Hormesis in Aging and Neurodegeneration—A Prodigy Awaiting Dissection

Abstract
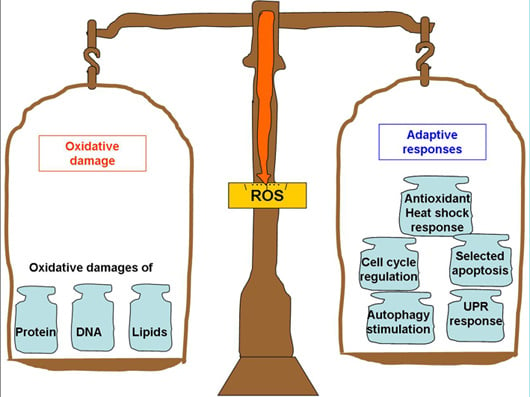
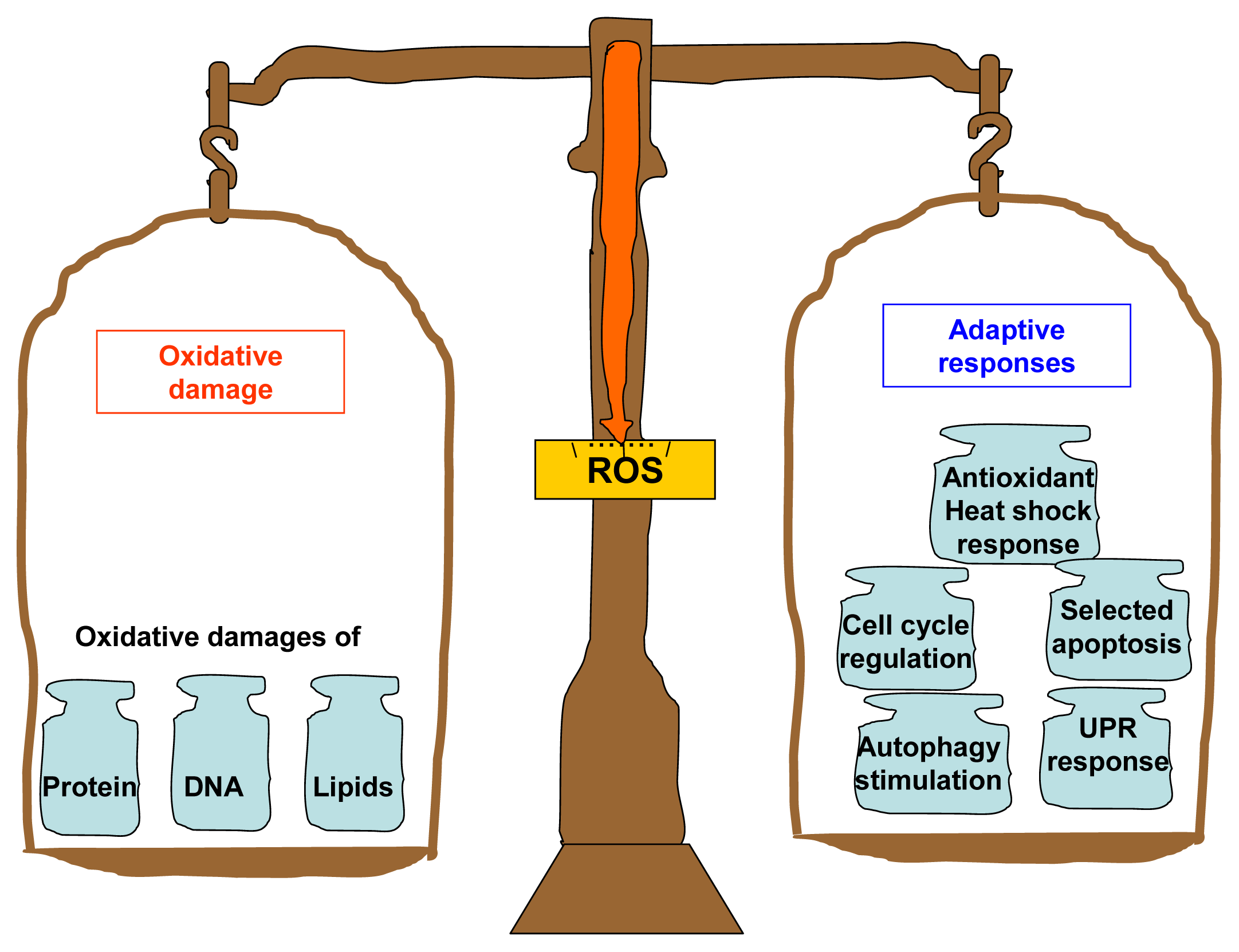
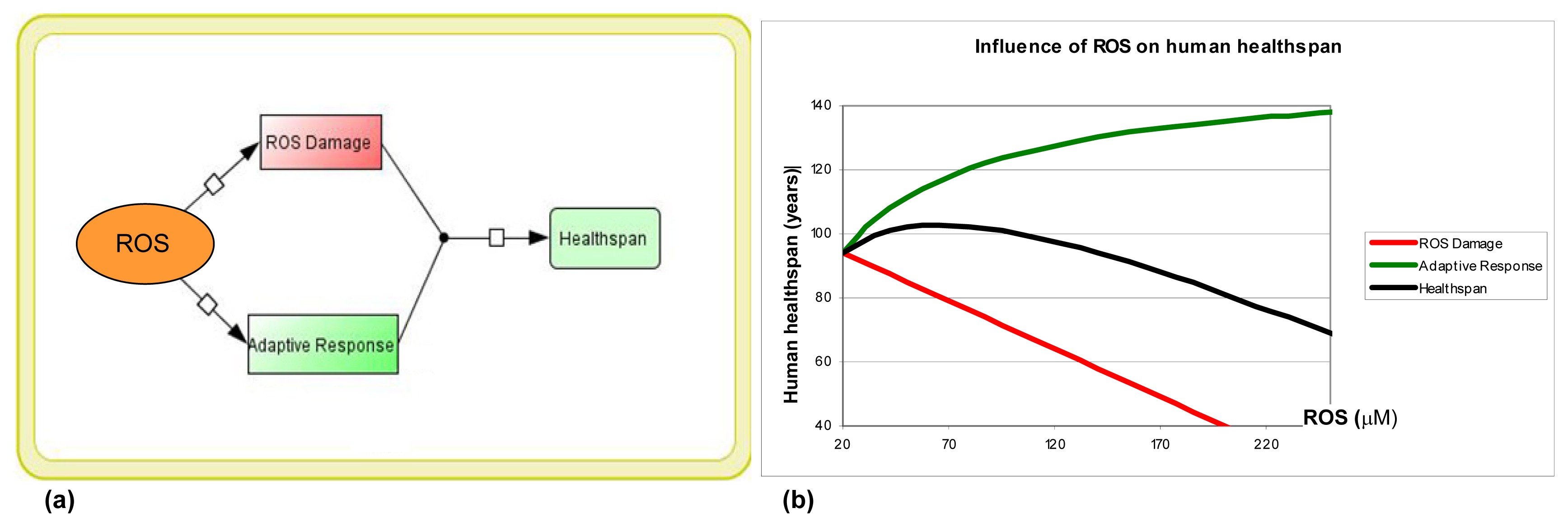
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
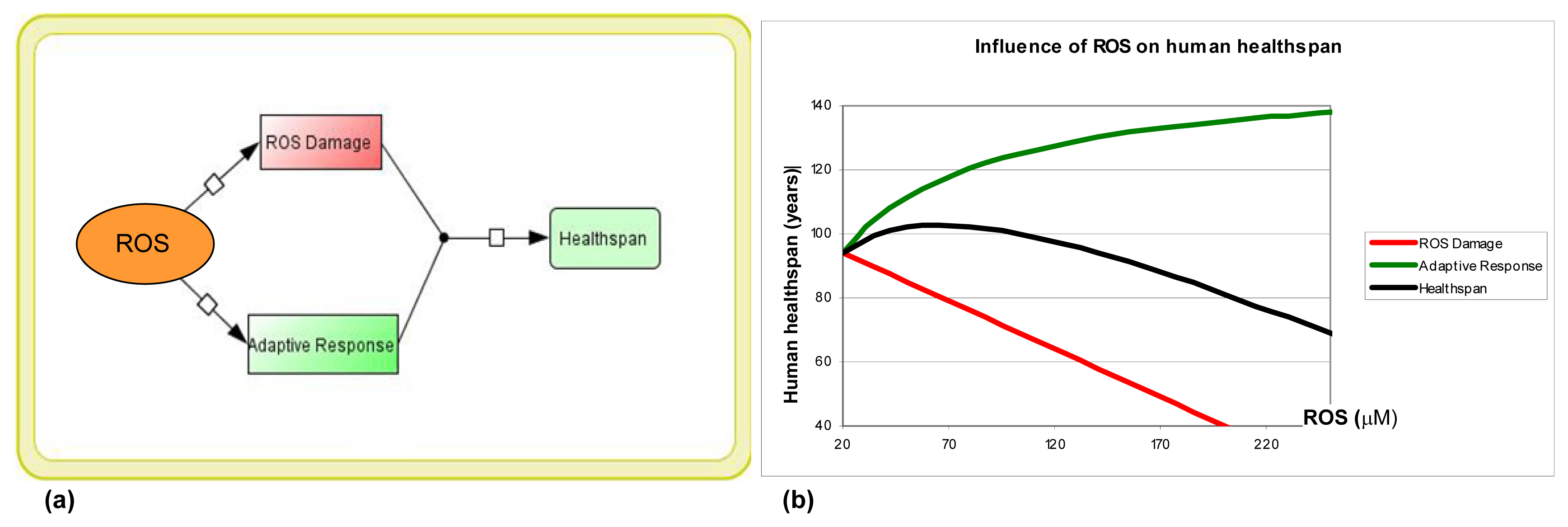
1. Introduction
2. ROS Hormesis in Aging and Neurodegenerative Diseases
2.1. ROS and Aging: Causal but Not Concomitant
2.2. Caloric Restriction Induced Lifespan Extension Is Mediated by ROS
2.3. The Neuroprotective Potential of ROS Hormesis
3. ROS-Mediated Adaptive Responses
3.1. Antioxidant and Heat Shock Responses
3.2. Cell Cycle Regulation and Selective Apoptosis
3.3. Crosstalk of ROS Hormesis and the Unfolded Protein Response
3.4. Autophagy Stimulation
4. Possible Additive Nature of ROS Hormesis
5. Potential Therapeutic Value of ROS Hormesis
6. Conclusions and Perspectives
Acknowledgments
Conflict of Interest
References
- Castro, P.V.; Khare, S.; Young, B.D.; Clarke, S.G. Caenorhabditis elegans battling starvation stress: Low levels of ethanol prolong lifespan in L1 larvae. PLoS One 2012, 7, e29984. [Google Scholar]
- Ritzmann, R.F.; Glasky, A.; Steinberg, A.; Melchior, C.L. The interaction of ethanol with the cognitive enhancers tacrine, physostigmine, and AIT-082. J. Gerontol 1994, 49, B51–B53. [Google Scholar]
- Briasoulis, A.; Agarwal, V.; Messerli, F.H. Alcohol consumption and the risk of hypertension in men and women: A systematic review and meta-analysis. J. Clin. Hypertens 2012, 14, 792–798. [Google Scholar]
- Marmot, M.G. Alcohol and coronary heart disease. Int. J. Epidemiol 2001, 30, 724–729. [Google Scholar]
- Maggio, R.; Riva, M.; Vaglini, F.; Fornai, F.; Racagni, G.; Corsini, G.U. Striatal increase of neurotrophic factors as a mechanism of nicotine protection in experimental parkinsonism. J. Neural Transm 1997, 104, 1113–1123. [Google Scholar]
- Quik, M. Smoking, nicotine and Parkinson’s disease. Trends Neurosci 2004, 27, 561–568. [Google Scholar]
- Baron, J.A. Beneficial effects of nicotine and cigarette smoking: The real, the possible and the spurious. Br. Med. Bull 1996, 52, 58–73. [Google Scholar]
- Calabrese, E.J. Historical blunders: How toxicology got the dose-response relationship half right. Cell. Mol. Biol 2005, 51, 643–654. [Google Scholar]
- Calabrese, E.J.; Baldwin, L.A. Defining hormesis. Hum. Exp. Toxicol 2002, 21, 91–97. [Google Scholar]
- Southam, C.M.; Ehrlich, J. Effects of Extract of western red cedar heartwood on certain wood-decaying fungi in culture. Phytopathology 1943, 33, 517–524. [Google Scholar]
- Kaiser, J. Hormesis. Sipping from a poisoned chalice. Science 2003, 302, 376–379. [Google Scholar]
- Cypser, J.R.; Tedesco, P.; Johnson, T.E. Hormesis and aging in Caenorhabditis elegans. Exp. Gerontol 2006, 41, 935–939. [Google Scholar]
- DeGracia, D.J.; Montie, H.L. Cerebral ischemia and the unfolded protein response. J. Neurochem 2004, 91. [Google Scholar] [CrossRef]
- Tuomisto, J.; Tuomisto, J.T. Is the fear of dioxin cancer more harmful than dioxin? Toxicol. Lett 2012, 210, 338–344. [Google Scholar]
- Feinendegen, L.E. Evidence for beneficial low level radiation effects and radiation hormesis. Br. J. Radiol 2005, 78, 3–7. [Google Scholar]
- Jaworowski, Z. Radiation hormesis—A remedy for fear. Hum. Exp. Toxicol 2010, 29, 263–270. [Google Scholar]
- Galimberti, D.; Scarpini, E. Alzheimer’s disease: From pathogenesis to disease-modifying approaches. CNS Neurol. Disord. Drug Targets 2011, 10, 163–174. [Google Scholar]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci 2011, 12, 65–72. [Google Scholar]
- Morley, J.E.; Farr, S.A. Hormesis and amyloid-beta protein: Physiology or pathology? J. Alzheimers Dis 2012, 29, 487–492. [Google Scholar]
- Calabrese, E.J.; Baldwin, L.A. Toxicology rethinks its central belief. Nature 2003, 421, 691–692. [Google Scholar]
- Thayer, K.A.; Melnick, R.; Burns, K.; Davis, D.; Huff, J. Fundamental flaws of hormesis for public health decisions. Environ. Health Perspect 2005, 113, 1271–1276. [Google Scholar]
- Calabrese, E.J. Hormesis: A revolution in toxicology, risk assessment and medicine. EMBO Rep 2004, 5, S37–S40. [Google Scholar]
- Craig, E.A. The heat shock response. CRC Crit. Rev. Biochem 1985, 18, 239–280. [Google Scholar]
- Lindquist, S. The heat-shock response. Annu. Rev. Biochem 1986, 55, 1151–1191. [Google Scholar]
- Calabrese, E.J.; Baldwin, L.A.; Holland, C.D. Hormesis: A highly generalizable and reproducible phenomenon with important implications for risk assessment. Risk Anal 1999, 19, 261–281. [Google Scholar]
- Pollycove, M.; Feinendegen, L.E. Radiation-induced versus endogenous DNA damage: Possible effect of inducible protective responses in mitigating endogenous damage. Hum. Exp. Toxicol 2003, 22, 290–306, ; discussion 307, 315–317, 319–323.. [Google Scholar]
- Michalek, R.D.; Nelson, K.J.; Holbrook, B.C.; Yi, J.S.; Stridiron, D.; Danie, L.W.; Fetrow, J.S.; King, S.B.; Poole, L.B.; Grayson, J.M. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J. Immunol 2007, 179, 6456–6467. [Google Scholar]
- Gems, D.; Partridge, L. Stress-response hormesis and aging: “That which does not kill us makes us stronger”. Cell Metab 2008, 7, 200–203. [Google Scholar]
- Hayflick, L. Biological aging is no longer an unsolved problem. Ann. N. Y. Acad. Sci. 2007, 1100. [Google Scholar] [CrossRef]
- Holliday, R. Aging is no longer an unsolved problem in biology. Ann. N. Y. Acad. Sci. 2006, 1067. [Google Scholar] [CrossRef]
- Rattan, S.I. Theories of biological aging: Genes, proteins, and free radicals. Free Radic. Res 2006, 40, 1230–1238. [Google Scholar]
- Bishop, N.A.; Guarente, L. Genetic links between diet and lifespan: Shared mechanisms from yeast to humans. Nat. Rev. Genet 2007, 8, 835–844. [Google Scholar]
- Pan, Y. Mitochondria, reactive oxygen species, and chronological aging: A message from yeast. Exp. Gerontol 2011, 46, 847–852. [Google Scholar]
- Joenje, H. Genetic toxicology of oxygen. Mutat. Res 1989, 219, 193–208. [Google Scholar]
- Afanas’ev, I. Reactive oxygen species signaling in cancer: Comparison with aging. Aging Dis 2011, 2, 219–230. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev 2007, 87, 315–424. [Google Scholar]
- Lagouge, M.; Larsson, N.G. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med 2013, 273, 529–543. [Google Scholar]
- Wiederkehr, A.; Wollheim, C.B. Minireview: Implication of mitochondria in insulin secretion and action. Endocrinology 2006, 147, 2643–2649. [Google Scholar]
- Ristow, M. Oxidative metabolism in cancer growth. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 339–345. [Google Scholar]
- Fukui, H.; Moraes, C.T. The mitochondrial impairment, oxidative stress and neurodegeneration connection: Reality or just an attractive hypothesis? Trends Neurosci 2008, 31, 251–256. [Google Scholar]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J 2008, 27, 306–314. [Google Scholar]
- Bratic, I.; Trifunovic, A. Mitochondrial energy metabolism and ageing. Biochim. Biophys. Acta 2010, 1797, 961–967. [Google Scholar]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res 2012, 111, 1198–1207. [Google Scholar]
- Ugidos, A.; Nystrom, T.; Caballero, A. Perspectives on the mitochondrial etiology of replicative aging in yeast. Exp. Gerontol 2010, 45, 512–515. [Google Scholar]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics 2003, 16, 29–37. [Google Scholar]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol 2010, 8, e1000556. [Google Scholar]
- Katsiki, N.; Manes, C. Is there a role for supplemented antioxidants in the prevention of atherosclerosis? Clin. Nutr 2009, 28, 3–9. [Google Scholar]
- Myung, S.K.; Kim, Y.; Ju, W.; Choi, H.J.; Bae, W.K. Effects of antioxidant supplements on cancer prevention: Meta-analysis of randomized controlled trials. Ann. Oncol 2010, 21, 166–179. [Google Scholar]
- Goto, S.; Radak, Z. Hormetic effects of reactive oxygen species by exercise: A view from animal studies for successful aging in human. Dose Response 2009, 8, 68–72. [Google Scholar]
- Liu, S.; Ajani, U.; Chae, C.; Hennekens, C.; Buring, J.E. Long-term beta-carotene supplementation and risk of type 2 diabetes mellitus: A randomized controlled trial. JAMA 1999, 282, 1073–1075. [Google Scholar]
- Song, Y.; Cook, N.R.; Albert, C.M.; van Denburgh, M.; Manson, J.E. Effects of vitamins C and E and beta-carotene on the risk of type 2 diabetes in women at high risk of cardiovascular disease: A randomized controlled trial. Am. J. Clin. Nutr 2009, 90, 429–437. [Google Scholar]
- Gomez-Cabrera, M.C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Vina, J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 2008, 87, 142–149. [Google Scholar]
- Khassaf, M.; McArdle, A.; Esanu, C.; Vasilaki, A.; McArdle, F.; Griffiths, R.D.; Brodie, D.A.; Jackson, M.J. Effect of vitamin C supplements on antioxidant defence and stress proteins in human lymphocytes and skeletal muscle. J. Physiol 2003, 549, 645–652. [Google Scholar]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar]
- Finley, L.W.; Haigis, M.C. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res. Rev 2009, 8, 173–188. [Google Scholar]
- Woo, D.K.; Shadel, G.S. Mitochondrial stress signals revise an old aging theory. Cell 2011, 144, 11–12. [Google Scholar]
- Rattan, S.I. Hormesis in aging. Ageing Res. Rev 2008, 7, 63–78. [Google Scholar]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Lingo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 2012, 489, 318–321. [Google Scholar]
- Castello, L.; Froio, T.; Cavallini, G.; Biasi, F.; Sapino, A.; Leonarduzzi, G.; Bergamini, E.; Poli, G.; Chiarpotto, E. Calorie restriction protects against age-related rat aorta sclerosis. FASEB J 2005, 19, 1863–1865. [Google Scholar]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar]
- Fontana, L.; Meyer, T.E.; Klein, S.; Holloszy, J.O. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc. Natl. Acad. Sci. USA 2004, 101, 6659–6663. [Google Scholar]
- Meyer, T.E.; Kovacs, S.J.; Ehsani, A.A.; Klein, S.; Holloszy, J.O.; Fontana, L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J. Am. Coll. Cardiol 2006, 47, 398–402. [Google Scholar]
- Arumugam, T.V.; Gleichmann, M.; Tang, S.C.; Mattson, M.P. Hormesis/preconditioning mechanisms, the nervous system and aging. Ageing Res. Rev 2006, 5, 165–178. [Google Scholar]
- Barja, G. Mitochondrial oxygen consumption and reactive oxygen species production are independently modulated: Implications for aging studies. Rejuvenation Res 2007, 10, 215–224. [Google Scholar]
- Hulbert, A.J.; Clancy, D.J.; Mair, W.; Braeckman, B.P.; Gems, D.; Partridge, L. Metabolic rate is not reduced by dietary-restriction or by lowered insulin/IGF-1 signalling and is not correlated with individual lifespan in Drosophila melanogaster. Exp. Gerontol 2004, 39, 1137–1143. [Google Scholar]
- Selman, C.; Phillips, T.; Staib, J.L.; Duncan, J.S.; Leeuwenburgh, C.; Speakman, J.R. Energy expenditure of calorically restricted rats is higher than predicted from their altered body composition. Mech. Ageing Dev 2005, 126, 783–793. [Google Scholar]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol 2010, 45, 410–418. [Google Scholar]
- Lin, S.J.; Kaeberlein, M.; Andalis, A.A.; Sturtz, L.A.; Defossez, P.A.; Culotta, V.C.; Fink, G.R.; Guarente, L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 2002, 418, 344–348. [Google Scholar]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 2007, 6, 280–293. [Google Scholar]
- Sharma, P.K.; Agrawal, V.; Roy, N. Mitochondria-mediated hormetic response in life span extension of calorie-restricted Saccharomyces cerevisiae. Age 2011, 33, 143–154. [Google Scholar]
- Ames, B.N. Increasing longevity by tuning up metabolism. To maximize human health and lifespan, scientists must abandon outdated models of micronutrients. EMBO Rep 2005, 6, S20–S24. [Google Scholar]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar]
- Cerqueira, F.M.; Cunha, F.M.; Laurindo, F.R.; Kowaltowski, A.J. Calorie restriction increases cerebral mitochondrial respiratory capacity in a NO*-mediated mechanism: Impact on neuronal survival. Free Radic. Biol. Med 2012, 52, 1236–1241. [Google Scholar]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol 1997, 82, 291–295. [Google Scholar]
- Mesquita, A.; Weinberger, M.; Silva, A.; Sampaio-Marques, B.; Almeida, B.; Leao, C.; Costa, V.; Rodrigues, F.; Burhans, W.C.; Ludovico, P. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc. Natl. Acad. Sci. USA 2010, 107, 15123–15128. [Google Scholar] [Green Version]
- Schulz, T.J.; Westermann, D.; Isken, F.; Voigt, A.; Laube, B.; Thierbach, R.; Kuhlow, D.; Zarse, K.; Schomburg, L.; Pfeiffer, A.F.H.; et al. Activation of mitochondrial energy metabolism protects against cardiac failure. Aging 2010, 2, 843–853. [Google Scholar]
- Warburton, D.E.; Nicol, C.W.; Bredin, S.S. Prescribing exercise as preventive therapy. CMAJ 2006, 174, 961–974. [Google Scholar]
- Hartwig, K.; Heidler, T.; Moch, J.; Daniel, H.; Wenzel, U. Feeding a ROS-generator to Caenorhabditis elegans leads to increased expression of small heat shock protein HSP-16.2 and hormesis. Genes Nutr 2009, 4, 59–67. [Google Scholar]
- Matus, S.; Castillo, K.; Hetz, C. Hormesis: Protecting neurons against cellular stress in Parkinson disease. Autophagy 2012, 8, 997–1001. [Google Scholar]
- Bonilla-Ramirez, L.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Low doses of paraquat and polyphenols prolong life span and locomotor activity in knock-down parkin Drosophila melanogaster exposed to oxidative stress stimuli: Implication in autosomal recessive juvenile Parkinsonism. Gene 2013, 512, 355–363. [Google Scholar]
- Harada, N.; Zhao, J.; Kurihara, H.; Nakagata, N.; Okajima, K. Resveratrol improves cognitive function in mice by increasing production of insulin-like growth factor-I in the hippocampus. J. Nutr. Biochem 2011, 22, 1150–1159. [Google Scholar]
- Park, H.R.; Kong, K.H.; Yu, B.P.; Mattson, M.P.; Lee, J. Resveratrol inhibits the proliferation of neural progenitor cells and hippocampal neurogenesis. J. Biol. Chem 2012, 287, 42588–42600. [Google Scholar]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem 2010, 285, 9100–9113. [Google Scholar]
- Borriello, A.; Bencivenga, D.; Caldarelli, I.; Tramontano, A.; Borgia, A.; Pirozzi, V.A.; Oliva, A.; Ragione, F.D. Resveratrol and cancer treatment: Is hormesis a yet unsolved matter? Curr. Pharm. Des. 2013, in press. [Google Scholar]
- Kouda, K.; Iki, M. Beneficial effects of mild stress (hormetic effects): Dietary restriction and health. J. Physiol. Anthropol 2010, 29, 127–132. [Google Scholar]
- Speciale, A.; Chirafisi, J.; Saija, A.; Cimino, F. Nutritional antioxidants and adaptive cell responses: An update. Curr. Mol. Med 2011, 11, 770–789. [Google Scholar]
- Menendez, J.A.; Joven, J.; Aragones, G.; Barrajon-Catalan, E.; Beltran-Debon, R.; Borras-Linares, I.; Camps, J.; Corominas-Faja, B.; Cufi, S.; Fernandez-Arroyo, S.; et al. Xenohormetic and anti-aging activity of secoiridoid polyphenols present in extra virgin olive oil: A new family of gerosuppressant agents. Cell Cycle 2013, 12, 555–578. [Google Scholar]
- Lima, D.C.; Cossa, A.C.; Perosa, S.R.; de Oliveira, E.M.; da Silva, J.A., Jr; da Silva, F.M.J.; da Silva, I.R.; da Graca, N.-M.M.; Cavalheiro, E.A. Neuroglobin is up-regulated in the cerebellum of pups exposed to maternal epileptic seizures. Int. J. Dev. Neurosci. 2011, 29, 891–897. [Google Scholar]
- Yu, Z.; Liu, N.; Liu, J.; Yang, K.; Wang, X. Neuroglobin, a Novel Target for Endogenous Neuroprotection against Stroke and Neurodegenerative Disorders. Int. J. Mol. Sci 2012, 13, 6995–7014. [Google Scholar]
- Liochev, S.I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 2013, 60. [Google Scholar] [CrossRef]
- Afanas’ev, I.B. On mechanism of superoxide signaling under physiological and pathophysiological conditions. Med. Hypotheses 2005, 64, 127–129. [Google Scholar]
- Feinendegen, L.E.; Bond, V.P.; Sondhaus, C.A.; Altman, K.I. Cellular signal adaptation with damage control at low doses versus the predominance of DNA damage at high doses. C. R. Acad. Sci. III 1999, 322, 245–251. [Google Scholar]
- Jones, S.A.; McArdle, F.; Jack, C.I.; Jackson, M.J. Effect of antioxidant supplementation on the adaptive response of human skin fibroblasts to UV-induced oxidative stress. Redox Rep 1999, 4, 291–299. [Google Scholar]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Deacylation and reacylation of neural membrane glycerophospholipids. J. Mol. Neurosci 2000, 14, 123–135. [Google Scholar]
- Semsei, I.; Rao, G.; Richardson, A. Changes in the expression of superoxide dismutase and catalase as a function of age and dietary restriction. Biochem. Biophys. Res. Commun 1989, 164, 620–625. [Google Scholar]
- Youngman, L.D.; Park, J.Y.; Ames, B.N. Protein oxidation associated with aging is reduced by dietary restriction of protein or calories. Proc. Natl. Acad. Sci. USA 1992, 89, 9112–9116. [Google Scholar]
- Fontana, L.; Vinciguerra, M.; Longo, V.D. Growth factors, nutrient signaling, and cardiovascular aging. Circ Res 2012, 110, 1139–1150. [Google Scholar]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol 2010. [Google Scholar] [CrossRef]
- Pirkkala, L.; Nykanen, P.; Sistonen, L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J 2001, 15, 1118–1131. [Google Scholar]
- Videla, L.A. Hormetic responses of thyroid hormone calorigenesis in the liver: Association with oxidative stress. IUBMB Life 2010, 62, 460–466. [Google Scholar]
- Ji, L.L.; Gomez-Cabrera, M.C.; Vina, J. Exercise and hormesis: Activation of cellular antioxidant signaling pathway. Ann. N. Y. Acad. Sci 2006, 1067, 425–435. [Google Scholar]
- Rao, G.; Xia, E.; Nadakavukaren, M.J.; Richardson, A. Effect of dietary restriction on the age-dependent changes in the expression of antioxidant enzymes in rat liver. J. Nutr 1990, 120, 602–609. [Google Scholar]
- Sreekumar, R.; Unnikrishnan, J.; Fu, A.; Nygren, J.; Short, K.R.; Schimke, J.; Barazzoni, R.; Nair, K.S. Impact of high-fat diet and antioxidant supplement on mitochondrial functions and gene transcripts in rat muscle. Am. J. Physiol. Endocrinol. Metab 2002, 282, E1055–E1061. [Google Scholar]
- Lee, S.J.; Hwang, A.B.; Kenyon, C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol 2010, 20, 2131–2136. [Google Scholar]
- Xie, M.; Roy, R. Increased levels of hydrogen peroxide induce a HIF-1-dependent modification of lipid metabolism in AMPK compromised C. elegans dauer larvae. Cell Metab 2012, 16, 322–335. [Google Scholar]
- Suzuki, K.; Kodama, S.; Watanabe, M. Extremely low-dose ionizing radiation causes activation of mitogen-activated protein kinase pathway and enhances proliferation of normal human diploid cells. Cancer Res 2001, 61, 5396–5401. [Google Scholar]
- Tsutsui, T.; Tanaka, Y.; Matsudo, Y.; Hasegawa, K.; Fujino, T.; Kodama, S.; Barrett, J.C. Extended lifespan and immortalization of human fibroblasts induced by X-ray irradiation. Mol. Carcinog 1997, 18, 7–18. [Google Scholar]
- Van Montfort, R.L.; Congreve, M.; Tisi, D.; Carr, R.; Jhoti, H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 2003, 423, 773–777. [Google Scholar]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Donate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar]
- Paulsen, C.E.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol 2012, 8, 57–64. [Google Scholar]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J. Biol. Chem 2004, 279, 25689–25695. [Google Scholar]
- Zuo, Y.; Xiang, B.; Yang, J.; Sun, X.; Wang, Y.; Cang, H.; Yi, J. Oxidative modification of caspase-9 facilitates its activation via disulfide-mediated interaction with Apaf-1. Cell Res 2009, 19, 449–457. [Google Scholar]
- Tucci, P. Caloric restriction: Is mammalian life extension linked to p53? Aging 2012, 4, 525–534. [Google Scholar]
- Salminen, A.; Kaarniranta, K. ER stress and hormetic regulation of the aging process. Ageing Res. Rev 2010, 9, 211–217. [Google Scholar]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem 2004, 279, 20108–20117. [Google Scholar]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol 2003, 23, 7198–7209. [Google Scholar]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Scheper, W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis 2012, 10, 212–215. [Google Scholar]
- Martins, I.; Galluzzi, L.; Kroemer, G. Hormesis, cell death and aging. Aging 2011, 3, 821–828. [Google Scholar]
- Szumiel, I. Radiation hormesis: Autophagy and other cellular mechanisms. Int. J. Radiat. Biol 2012, 88, 619–628. [Google Scholar]
- Eskelinen, E.L. New insights into the mechanisms of macroautophagy in mammalian cells. Int. Rev. Cell Mol. Biol 2008, 266, 207–247. [Google Scholar]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007, 26, 1749–1760. [Google Scholar]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci 2013, 14, 3834–3859. [Google Scholar]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 2010, 11, 35–46. [Google Scholar]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol 2009, 11, 1305–1314. [Google Scholar]
- Morselli, E.; Marino, G.; Bennetzen, M.V.; Eisenberg, T.; Megalou, E.; Schroeder, S.; Cabrera, S.; Bénit, P.; Rustin, P.; Criollo, A.; et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol 2011, 192, 615–629. [Google Scholar] [Green Version]
- Mattson, M.P.; Son, T.G.; Camandola, S. Viewpoint: Mechanisms of action and therapeutic potential of neurohormetic phytochemicals. Dose Response 2007, 5, 174–186. [Google Scholar]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med 2011, 51, 327–326. [Google Scholar]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 2009, 28, 3015–3026. [Google Scholar]
- Yokoo, S.; Furumoto, K.; Hiyama, E.; Miwa, N. Slow-down of age-dependent telomere shortening is executed in human skin keratinocytes by hormesis-like-effects of trace hydrogen peroxide or by anti-oxidative effects of pro-vitamin C in common concurrently with reduction of intracellular oxidative stress. J Cell. Biochem 2004, 93, 588–597. [Google Scholar]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar]
- Otani, A.; Kojima, H.; Guo, C.; Oishi, A.; Yoshimura, N. Low-dose-rate, low-dose irradiation delays neurodegeneration in a model of retinitis pigmentosa. Am. J. Pathol 2012, 180, 328–336. [Google Scholar]
- Li, N.; Stojanovski, S.; Maechler, P. Mitochondrial hormesis in pancreatic beta cells: Does uncoupling protein 2 play a role? Oxid. Med. Cell. Longev. 2012. [Google Scholar] [CrossRef]
- Duerrschmidt, N.; Hagen, A.; Gaertner, C.; Wermke, A.; Nowicki, M.; Spanel-Borowski, K.; Stepan, H.; Mohr, F.W.; Dhein, S. Nicotine effects on human endothelial intercellular communication via alpha4beta2 and alpha3beta2 nicotinic acetylcholine receptor subtypes. Naunyn Schmiedebergs Arch. Pharmacol 2012, 385, 621–632. [Google Scholar]
- Piri, M.; Nasehi, M.; Shahab, Z.; Zarrindast, M.R. The effects of nicotine on nitric oxide induced anxiogenic-like behaviors in the dorsal hippocampus. Neurosci. Lett 2012, 528, 93–98. [Google Scholar]
- Calabrese, E.J.; Mattson, M.P.; Calabrese, V. Dose response biology: The case of resveratrol. Hum. Exp. Toxicol 2010, 29, 1034–1037. [Google Scholar]
- Brink, T.C.; Demetrius, L.; Lehrach, H.; Adjaye, J. Age-related transcriptional changes in gene expression in different organs of mice support the metabolic stability theory of aging. Biogerontology 2009, 10, 549–564. [Google Scholar]
- Mao, L. Genetic Background Specific Hypoxia Resistance in Rat is Correlated with Balanced Activation of a Cross-Chromosomal Genetic Network Centering on Physiological Homeostasis. Front. Genet 2012. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Cuzzocrea, S.; Iavicoli, I.; Rizzarelli, E.; Calabrese, E.J. Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol. Aspects Med 2011, 32, 279–304. [Google Scholar]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal 2010, 13, 1763–1811. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mao, L.; Franke, J. Hormesis in Aging and Neurodegeneration—A Prodigy Awaiting Dissection. Int. J. Mol. Sci. 2013, 14, 13109-13128. https://doi.org/10.3390/ijms140713109
Mao L, Franke J. Hormesis in Aging and Neurodegeneration—A Prodigy Awaiting Dissection. International Journal of Molecular Sciences. 2013; 14(7):13109-13128. https://doi.org/10.3390/ijms140713109
Chicago/Turabian StyleMao, Lei, and Jacqueline Franke. 2013. "Hormesis in Aging and Neurodegeneration—A Prodigy Awaiting Dissection" International Journal of Molecular Sciences 14, no. 7: 13109-13128. https://doi.org/10.3390/ijms140713109
APA StyleMao, L., & Franke, J. (2013). Hormesis in Aging and Neurodegeneration—A Prodigy Awaiting Dissection. International Journal of Molecular Sciences, 14(7), 13109-13128. https://doi.org/10.3390/ijms140713109