol. Physiol 2010, 299, L861–L871. [Google Scholar]
- Wang, M.; Li, W.; Chang, G.; Ye, C.; Ou, J.; Li, X.; Liu, Y.; Cheang, T.; Huang, X.; Wang, S. MicroRNA-21 regulates vascular smooth muscle cell function via targeting tropomyosin 1 in arteriosclerosis obliterans of lower extremities. Arterioscler. Thromb. Vasc. Biol 2011, 31, 2044–2053. [Google Scholar]
- Song, J.T.; Hu, B.; Qu, H.Y.; Bi, C.L.; Huang, X.Z.; Zhang, M. Mechanical stretch modulates MicroRNA 21 expression, participating in proliferation and apoptosis in cultured human aortic smooth muscle cells. PLoS One 2012, 7, e47657. [Google Scholar]
- Chapman, G.B.; Durante, W.; Hellums, J.D.; Schafer, A.I. Physiological cyclic stretch causes cell cycle arrest in cultured vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol 2000, 278, H748–H754. [Google Scholar]
- Cheng, W.; Wang, B.; Chen, S.; Chang, H.; Shyu, K. Mechanical stretch induces the apoptosis regulator PUMA in vascular smooth muscle cells. Cardiovasc. Res 2012, 93, 181–189. [Google Scholar]
- Li, C.; Wernig, F.; Leitges, M.; Hu, Y.; Xu, Q. Mechanical stress-activated PKCdelta regulates smooth muscle cell migration. FASEB J 2003, 17, 2106–2108. [Google Scholar]
- Weber, M.; Baker, M.B.; Moore, J.P.; Searles, C.D. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem. Biophys. Res. Commun 2010, 393, 643–648. [Google Scholar]
- Zhu, S.; Deng, S.; Ma, Q.; Zhang, T.; Jia, C.; Zhuo, D.; Yang, F.; Wei, J.; Wang, L.; Dykxhoorn, D.M.; et al. microRNA-10A* and microRNA-21 modulate endothelial progenitor cell senescence via suppressing Hmga2. Circ. Res 2013, 112, 152–164. [Google Scholar]
- van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar]
- Kwiecinski, M.; Noetel, A.; Elfimova, N.; Trebicka, J.; Schievenbusch, S.; Strack, I.; Molnar, L.; von Brandenstein, M.; Töx, U.; Nischt, R.; et al. Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis in hepatic stellate cells by miRNA-29 induction. PLoS One 2011, 6, e24568. [Google Scholar]
- Wang, B.; Komers, R.; Carew, R.; Winbanks, C.E.; Xu, B.; Herman-Edelstein, M.; Koh, P.; Thomas, M.; Jandeleit-Dahm, K.; Gregorevic, P.; et al. Suppression of microRNA-29 expression by TGF-β1 promotes collagen expression and renal fibrosis. J. Am. Soc. Nephrol 2012, 23, 252–265. [Google Scholar]
- Maurer, B.; Stanczyk, J.; Jüngel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum 2010, 62, 1733–1743. [Google Scholar]
- Ogawa, T.; Iizuka, M.; Sekiya, Y.; Yoshizato, K.; Ikeda, K.; Kawada, N. Suppression of type I collagen production by microRNA-29b in cultured human stellate cells. Biochem. Biophys. Res. Commun 2010, 391, 316–321. [Google Scholar]
- Boettger, T.; Beetz, N.; Kostin, S.; Schneider, J.; Krüger, M.; Hein, L.; Braun, T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J. Clin. Invest 2009, 119, 2634–2647. [Google Scholar]
- Cheng, Y.; Liu, X.; Yang, J.; Lin, Y.; Xu, D.Z.; Lu, Q.; Deitch, E.A.; Huo, Y.; Delphin, E.S.; Zhang, C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ. Res 2009, 105, 158–166. [Google Scholar]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar]
- Xin, M.; Small, E.M.; Sutherland, L.B.; Qi, X.; McAnally, J.; Plato, C.F.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 2009, 23, 2166–2178. [Google Scholar]
- Quintavalle, M.; Elia, L.; Condorelli, G.; Courtneidge, S.A. MicroRNA control of podosome formation in vascular smooth muscle cells in vivo and in vitro. J. Cell Biol 2010, 189, 13–22. [Google Scholar]
- Hergenreider, E.; Heydt, S.; Tréguer, K.; Boettger, T.; Horrevoets, A.J.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol 2012, 14, 249–256. [Google Scholar]
- Golledge, A.L.; Walker, P.; Norman, P.E.; Golledge, J. A systematic review of studies examining inflammation associated cytokines in human abdominal aortic aneurysm samples. Dis. Markers 2009, 26, 181–188. [Google Scholar]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar]
- Asgeirsdóttir, S.A.; van Solingen, C.; Kurniati, N.F.; Zwiers, P.J.; Heeringa, P.; van Meurs, M.; Satchell, S.C.; Saleem, M.A.; Mathieson, P.W.; Banas, B.; et al. MicroRNA-126 contributes to renal microvascular heterogeneity of VCAM-1 protein expression in acute inflammation. Am. J. Physiol. Renal Physiol 2012, 302, F1630–F1639. [Google Scholar]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Köppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal 2009, 2, ra81. [Google Scholar]
- Vasa-Nicotera, M.; Chen, H.; Tucci, P.; Yang, A.L.; Saintigny, G.; Menghini, R.; Mahè, C.; Agostini, M.; Knight, R.A.; Melino, G.; et al. miR-146a is modulated in human endothelial cell with aging. Atherosclerosis 2011, 217, 326–330. [Google Scholar]
- Sun, S.G.; Zheng, B.; Han, M.; Fang, X.M.; Li, H.X.; Miao, S.B.; Su, M.; Han, Y.; Shi, H.J.; Wen, J.K. miR-146a and Kruppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO Rep 2011, 12, 56–62. [Google Scholar]
- Suárez, Y.; Wang, C.; Manes, T.D.; Pober, J.S. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: Feedback control of inflammation. J. Immunol 2010, 184, 21–25. [Google Scholar]
- Zhu, N.; Zhang, D.; Chen, S.; Liu, X.; Lin, L.; Huang, X.; Guo, Z.; Liu, J.; Wang, Y.; Yuan, W.; Qin, Y. Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis 2011, 215, 286–293. [Google Scholar]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Invest 2012, 122, 4190–4202. [Google Scholar]
- Donners, M.M.; Wolfs, I.M.; Stöger, L.J.; van der Vorst, E.P.; Pöttgens, C.C.; Heymans, S.; Schroen, B.; Gijbels, M.J.; de Winther, M.P. Hematopoietic miR155 deficiency enhances atherosclerosis and decreases plaque stability in hyperlipidemic mice. PLoS One 2012, 7, e35877. [Google Scholar]
- Pahl, M.C.; Derr, K.; Gäbel, G.; Hinterseher, I.; Elmore, J.R.; Schworer, C.M.; Peeler, T.C.; Franklin, D.P.; Gray, J.L.; Carey, D.J.; et al. MicroRNA expression signature in human abdominal aortic aneurysms. BMC Med. Genomics 2012, 5, 25. [Google Scholar]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar]
- Van Rooij, E.; Olson, E.N. MicroRNA therapeutics for cardiovascular disease: Opportunities and obstacles. Nat. Rev. Drug Discov 2012, 11, 860–872. [Google Scholar]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar]
- Van Rooij, E.; Purcell, A.L.; Levin, A.A. Developing microRNA therapeutics. Circ. Res 2012, 110, 496–507. [Google Scholar]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med 2013, 368, 1685–1694. [Google Scholar]
- Mishra, P.K.; Tyagi, N.; Kumar, M.; Tyagi, S.C. MicroRNAs as a therapeutic target for cardiovascular diseases. J. Cell Mol. Med 2009, 13, 778–789. [Google Scholar]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, J.T.; Maitra, A. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther 2011, 10, 1470–1480. [Google Scholar]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther 2011, 19, 1116–1122. [Google Scholar]
- Moxon, J.V.; Parr, A.; Emeto, T.I.; Walker, P.; Norman, P.E.; Golledge, J. Diagnosis and monitoring of abdominal aortic aneurysm: Current status and future prospects. Curr. Probl. Cardiol 2010, 35, 512–548. [Google Scholar]
- D’Alessandra, Y.; Devanna, P.; Limana, F.; Straino, S.; di Carlo, A.; Brambilla, P.G.; Rubino, M.; Carena, M.C.; Spazzafumo, L.; de Simone, M.; et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J 2010, 31, 2765–2773. [Google Scholar]
- Fichtlscherer, S.; de Rosa, S.; Fox, H.; Schwietz, T.; Fischer, A.; Liebetrau, C.; Weber, M.; Hamm, C.W.; Röxe, T.; Müller-Ardogan, M.; et al. Circulating microRNAs in patients with coronary artery disease. Circ. Res 2010, 107, 677–684. [Google Scholar]
- Tijsen, A.J.; Creemers, E.E.; Moerland, P.D.; de Windt, L.J.; van der Wal, A.C.; Kok, W.E.; Pinto, Y.M. MiR423-5p as a circulating biomarker for heart failure. Circ. Res 2010, 106, 1035–1039. [Google Scholar]
- Zampetaki, A.; Willeit, P.; Tilling, L.; Drozdov, I.; Prokopi, M.; Renard, J.M.; Mayr, A.; Weger, S.; Schett, G.; Shah, A.; et al. Prospective study on circulating MicroRNAs and risk of myocardial infarction. J. Am. Coll Cardiol 2012, 60, 290–299. [Google Scholar]
- Engelhardt, S. Small RNA biomarkers come of age. J. Am. Coll Cardiol 2012, 60, 300–303. [Google Scholar]
- Zampetaki, A.; Mayr, M. Analytical challenges and technical limitations in assessing circulating miRNAs. Thromb. Haemost 2012, 108, 592–598. [Google Scholar]
- Kin, K.; Miyagawa, S.; Fukushima, S.; Shirakawa, Y.; Torikai, K.; Shimamura, K.; Daimon, T.; Kawahara, Y.; Kuratani, T.; Sawa, Y. Tissue- and plasma-specific microRNA signatures for atherosclerotic abdominal aortic aneurysm. J. Am. Heart Assoc 2012, 1, e000745. [Google Scholar]
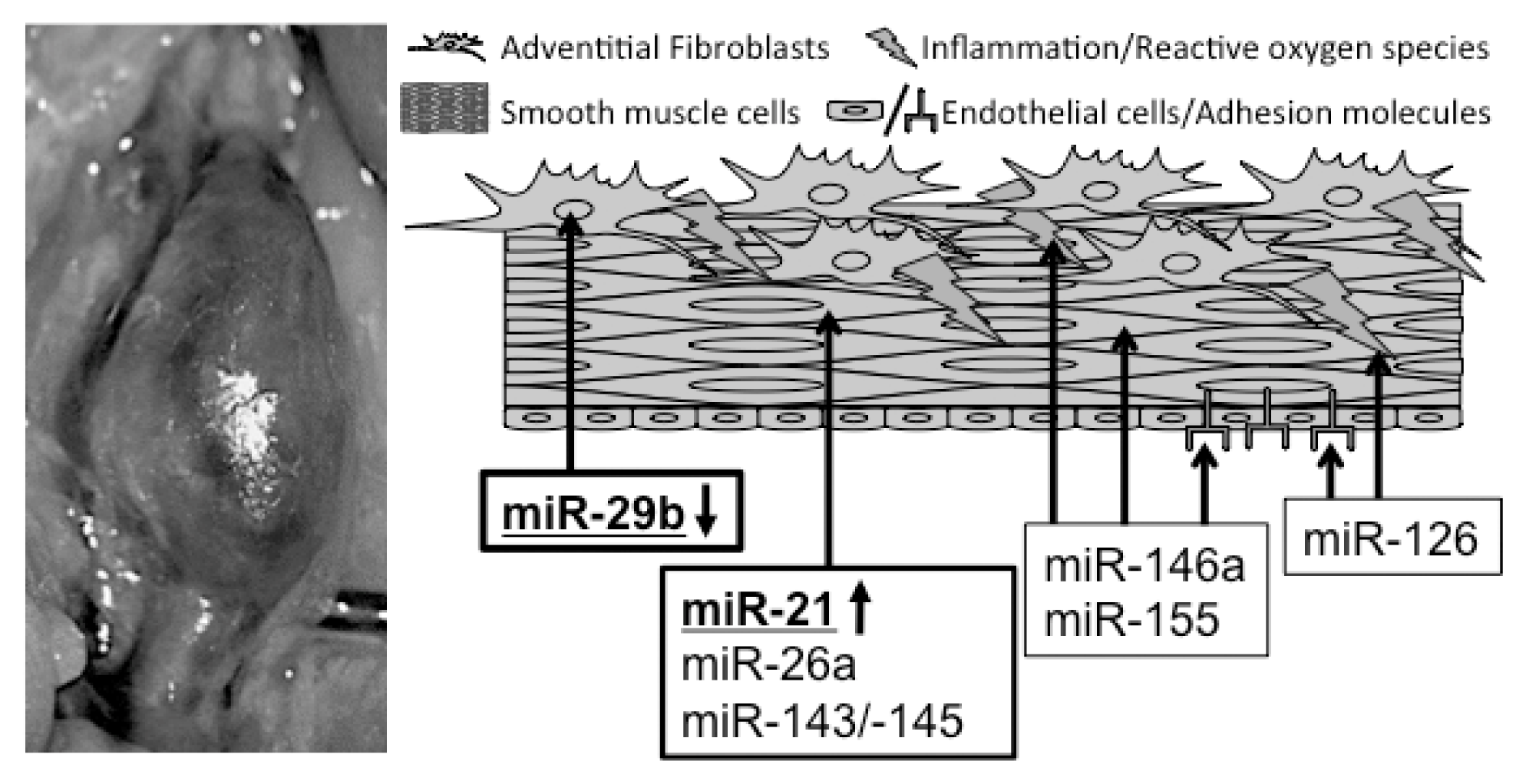

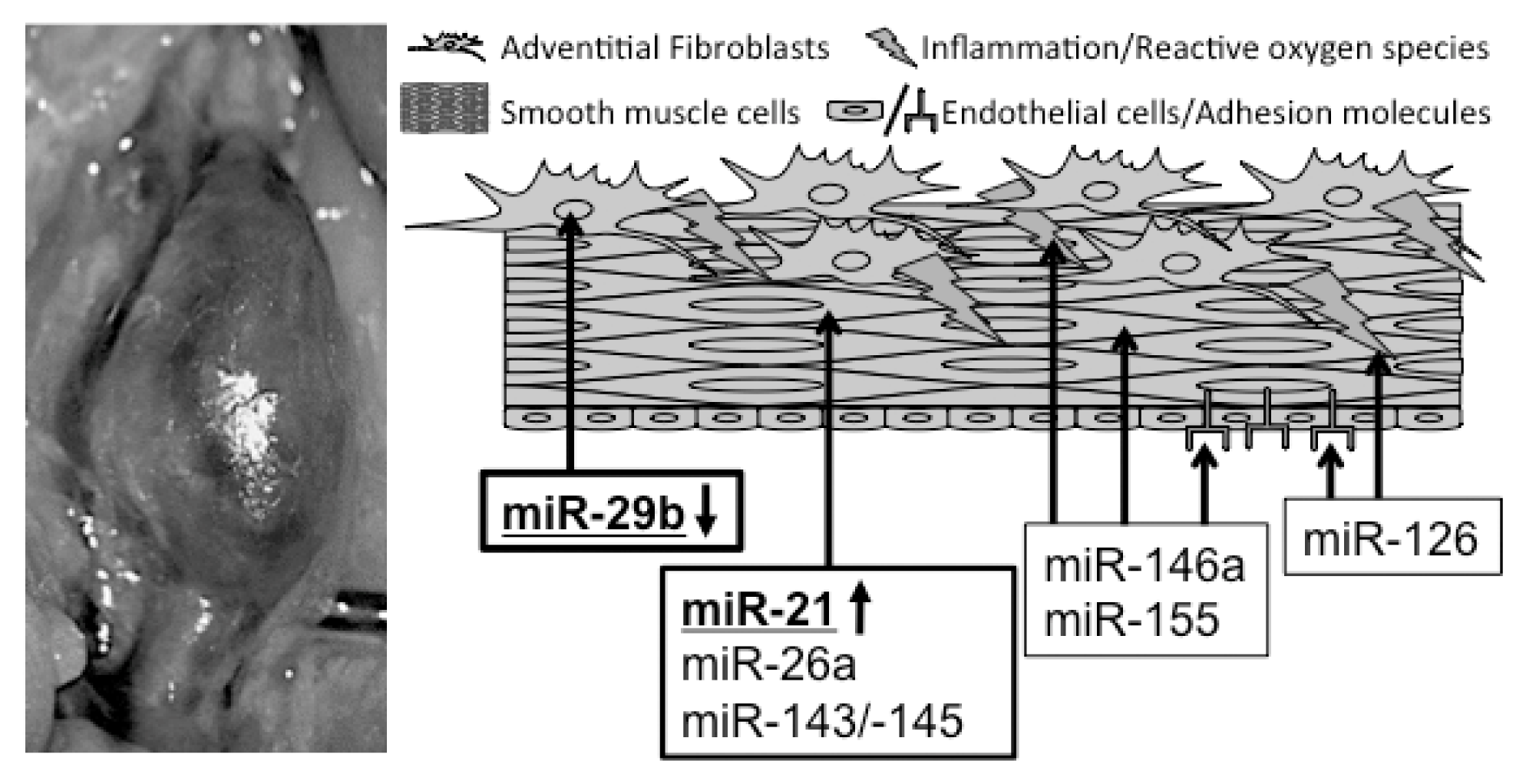{kind=link}
| microRNA | Model of AAA induction | Effect on AAA progression |
|---|---|---|
| miR-21 | PPE-infusion in C57BL/6 mice and AngII-infusion in ApoE−/− mice [59] | Regulates proliferation and apoptosis in ASMCs via PTEN/PI3K/AKT; induction of miR-21 through NFκB |
| miR-26a | PPE-infusion in C57BL/6 mice and AngII-infusion in ApoE−/− mice [60] | Inhibition of ASMC-differentiation via SMAD-1 and SMAD-4 depression |
| miR-29b | AngII in 1.5-year-old C57BL/6 [61]; PPE-infusion in C57BL/6 mice and AngII in ApoE−/− mice [62] | Modulating the fibrotic response in aortic wall through several collagen isoforms; repression of miR-29b in AFBs through TGF-β |
| miR-143/145 | miR-143/145 knockout and ApoE−/− mice [63] | Regulation of ASMC homeostasis and differentiation |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maegdefessel, L.; Spin, J.M.; Adam, M.; Raaz, U.; Toh, R.; Nakagami, F.; Tsao, P.S. Micromanaging Abdominal Aortic Aneurysms. Int. J. Mol. Sci. 2013, 14, 14374-14394. https://doi.org/10.3390/ijms140714374
Maegdefessel L, Spin JM, Adam M, Raaz U, Toh R, Nakagami F, Tsao PS. Micromanaging Abdominal Aortic Aneurysms. International Journal of Molecular Sciences. 2013; 14(7):14374-14394. https://doi.org/10.3390/ijms140714374
Chicago/Turabian StyleMaegdefessel, Lars, Joshua M. Spin, Matti Adam, Uwe Raaz, Ryuji Toh, Futoshi Nakagami, and Philip S. Tsao. 2013. "Micromanaging Abdominal Aortic Aneurysms" International Journal of Molecular Sciences 14, no. 7: 14374-14394. https://doi.org/10.3390/ijms140714374