The Intertwining of Transposable Elements and Non-Coding RNAs


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Non-coding RNAs
1.2. Transposable Elements
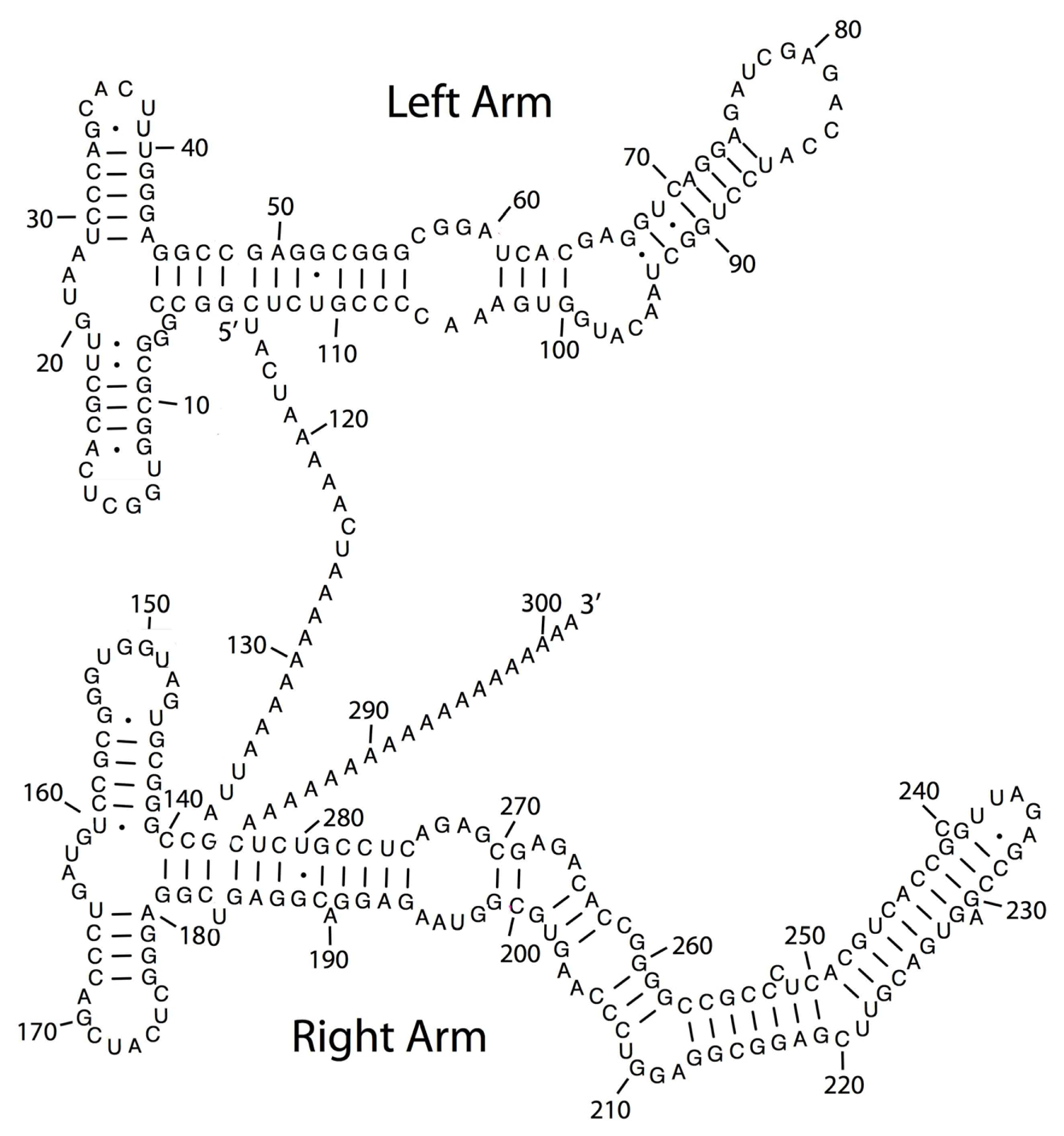
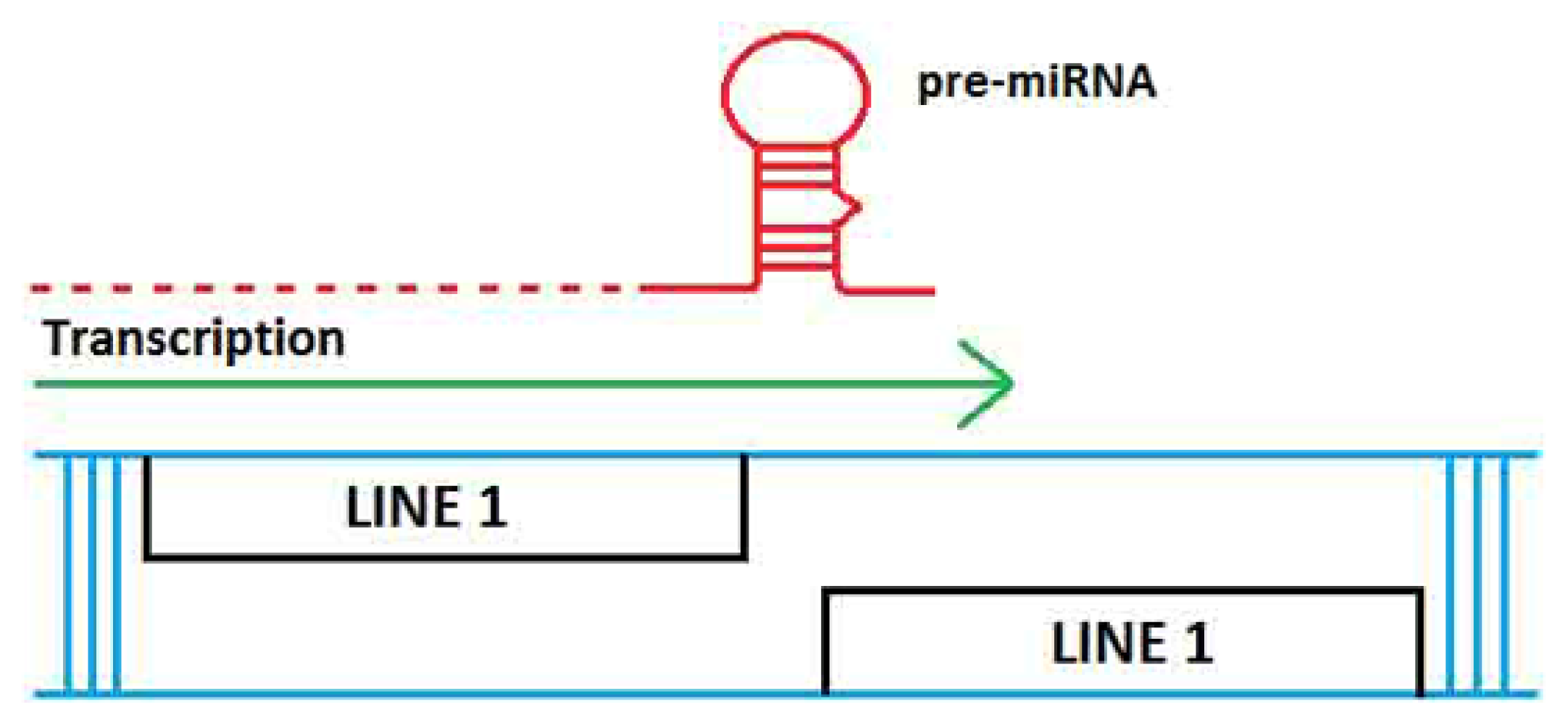
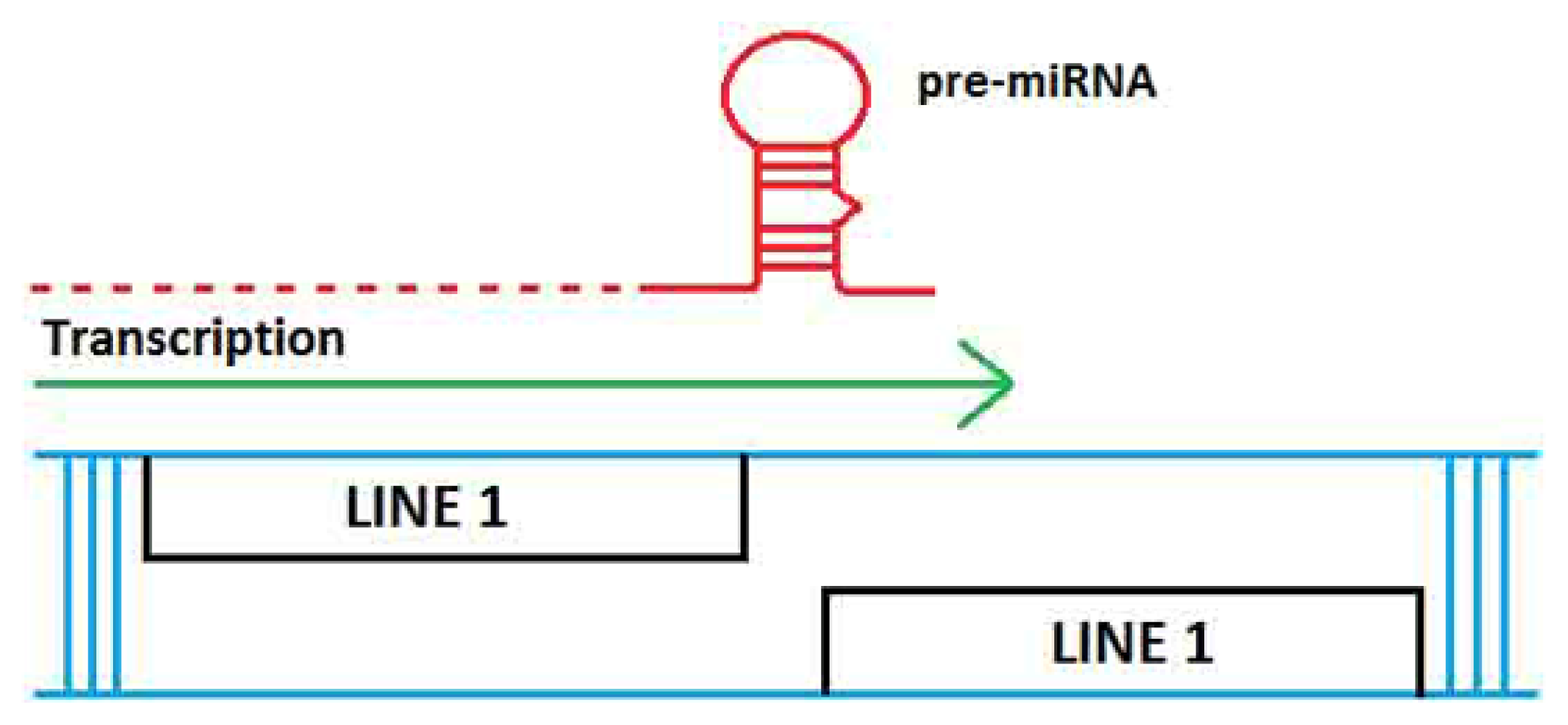
2. TE Origins of miRNAs
3. Interaction of TEs with ncRNAs—Functional and Disease-Related Significance
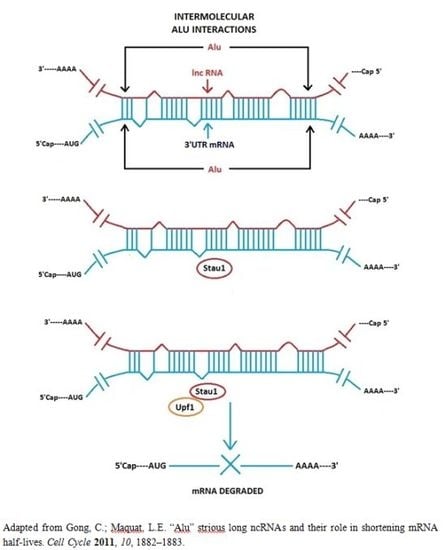
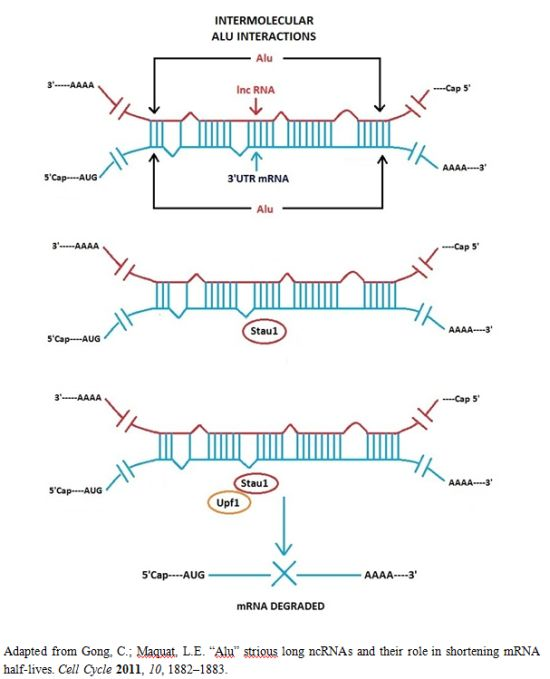
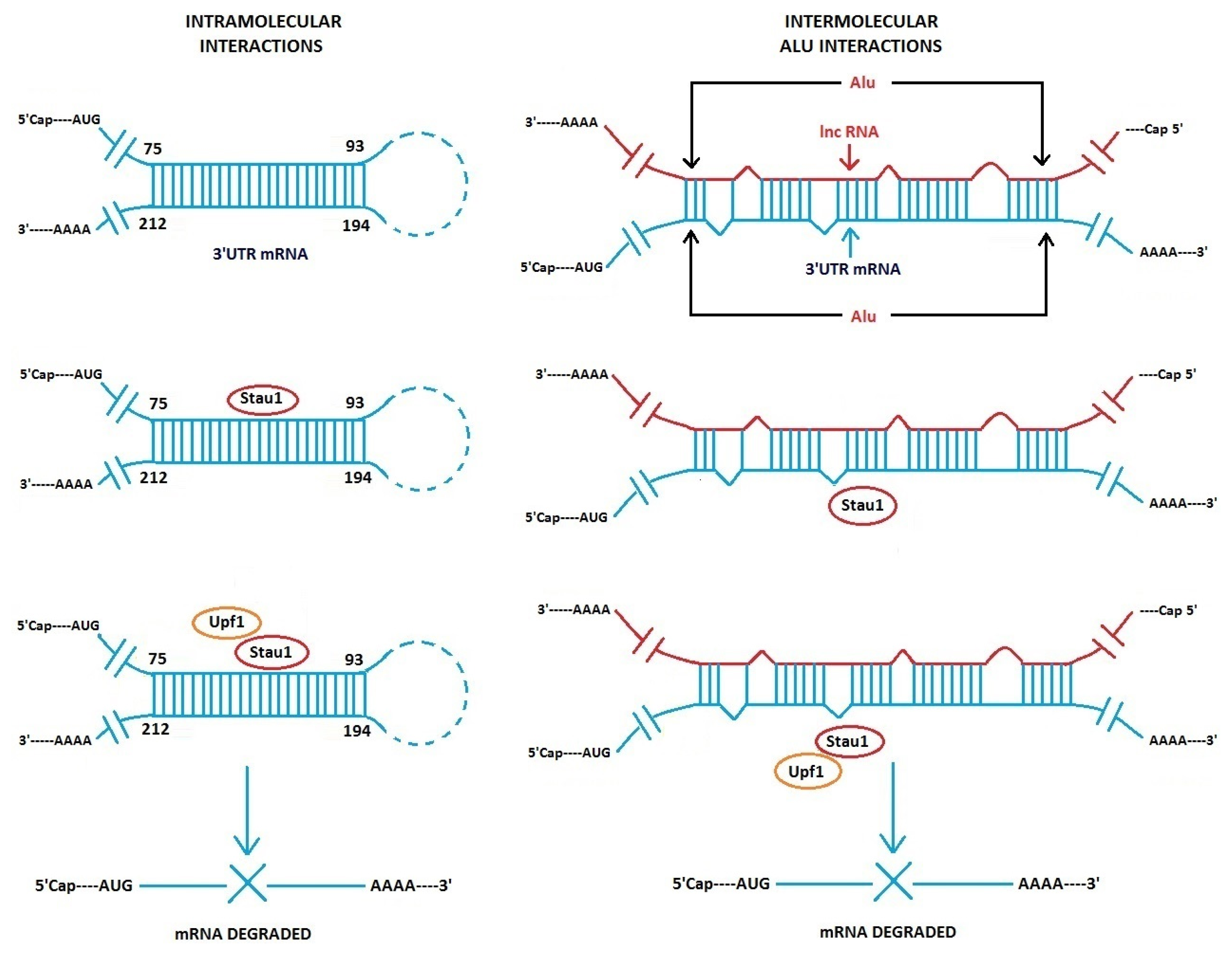
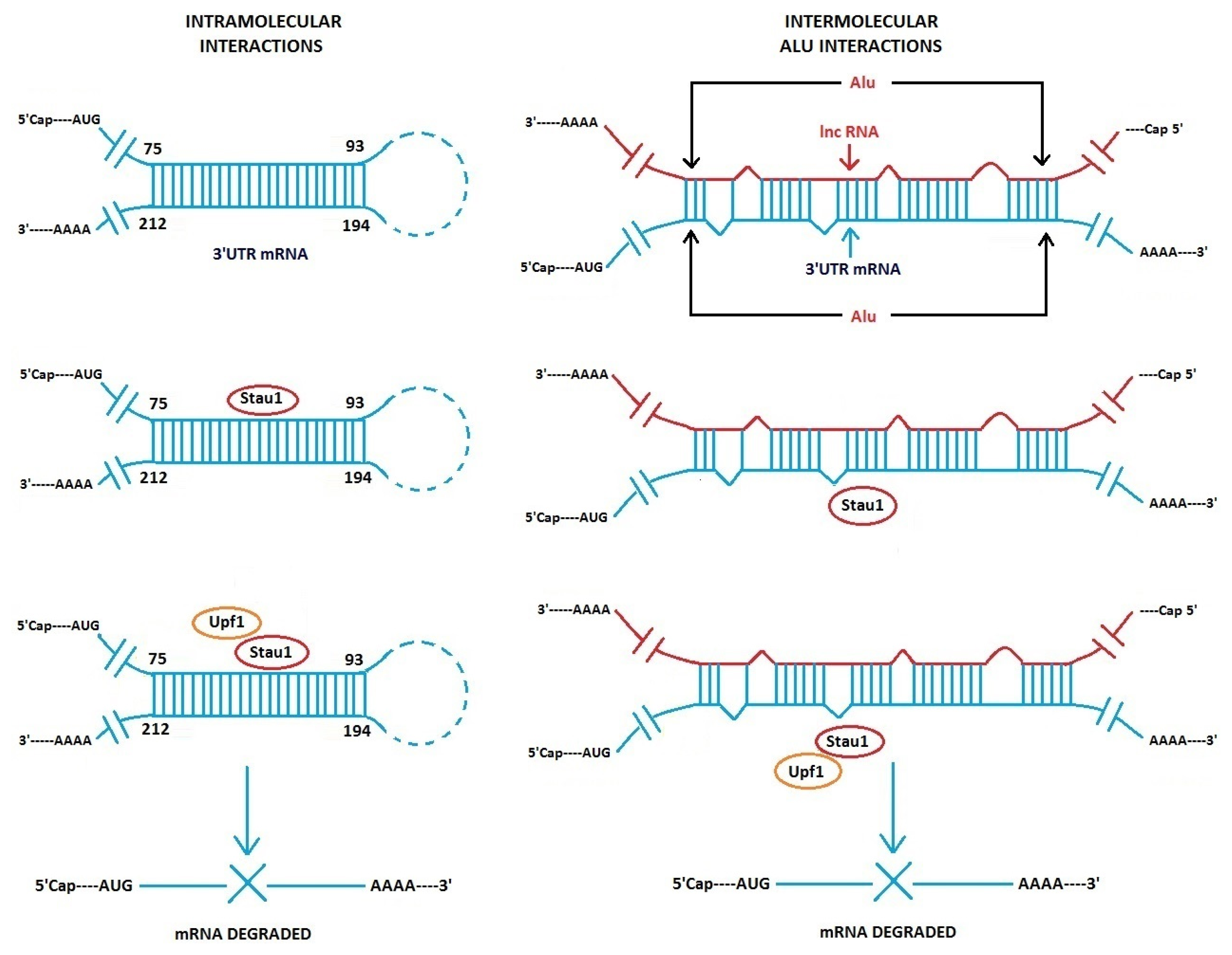
3.1. Alu Element Embedded in Long ncRNAs and mRNAs—Crucial Role in Target mRNA Decay
3.2. Long Non-Coding Antisense RNA Controls mRNA Translation—Importance of Embedded SINE/Alu Repeats
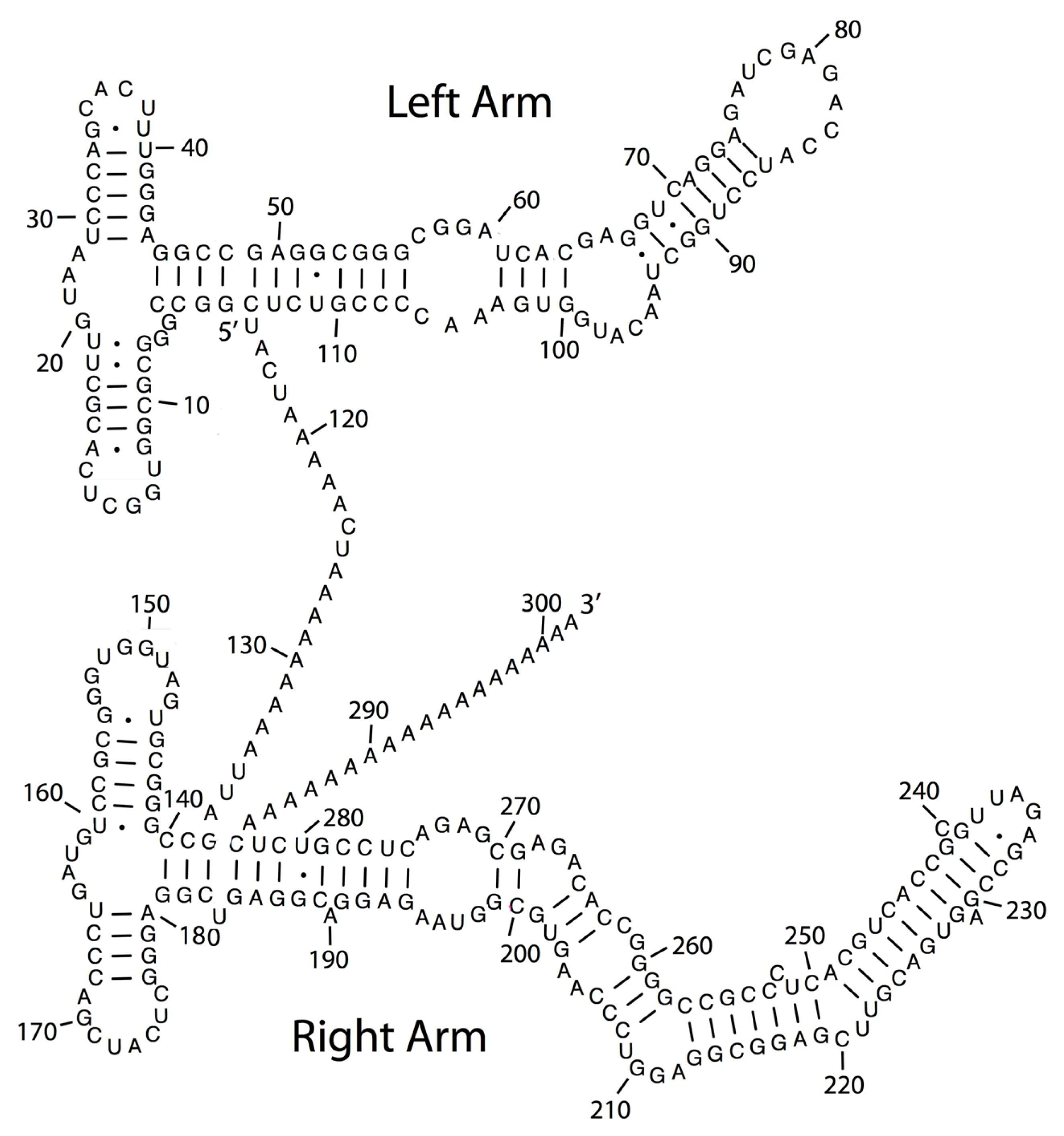
3.3. Point Mutation in LINE-1/Alu Element Embedded in a lncRNA Results in Lethal Brain Disease
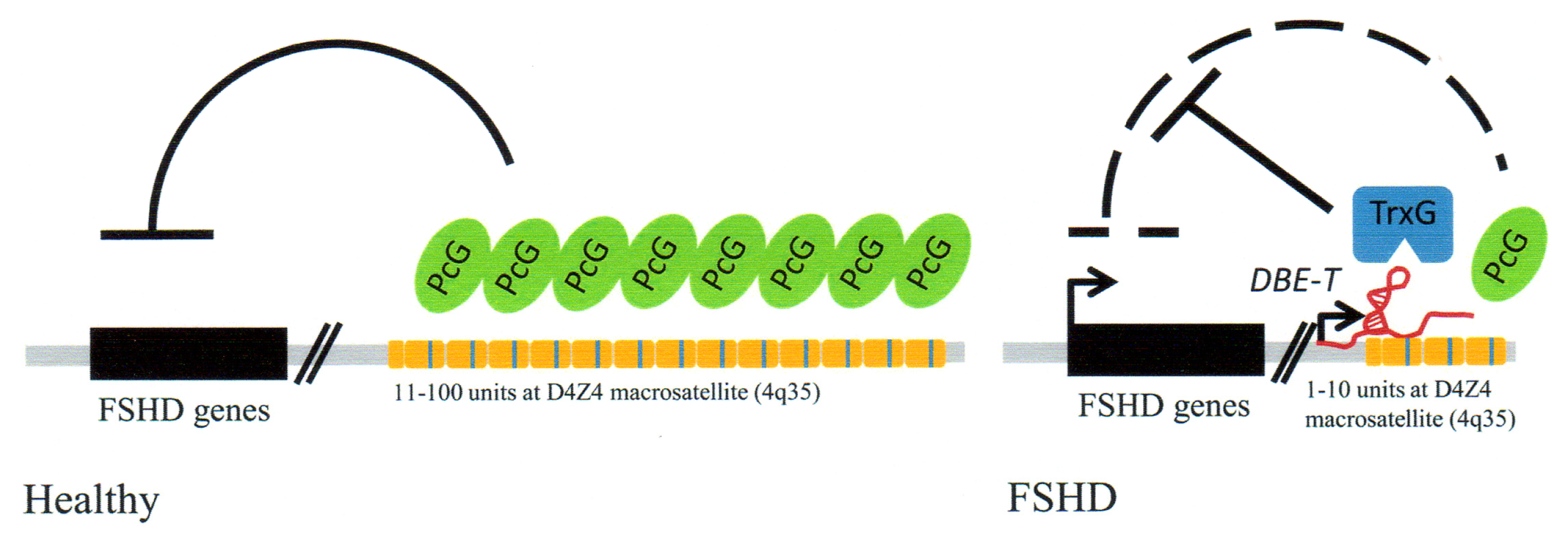
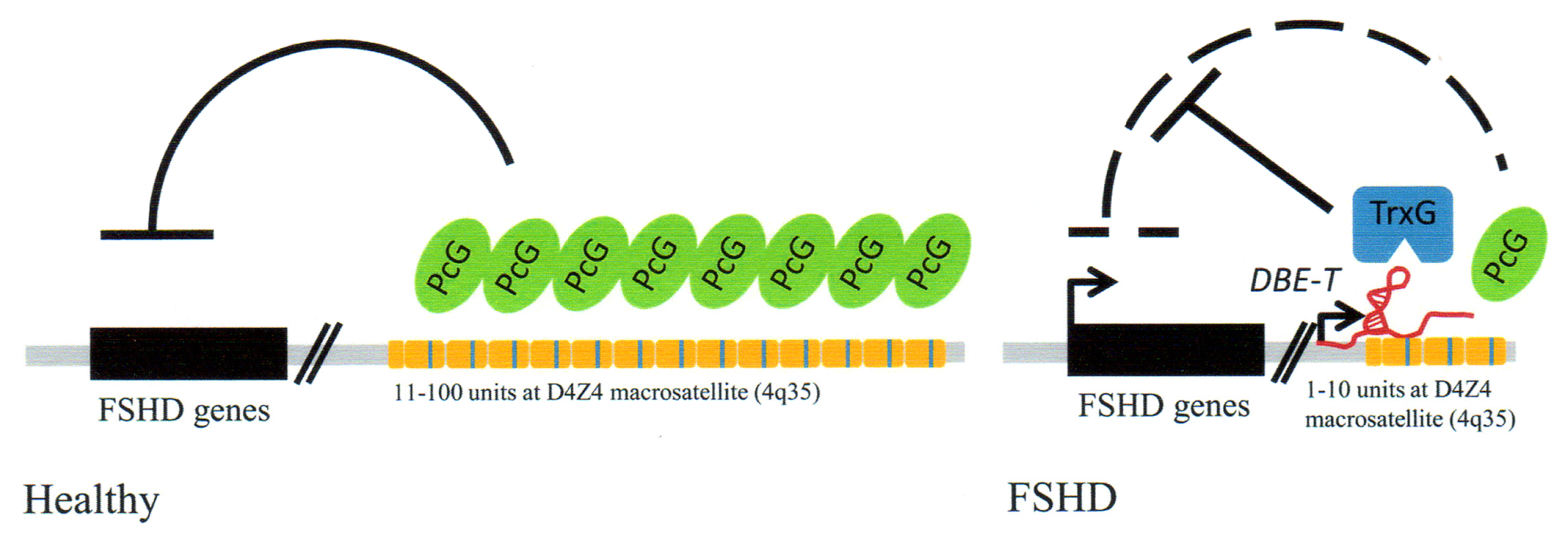
3.4. Facioscapulohumeral Muscular Dystrophy (FSHD)-Involvement of Tandem Repeats and Long ncRNA
3.5. Embedded Alu Sequences Can Take Part in Alternative Splicing and A to I Editing in Human mRNAs
3.6. Human Endogenous Retrovirus (HERV) LTR Transcripts
3.8. Regulatory Non-Coding Circular RNAs
3.9. piRNAs—Known Regulators of TEs
3.10. SINE/Alu Transcripts Function as ncRNAs in Gene Regulation at the Transcription Level
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Mattick, J.S. Rocking the foundations of molecular genetics. Proc. Natl. Acad. Sci. USA 2012, 109, 16400–16401. [Google Scholar]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet 2009, 10, 57–63. [Google Scholar]
- Habegger, L.; Sboner, A.; Gianoulis, T.A.; Rozowsky, J.; Agarwal, A.; Snyder, M.; Gerstein, M. RSEQtools: A modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics 2011, 27, 281–283. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 2012, 22, 1775–1789. [Google Scholar]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.N.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar]
- Di Leva, G.; Garofalo, M. Non-Coding RNAs and Cancer. In Oncogene and Cancer—From Bench to Clinic; Siregar, Y., Ed.; InTech: New York, NY, USA, 2013; Volume Chapter 14, pp. 317–358. [Google Scholar]
- Mizuno, T.; Chou, M.Y.; Inouye, M. A unique mechanism regulating gene expression: Translational inhibition by a complementary RNA transcript (micRNA). Proc. Natl. Acad. Sci. USA 1984, 81, 1966–1970. [Google Scholar]
- Andersen, J.; Forst, S.A.; Zhao, K.; Inouye, M.; Delihas, N. The function of micF RNA: micF RNA is a major factor in the thermal regulation of OmpF protein in Escherichia coli. J. Biol. Chem 1989, 264, 17961–17970. [Google Scholar]
- Schmidt, M.; Zheng, P.; Delihas, N. Secondary structures of Escherichia coli antisense micF RNA, the 5′-end of the target ompF mRNA, and the RNA/RNA Duplex. Biochemistry 1995, 34, 3621–3631. [Google Scholar]
- Cowley, M.; Oakey, R.J. Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet 2013, 9, e1003234. [Google Scholar]
- Goodier, J.L.; Kazazian, H.H., Jr. Retrotransposons revisited: The restraint and rehabilitation of parasites. Cell 2008, 135, 23–35. [Google Scholar]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Doudna, J.A. The Doudna Lab. Translational control by mRNA secondary structure—Alu-element regulated miRNA interactions. Available online: http://doudna.berkeley.edu/ (on accessed 24 June 2013).
- De Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 2011, 7, e1002384. [Google Scholar]
- Casa, V.; Gabellini, D. A repetitive elements perspective in Polycomb epigenetics. Front. Genet 2012, 3, 199. [Google Scholar]
- Ahmed, M.; Liang, P. Transposable elements are a significant contributor to tandem repeats in the human genome. Comp. Funct. Genomics 2012, 12, 947089. [Google Scholar]
- Smalheiser, N.R.; Torvik, V.I. Mammalian microRNAs derived from genomic repeats. Trends Genet 2005, 21, 322–326. [Google Scholar]
- Piriyapongsa, J.; Jordan, I.K. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2007, 2, e203. [Google Scholar]
- Piriyapongsa, J.; Mariño-Ramírez, L.; Jordan, I.K. Origin and evolution of human microRNAs from transposable elements. Genetics 2007, 176, 1323–1337. [Google Scholar]
- Devor, E.J.; Peek, A.S.; Lanier, W.; Samollow, P.B. Marsupial-specific microRNAs evolved from marsupial-specific TEs. Gene 2009, 448, 187–191. [Google Scholar]
- Yuan, Z.; Sun, X.; Jiang, D.; Ding, Y.; Lu, Z.; Gong, L.; Liu, H.; Xie, J. Origin and evolution of a placental-specific microRNA family in the human genome. BMC Evol. Biol 2010, 10, 346. [Google Scholar]
- Kapitonov, V.V.; Jurka, J. MER53, a non-autonomous DNA transposon associated with a variety of functionally related defense genes in the human genome. DNA Seq 1998, 8, 277–288. [Google Scholar]
- Yuan, Z.; Sun, X.; Liu, H.; Xie, J. MicroRNA genes derived from repetitive elements and expanded by segmental duplication events in mammalian genomes. PLoS One 2011, 6, e17666. [Google Scholar]
- Borchert, G.M.; Holton, N.W.; Williams, J.D.; Hernan, W.L.; Bishop, I.P.; Dembosky, J.A.; Elste, J.E.; Gregoire, N.S.; Kim, J.A.; Koehler, W.W.; et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob. Genet. Element 2011, 1, 8–17. [Google Scholar]
- Tempel, S.; Pollet, N.; Tahi, F. ncRNAclassifier: A tool for detection and classification of transposable element sequences in RNA hairpins. BMC Bioinforma 2012, 13, 246. [Google Scholar]
- Ahn, K.; Gim, J.A.; Ha, H.S.; Han, K.; Kim, H.S. The novel MER transposon-derived miRNAs in human genome. Gene 2013, 512, 422–428. [Google Scholar]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar]
- Gong, C.; Maquat, L.E. “Alu” strious long ncRNAs and their role in shortening mRNA half-lives”. Cell Cycle 2011, 10, 1882–1883. [Google Scholar]
- Kim, Y.K.; Furic, L.; Parisien, M.; Major, F.; DesGroseiller, L.; Maquat, L.E. Staufen1 regulates diverse classes of mammalian transcripts. EMBO J 2007, 26, 2670–2681. [Google Scholar]
- Capshew, C.R.; Dusenbury, K.L.; Hundley, H.A. Inverted Alu dsRNA structures do not affect localization but can alter translation efficiency of human mRNAs independent of RNA editing. Nucleic Acids Res 2012, 40, 8637–8645. [Google Scholar]
- Gleghorn, M.L.; Gong, C.; Kielkopf, C.L.; Maquat, L.E. Staufen1 dimerizes through a conserved motif and a degenerate dsRNA-binding domain to promote mRNA decay. Nat. Struct. Mol. Biol 2013, 20, 515–524. [Google Scholar]
- Park, E.; Gleghorn, M.L.; Maquat, L.E. Staufen2 functions in Staufen1-mediated mRNA decay by binding to itself and its paralog and promoting UPF1 helicase but not ATPase activity. Proc. Natl. Acad. Sci. USA 2013, 110, 405–412. [Google Scholar]
- Wang, J.; Gong, C.; Maquat, L.E. Control of myogenesis by rodent SINE-containing lncRNAs. Genes Dev 2013, 27, 793–804. [Google Scholar]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem 2004, 279, 13256–13264. [Google Scholar]
- Barrachina, M.; Castaño, E.; Dalfó, E.; Maes, T.; Buesa, C.; Ferrer, I. Reduced ubiquitin C-terminal hydrolase-1 expression levels in dementia with Lewy bodies. Neurobiol. Dis 2006, 22, 265–273. [Google Scholar]
- Cartault, F.; Munier, P.; Benko, E.; Desguerre, I.; Hanein, S.; Boddaert, N.; Bandiera, S.; Vellayoudom, J.; Krejbich-Trotot, P.; Bintner, M. Mutation in a primate-conserved retrotransposon reveals a noncoding RNA as a mediator of infantile encephalopathy. Proc. Natl. Acad. Sci. USA 2012, 109, 4980–4985. [Google Scholar]
- Hewitt, J.E.; Lyle, R.; Clark, L.N.; Valleley, E.M.; Wright, T.J.; Wijmenga, C.; van Deutekom, J.C.; Francis, F.; Sharpe, P.T.; Hofker, M.; et al. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum. Mol. Genet 1994, 8, 1287–1295. [Google Scholar]
- Clapp, J.; Mitchell, L.M.; Bolland, D.J.; Fantes, J.; Corcoran, A.E.; Scotting, P.J.; Armour, J.A.; Hewitt, J.E. Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet 2007, 81, 264–279. [Google Scholar]
- Snider, L.; Asawachaicharn, A.; Tyler, A.E.; Geng, L.N.; Petek, L.M.; Maves, L.; Miller, D.G.; Lemmers, R.J.; Winokur, S.T.; Tawil, R. RNAtranscripts, miRNA-sizedfragments and proteinsproduced from D4Z4units: New candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum. Mol. Genet 2009, 18, 2414–2430. [Google Scholar]
- Cabianca, D.S.; Gabellini, D. FSHD: Copy number variations on the theme of muscular dystrophy. J. Cell Biol 2010, 191, 1049–1060. [Google Scholar]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 2012, 819–831. [Google Scholar]
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R.; et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet 1992, 2, 26–30. [Google Scholar]
- Emanuel, B.S. Molecular mechanisms and diagnosis of chromosome 22q11.2 rearrangements. Dev. Disabil. Res. Rev 2008, 14, 11–18. [Google Scholar]
- Ponicsan, S.L.; Kugel, J.F.; Goodrich, J.A. Genomic gems: SINE RNAs regulate mRNA production. Curr. Opin. Genet. Dev 2010, 20, 149–155. [Google Scholar]
- Sorek, R.; Ast, G.; Graur, D. Alu-containing exons are alternatively spliced. Genome Res 2002, 12, 1060–1067. [Google Scholar]
- Ram, O.; Schwartz, S.; Ast, G. Multifactorial interplay controls the splicing profile of Alu-derived exons. Mol. Cell Biol 2008, 28, 3513–3525. [Google Scholar]
- Gal-Mark, N.; Schwartz, S.; Ast, G. Alternative splicing of Alu exons—Two arms are better than one. Nucleic Acids Res 2008, 36, 2012–2023. [Google Scholar]
- Schwartz, S.; Gal-Mark, N.; Kfir, N.; Oren, R.; Kim, E.; Ast, G. Alu exonization events reveal features required for precise recognition of exons by the splicing machinery. PLoS Comput. Biol 2009, 5, e1000300. [Google Scholar]
- Dagan, T.; Sorek, R.; Sharon, E.; Ast, G.; Graur, D. AluGene: A database of Alu elements incorporated within protein-coding genes. Nucleic Acids Res 2004, 32, D489–D492. [Google Scholar]
- Lev-Maor, G.; Ram, O.; Kim, E.; Sela, N.; Goren, A.; Levanon, E.Y.; Ast, G. Intronic Alus influence alternative splicing. PLoS Genet 2008, 4, e1000204. [Google Scholar]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of alu-containing mRNAs in the human transcriptome. PLoS Biol 2004, 2, e391. [Google Scholar]
- Xu, L.; Elkahloun, A.G.; Candotti, F.; Grajkowski, A.; Beaucage, S.L.; Petricoin, E.F.; Calvert, V.; Juhl, H.; Mills, F.; Mason, K. A novel function of RNAs arising from the long terminal repeat of human endogenous retrovirus 9 in cell cycle arrest. J. Virol 2013, 87, 25–36. [Google Scholar]
- Kelley, D.; Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol 2012, 13, R107. [Google Scholar]
- Cocquerelle, C.; Mascrez, B.; Hetuin, D.; Bailleul, B. Mis-splicing yields circular RNA molecules. FASEB J 1993, 7, 155–160. [Google Scholar]
- Capel, B.; Swain, A.; Nicolis, S.; Hacker, A.; Walter, M.; Koopman, P.; Goodfellow, P.; Lovell-Badge, R. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 1993, 73, 1019–1030. [Google Scholar]
- Hsu, M.-T.; Coca-Prados, M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature 1979, 280, 339–340. [Google Scholar]
- Nigro, J.M.; Cho, K.R.; Fearon, E.R.; Kern, S.E.; Ruppert, J.M.; Oliner, J.D.; Kinzler, K.W.; Vogelstein, B. Scrambled exons. Cell 1991, 64, 607–613. [Google Scholar]
- Zaphiropoulos, P.G. Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: Correlation with exon skipping. Proc. Natl. Acad. Sci. USA 1996, 93, 6536–6541. [Google Scholar]
- Li, X.-F.; Lytton, J. A circularized sodium-calcium exchanger exon 2 transcript. J. Biol. Chem. 1999, 274, 8153–8160. [Google Scholar]
- Gualandi, F.; Trabanelli, C.; Rimessi, P.; Calzolari, E.; Toffolatti, L.; Patarnello, T.; Kunz, G.; Muntoni, F.; Ferlini, A. Multiple exon skipping and RNA circularisation contribute to the severe phenotypic expression of exon 5 dystrophin deletion. J. Med. Genet 2003, 40, e100. [Google Scholar]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 2012, 7, e30733. [Google Scholar]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar]
- Lin, H.; Spradling, A.C. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development 1997, 124, 2463–2476. [Google Scholar]
- Karginov, F.V.; Hannon, G.J. The CRISPR system: Small RNA-guided defense in bacteria and archaea. Mol. Cell 2010, 37, 7–19. [Google Scholar]
- Wiedenheft, B.; van Duijn, E.; Bultema, J.B.; Waghmare, S.P.; Zhou, K.; Barendregt, A.; Westphal, W.; Heck, A.J.; Boekema, E.J.; Dickman, M.J.; et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 10092–10097. [Google Scholar]
- Sashital, D.G.; Jinek, M.; Doudna, J.A. An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat. Struct. Mol. Biol 2011, 18, 680–687. [Google Scholar]
- Brennecke, J.; Aravin, A.A.; Stark, A.; Dus, M.; Kellis, M.; Sachidanandam, R.; Hannon, G.J. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 2007, 128, 1089–1103. [Google Scholar]
- Malone, C.D.; Hannon, G.J. Small RNAs as guardians of the genome. Cell 2009, 136, 656–668. [Google Scholar]
- Simonelig, M. Developmental functions of piRNAs and transposable elements: A Drosophila point-of-view. RNA Biol 2011, 8, 754–759. [Google Scholar]
- Bagijn, M.P.; Goldstein, L.D.; Sapetschnig, A.; Weick, E.M.; Bouasker, S.; Lehrbach, N.J.; Simard, M.J.; Miska, E.A. Function, targets, and evolution of Caenorhabditis elegans piRNAs. Science 2012, 337, 574–578. [Google Scholar]
- Le Thomas, A.; Rogers, A.K.; Webster, A.; Marinov, G.K.; Liao, S.E.; Perkins, E.M.; Hur, J.K.; Aravin, A.A.; Tóth, K.F. Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev 2013, 27, 390–399. [Google Scholar]
- Sigurdsson, M.I.; Smith, A.V.; Bjornsson, H.T.; Jonsson, J.J. The distribution of a germline methylation marker suggests a regional mechanism of LINE-1 silencing by the piRNA-PIWI system. BMC Genet 2012, 13, 31. [Google Scholar] [Green Version]
- Watanabe, T.; Tomizawa, S.; Mitsuya, K.; Totoki, Y.; Yamamoto, Y.; Kuramochi-Miyagawa, S.; Iida, N.; Hoki, Y.; Murphy, P.J.; Toyoda, A.; et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 2011, 332, 848–852. [Google Scholar]
- Espinoza, C.A.; Goodrich, J.A.; Kugel, J.F. Characterization of the structure, function, and mechanism of B2 RNA, an ncRNA repressor of RNA polymerase II transcription. RNA 2007, 13, 583–596. [Google Scholar]
- Yakovchuk, P.; Goodrich, J.A.; Kugel, J.F. B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 5569–5574. [Google Scholar]
- Yakovchuk, P.; Goodrich, J.A.; Kugel, J.F. B2 RNA represses TFIIH phosphorylation of RNA polymerase II. Transcription 2011, 2, 45–49. [Google Scholar]
- Blackwell, B.J.; Lopez, M.F.; Wang, J.; Krastins, B.; Sarracino, D.; Tollervey, J.R.; Dobke, M.; Jordan, I.K.; Lunyak, V.V. Protein interactions with piALU RNA indicates putative participation of retroRNA in the cell cycle, DNA repair and chromatin assembly. Mob. Genet. Elements 2012, 2, 26–35. [Google Scholar]
- Wassarman, K.M.; Storz, G. 6S RNA regulates E. coli RNA polymerase activity. Cell 2000, 101, 613–623. [Google Scholar]
- Wassarman, K.M. 6S RNA: A regulator of transcription. Mol. Microbiol 2007, 65, 1425–1431. [Google Scholar]
- Decker, K.B.; Hinton, D.M. The secret to 6S: Regulating RNA polymerase by ribo-sequestration. Mol. Microbiol 2009, 73, 137–140. [Google Scholar]
- Holmqvist, E.; Unoson, C.; Reimegård, J.; Wagner, E.G. A mixed double negative feedback loop between the sRNA MicF and the global regulator. Lrp. Mol. Microbiol 2012, 4, 414–427. [Google Scholar]
- Corcoran, C.P.; Podkaminski, D.; Papenfort, K.; Urban, J.H.; Hinton, J.C.; Vogel, J. Superfolder GFP reporters validate diverse new mRNA targets of the classic porin regulator, MicF RNA. Mol. Microbiol 2012, 84, 428–445. [Google Scholar]
- Brownlee, G.G. Sequence of 6S RNA of E. coli. Nat. New Biol 1971, 229, 147–149. [Google Scholar]
- Hauptman, N.; Glavač, D. Long non-coding RNA in cancer. Int. J. Mol. Sci 2013, 14, 4655–4669. [Google Scholar]
- Gutschner, T.; Diederichs, S. The Hallmarks of Cancer: A long non-coding RNA point of view. RNA Biol 2012, 9, 703–719. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hadjiargyrou, M.; Delihas, N. The Intertwining of Transposable Elements and Non-Coding RNAs. Int. J. Mol. Sci. 2013, 14, 13307-13328. https://doi.org/10.3390/ijms140713307
Hadjiargyrou M, Delihas N. The Intertwining of Transposable Elements and Non-Coding RNAs. International Journal of Molecular Sciences. 2013; 14(7):13307-13328. https://doi.org/10.3390/ijms140713307
Chicago/Turabian StyleHadjiargyrou, Michael, and Nicholas Delihas. 2013. "The Intertwining of Transposable Elements and Non-Coding RNAs" International Journal of Molecular Sciences 14, no. 7: 13307-13328. https://doi.org/10.3390/ijms140713307