MicroRNA Regulation in Renal Pathophysiology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
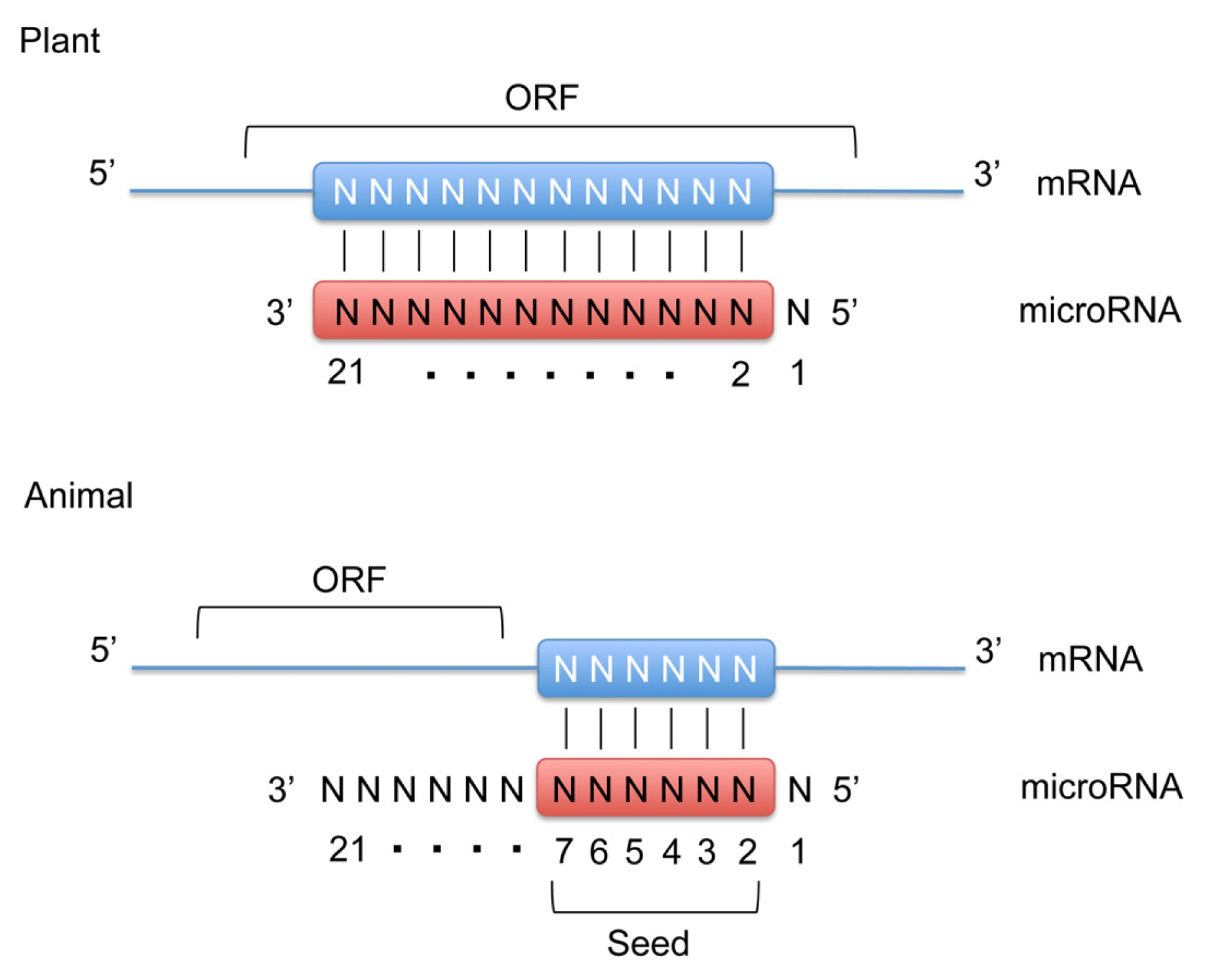
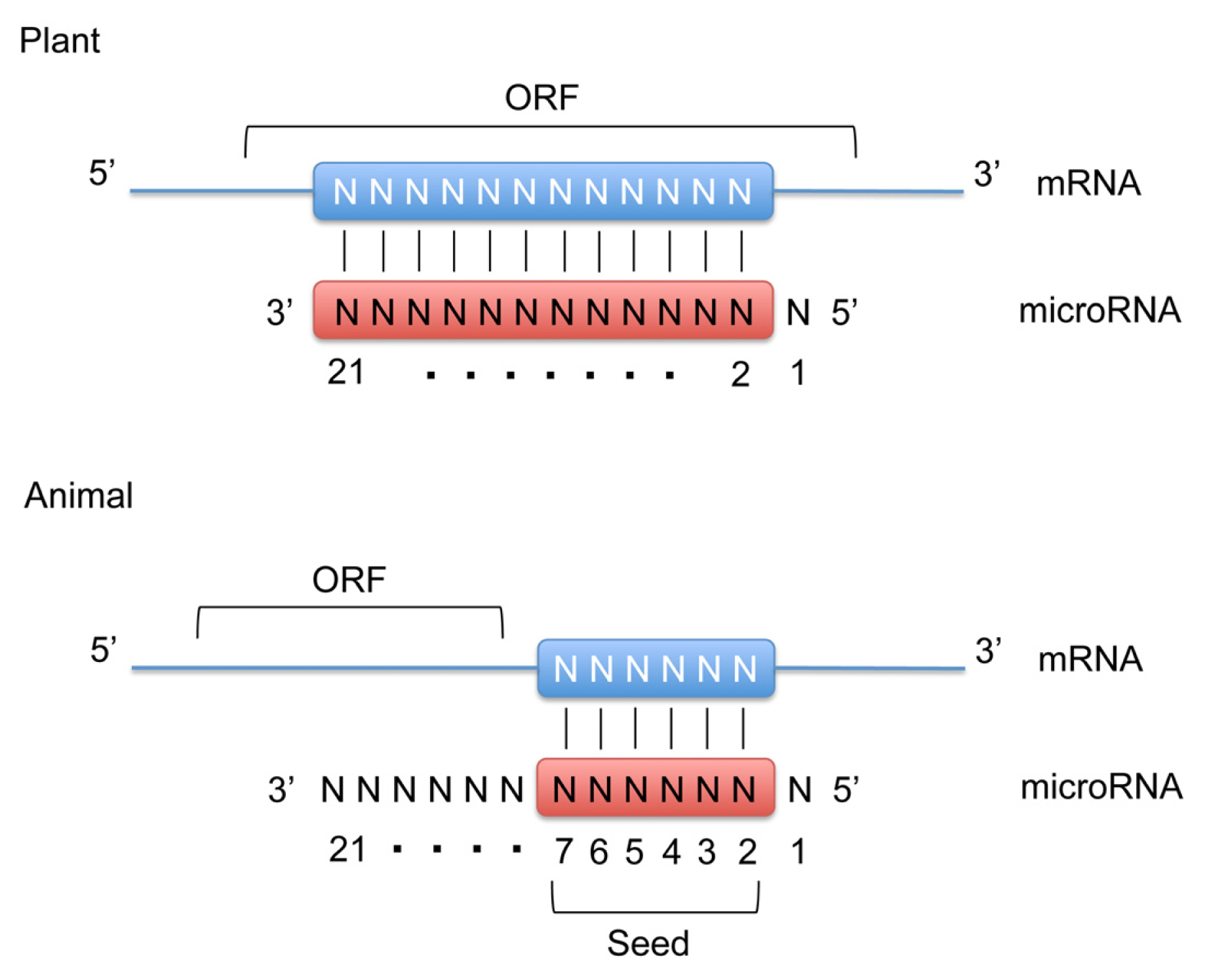
:1. Introduction
2. MicroRNA in Glomerular Diseases
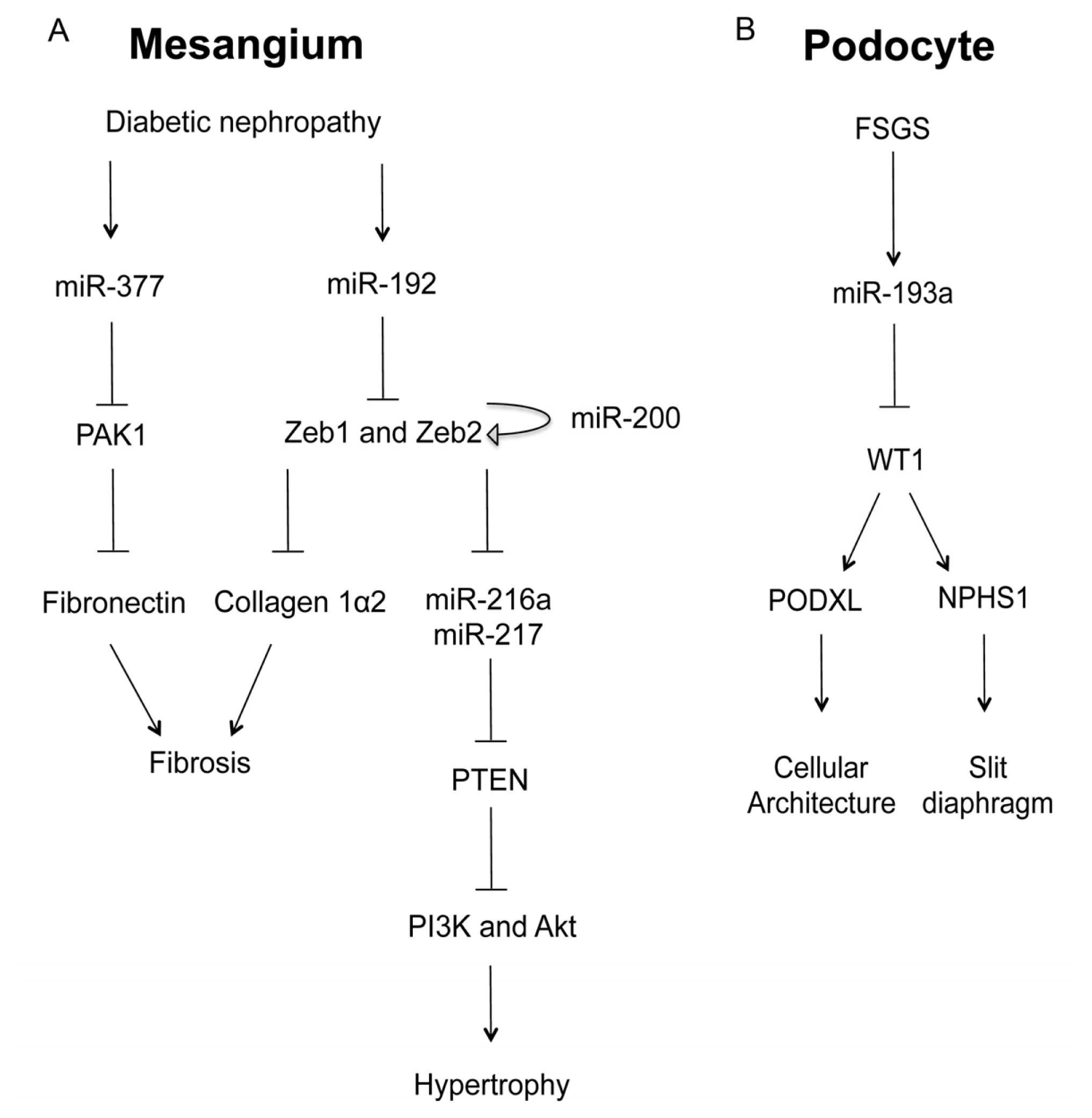
2.1. MicroRNA in Glomerular Mesangial Cell
2.2. MicroRNA in Glomerular Podocyte
3. MicroRNA in the Proximal Nephron
4. MicroRNA in the Loop of Henle
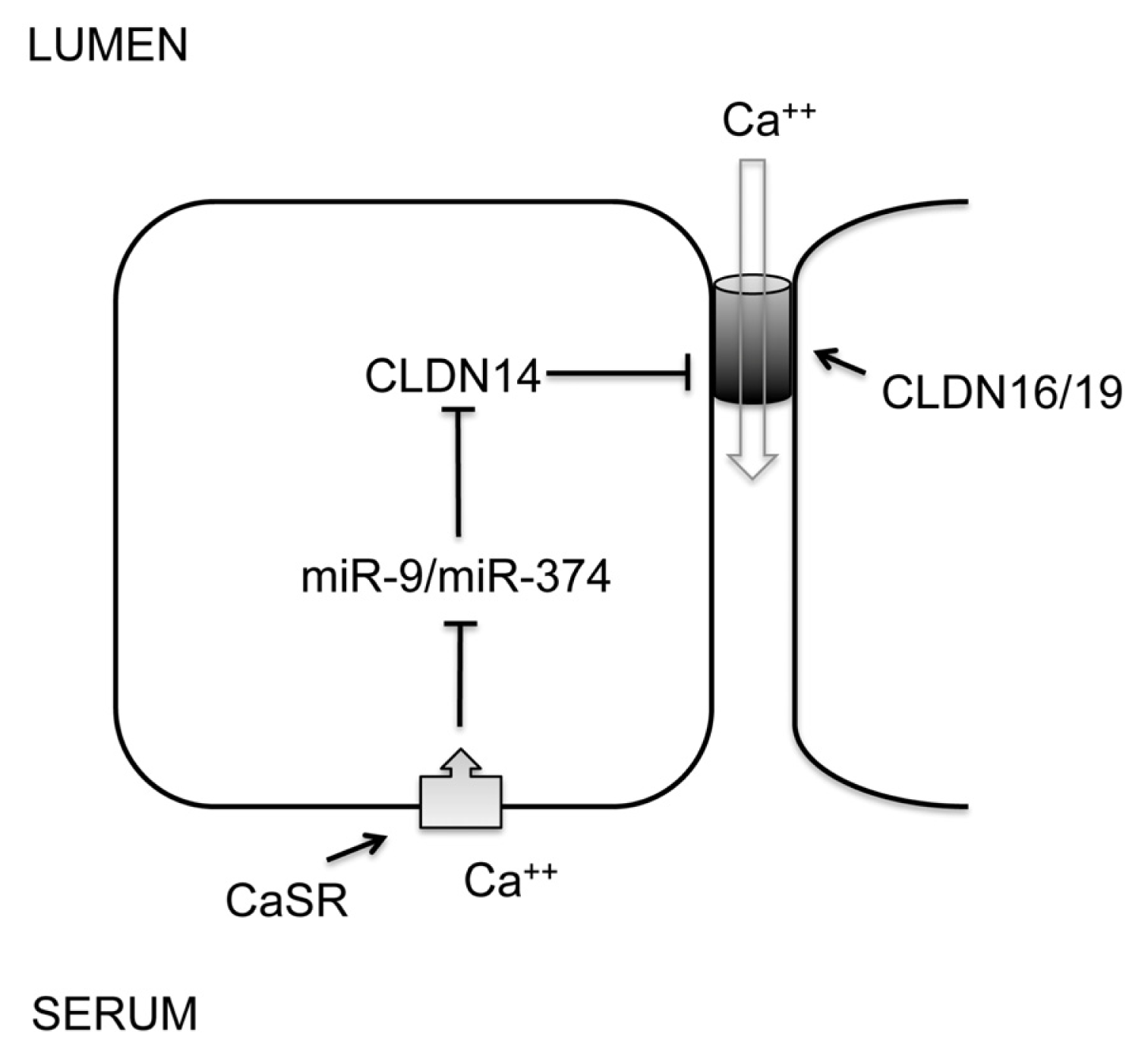
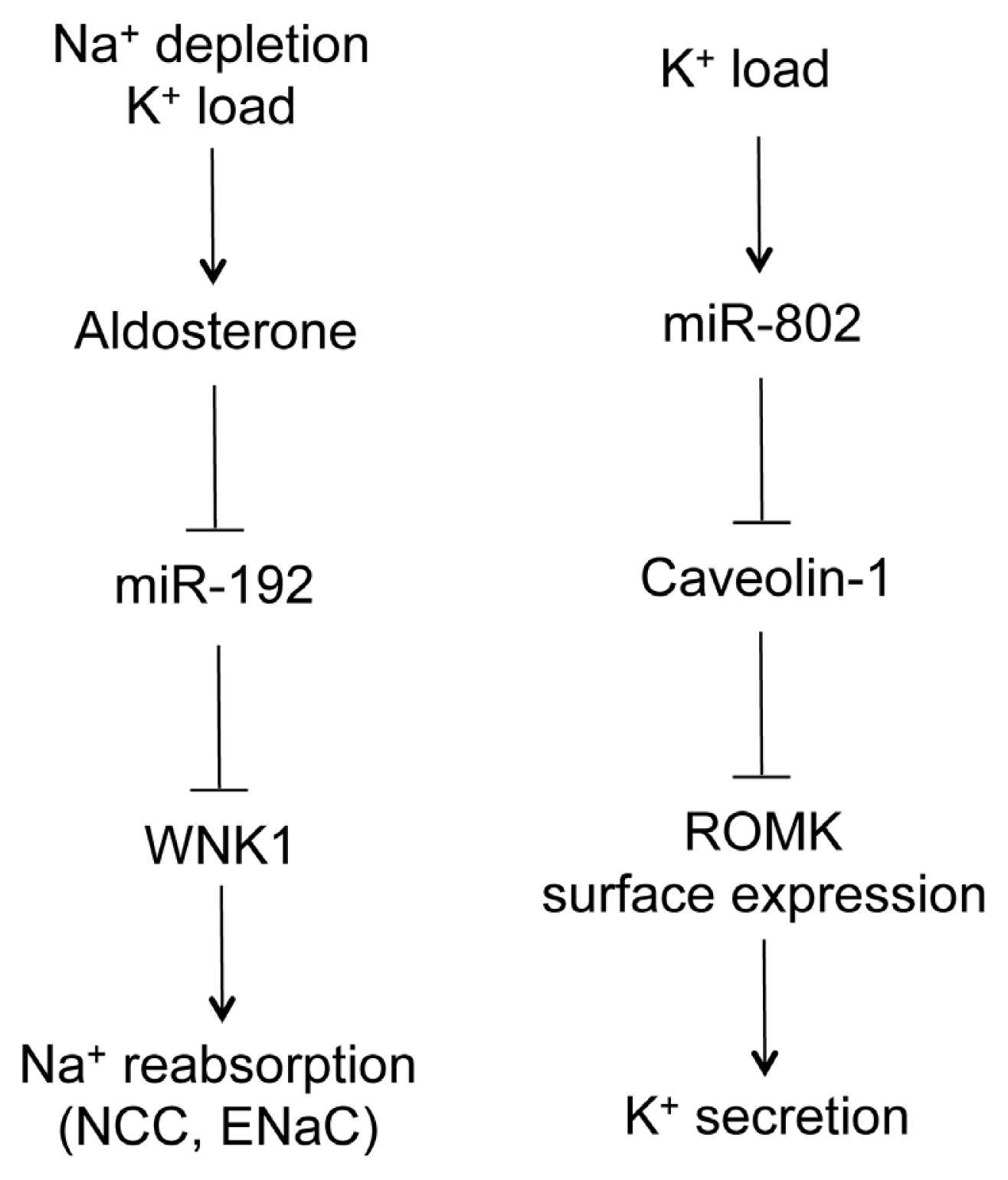
5. MicroRNA in the Distal Nephron
6. MicroRNA as Therapeutic Candidates
7. Conclusions
Acknowledgements
Conflict of interests
References
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar]
- Rajewsky, N. microRNA target predictions in animals. Nat. Genet 2006, 38, S8–S13. [Google Scholar]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet 2010, 11, 597–610. [Google Scholar]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNA. Cell 2009, 136, 669–687. [Google Scholar]
- Ho, J.; Pandey, P.; Schatton, T.; Sims-Lucas, S.; Khalid, M.; Frank, M.H.; Hartwig, S.; Kreidberg, J.A. The pro-apoptotic protein Bim is a microRNA target in kidney progenitors. J. Am. Soc. Nephrol 2011, 22, 1053–1063. [Google Scholar]
- Nagalakshmi, V.K.; Ren, Q.; Pugh, M.M.; Valerius, M.T.; McMahon, A.P.; Yu, J. Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int 2011, 79, 317–330. [Google Scholar]
- Ho, J.; Ng, K.H.; Rosen, S.; Dostal, A.; Gregory, R.I.; Kreidberg, J.A. Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J. Am. Soc. Nephrol 2008, 19, 2069–2075. [Google Scholar]
- Harvey, S.J.; Jarad, G.; Cunningham, J.; Goldberg, S.; Schermer, B.; Harfe, B.D.; McManus, M.T.; Benzing, T.; Miner, J.H. Podocyte-specificdeletionofdicer alters cytoskeletal dynamics and causes glomerular disease. J. Am. Soc. Nephrol 2008, 19, 2150–2158. [Google Scholar]
- Shi, S.; Yu, L.; Chiu, C.; Sun, Y.; Chen, J.; Khitrov, G.; Merkenschlager, M.; Holzman, L.B.; Zhang, W.; Mundel, P.; et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J. Am. Soc. Nephrol 2008, 19, 2159–2169. [Google Scholar]
- Zhdanova, O.; Srivastava, S.; Di, L.; Li, Z.; Tchelebi, L.; Dworkin, S.; Johnstone, D.B.; Zavadil, J.; Chong, M.M.; Littman, D.R.; et al. The inducible deletion of Drosha and microRNAs in mature podocytes results in a collapsing glomerulopathy. Kidney Int 2011, 80, 719–730. [Google Scholar]
- Sequeira-Lopez, M.L.; Weatherford, E.T.; Borges, G.R.; Monteagudo, M.C.; Pentz, E.S.; Harfe, B.D.; Carretero, O.; Sigmund, C.D.; Gomez, R.A. The microRNA-processing enzyme dicer maintains juxtaglomerular cells. J. Am. Soc. Nephrol 2010, 21, 460–467. [Google Scholar]
- Wei, Q.; Bhatt, K.; He, H.Z.; Mi, Q.S.; Haase, V.H.; Dong, Z. Targeted deletion of Dicer from proximal tubules protects against renal ischemia reperfusion injury. J. Am. Soc. Nephrol 2010, 21, 756–761. [Google Scholar]
- Sun, Y.; Koo, S.; White, N.; Peralta, E.; Esau, C.; Dean, N.M.; Perera, R.J. Development of a micro-array to detect human and mouse microRNAs and characterization of expression in human organs. Nucleic Acids Res 2004, 32, e188. [Google Scholar]
- Tian, Z.; Greene, A.S.; Pietrusz, J.L.; Matus, I.R.; Liang, M. MicroRNA-target pairs in the rat kidney identified by microRNA microarray, proteomic, and bioinformatics analysis. Genome Res 2008, 18, 404–411. [Google Scholar]
- Kato, M.; Zhang, J.; Wang, M.; Lanting, L.; Yuan, H.; Rossi, J.J.; Natarajan, R. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. USA 2007, 104, 3432–3437. [Google Scholar]
- Kato, M.; Putta, S.; Wang, M.; Yuan, H.; Lanting, L.; Nair, I.; Gunn, A.; Nakagawa, Y.; Shimano, H.; Todorov, I.; et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat. Cell Biol 2009, 11, 881–889. [Google Scholar]
- Jiang, B.H.; Liu, L.Z. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv. Cancer Res 2009, 102, 19–65. [Google Scholar]
- Wang, Q.; Wang, Y.; Minto, A.W.; Wang, J.; Shi, Q.; Li, X.; Quigg, R.J. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J 2008, 22, 4126–4135. [Google Scholar]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 2008, 68, 7846–7854. [Google Scholar]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 2008, 9, 582–589. [Google Scholar]
- Kato, M.; Arce, L.; Wang, M.; Putta, S.; Lanting, L.; Natarajan, R. A microRNA circuit mediates transforming growth factor-β1 autoregulation in renal glomerular mesangial cells. Kidney Int 2011, 80, 358–368. [Google Scholar]
- Wang, G.; Kwan, B.C.; Lai, F.M.; Choi, P.C.; Chow, K.M.; Li, P.K.; Szeto, C.C. Intrarenal expression of microRNAs in patients with IgA nephropathy. Lab. Invest 2010, 90, 98–103. [Google Scholar]
- Gebeshuber, C.A.; Kornauth, C.; Dong, L.; Sierig, R.; Seibler, J.; Reiss, M.; Tauber, S.; Bilban, M.; Wang, S.; Kain, R.; et al. Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1. Nat. Med 2013, 19, 481–487. [Google Scholar]
- Chen, Y.Q.; Wang, X.X.; Yao, X.M.; Zhang, D.L.; Yang, X.F.; Tian, S.F.; Wang, N.S. MicroRNA-195 promotes apoptosis in mouse podocytes via enhanced caspase activity driven by BCL2 insufficiency. Am. J. Nephrol 2011, 34, 549–559. [Google Scholar]
- Wang, B.; Komers, R.; Carew, R.; Winbanks, C.E.; Xu, B.; Herman-Edelstein, M.; Koh, P.; Thomas, M.; Jandeleit-Dahm, K.; Gregorevic, P.; et al. Suppression of microRNA-29 expression by TGF-β1 promotes collagen expression and renal fibrosis. J. Am. Soc. Nephrol 2012, 23, 252–265. [Google Scholar]
- Schrier, R.W.; Wang, W.; Poole, B.; Mitra, A. Acute renal failure: Definitions, diagnosis, pathogenesis, and therapy. J. Clin. Invest 2004, 114, 5–14. [Google Scholar]
- Godwin, J.G.; Ge, X.; Stephan, K.; Jurisch, A.; Tullius, S.G.; Iacomini, J. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc. Natl. Acad. Sci. USA 2010, 107, 14339–14344. [Google Scholar]
- Bhatt, K.; Zhou, L.; Mi, Q.S.; Huang, S.; She, J.X.; Dong, Z. MicroRNA-34a is induced via p53 during cisplatin nephrotoxicity and contributes to cell survival. Mol. Med 2010, 16, 409–416. [Google Scholar]
- Krupa, A.; Jenkins, R.; Luo, D.D.; Lewis, A.; Phillips, A.; Fraser, D. Loss of microRNA-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Nephrol 2010, 21, 438–447. [Google Scholar]
- Pollak, M.R.; Brown, E.M.; Chou, Y.H.; Hebert, S.C.; Marx, S.J.; Steinmann, B.; Levi, T.; Seidman, C.E.; Seidman, J.G. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 1993, 75, 1297–1303. [Google Scholar]
- Simon, D.B.; Lu, Y.; Choate, K.A.; Velazquez, H.; Al-Sabban, E.; Praga, M.; Casari, G.; Bettinelli, A.; Colussi, G.; Rodriguez-Soriano, J.; et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 1999, 285, 103–106. [Google Scholar]
- Konrad, M.; Schaller, A.; Seelow, D.; Pandey, A.V.; Waldegger, S.; Lesslauer, A.; Vitzthum, H.; Suzuki, Y.; Luk, J.M.; Becker, C.; et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am. J. Hum. Genet 2006, 79, 949–957. [Google Scholar]
- Thorleifsson, G.; Holm, H.; Edvardsson, V.; Walters, G.B.; Styrkarsdottir, U.; Gudbjartsson, D.F.; Sulem, P.; Halldorsson, B.V.; de Vegt, F.; d’Ancona, F.C.; et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat. Genet 2009, 41, 926–930. [Google Scholar]
- Hou, J.; Renigunta, A.; Konrad, M.; Gomes, A.S.; Schneeberger, E.E.; Paul, D.L.; Waldegger, S.; Goodenough, D.A. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J. Clin. Invest 2008, 118, 619–628. [Google Scholar]
- Gong, Y.; Renigunta, V.; Himmerkus, N.; Zhang, J.; Renigunta, A.; Bleich, M.; Hou, J. Claudin-14 regulates renal Ca++ transport in response to CaSR signalling via a novel microRNA pathway. EMBO J 2012, 31, 1999–2012. [Google Scholar]
- Weber, S.; Schneider, L.; Peters, M.; Misselwitz, J.; Rönnefarth, G.; Böswald, M.; Bonzel, K.E.; Seeman, T.; Suláková, T.; Kuwertz-Bröking, E.; et al. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J. Am. Soc. Nephrol 2001, 12, 1872–1881. [Google Scholar]
- Hou, J.; Shan, Q.; Wang, T.; Gomes, A.S.; Yan, Q.; Paul, D.L.; Bleich, M.; Goodenough, D.A. Transgenic RNAi depletion of claudin-16 and the renal handling of magnesium. J. Biol. Chem 2007, 282, 17114–17122. [Google Scholar]
- Hou, J.; Renigunta, A.; Gomes, A.S.; Hou, M.; Paul, D.L.; Waldegger, S.; Goodenough, D.A. Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc. Natl. Acad. Sci. USA 2009, 106, 15350–15355. [Google Scholar]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol 2010, 12, 247–256. [Google Scholar]
- Hu, H.; Du, L.; Nagabayashi, G.; Seeger, R.C.; Gatti, R.A. ATM is down-regulated by N-Myc-regulated microRNA-421. Proc. Natl. Acad. Sci. USA 2010, 107, 1506–1511. [Google Scholar]
- Brennan, S.C.; Thiem, U.; Roth, S.; Aggarwal, A.; Fetahu, I.S.; Tennakoon, S.; Gomes, A.R.; Brandi, M.L.; Bruggeman, F.; Mentaverri, R.; et al. Calcium sensing receptor signalling in physiology and cancer. Biochim. Biophys. Acta 2012, 1833, 1732–1744. [Google Scholar]
- Buchert, M.; Papin, M.; Bonnans, C.; Darido, C.; Raye, W.S.; Garambois, V.; Pélegrin, A.; Bourgaux, J.F.; Pannequin, J.; Joubert, D.; et al. Symplekin promotes tumorigenicity by up-regulating claudin-2 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2628–2633. [Google Scholar]
- Nübel, T.; Preobraschenski, J.; Tuncay, H.; Weiss, T.; Kuhn, S.; Ladwein, M.; Langbein, L.; Zöller, M. Claudin-7 regulates EpCAM-mediated functions in tumor progression. Mol. Cancer Res 2009, 7, 285–299. [Google Scholar]
- Mladinov, D.; Liu, Y.; Mattson, D.L.; Liang, M. MicroRNAs contribute to the maintenance of cell-type-specific physiological characteristics: miR-192 targets Na+/K+-ATPase β1. Nucleic Acids Res 2013, 41, 1273–1283. [Google Scholar]
- Guyton, A.C. Blood pressure control—Special role of the kidneys and body fluids. Science 1991, 252, 1813–1816. [Google Scholar]
- Elvira-Matelot, E.; Zhou, X.O.; Farman, N.; Beaurain, G.; Henrion-Caude, A.; Hadchouel, J.; Jeunemaitre, X. Regulation of WNK1 expression by miR-192 and aldosterone. J. Am. Soc. Nephrol 2010, 21, 1724–1731. [Google Scholar]
- McCormick, J.A.; Yang, C.L.; Ellison, D.H. WNK kinases and renal sodium transport in health and disease: An integrated view. Hypertension 2008, 51, 588–596. [Google Scholar]
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; et al. Human hypertension caused by mutations in WNK kinases. Science 2001, 293, 1107–1112. [Google Scholar]
- Lin, D.H.; Yue, P.; Pan, C.; Sun, P.; Wang, W.H. MicroRNA 802 stimulates ROMK channels by suppressing caveolin-1. J. Am. Soc. Nephrol 2011, 22, 1087–1098. [Google Scholar]
- Huang, W.; Liu, H.; Wang, T.; Zhang, T.; Kuang, J.; Luo, Y.; Chung, S.S.; Yuan, L.; Yang, J.Y. Tonicity-responsive microRNAs contribute to the maximal induction of osmoregulatory transcription factor OREBP in response to high-NaCl hypertonicity. Nucleic Acids Res 2011, 39, 475–485. [Google Scholar]
- Burg, M.B.; Ferraris, J.D.; Dimitrieva, N.I. Cellular response to hyperosmotic stresses. Physiol. Rev 2007, 87, 1441–1474. [Google Scholar]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar]
- Obad, S.; dos Santos, C.O.; Petri, A.; Heidenblad, M.; Broom, O.; Ruse, C.; Fu, C.; Lindow, M.; Stenvang, J.; Straarup, E.M.; et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet 2011, 43, 371–378. [Google Scholar]
- Castoldi, M.; Vujic Spasic, M.; Altamura, S.; Elmén, J.; Lindow, M.; Kiss, J.; Stolte, J.; Sparla, R.; D’Alessandro, L.A.; Klingmüller, U.; et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J. Clin. Invest 2011, 121, 1386–1396. [Google Scholar]
- Yi, R.; Poy, M.N.; Stoffel, M.; Fuchs, E. A skin microRNA promotes differentiation by repressing “stemness”. Nature 2008, 452, 225–229. [Google Scholar]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med 2010, 207, 1589–1597. [Google Scholar]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar]
- Putta, S.; Lanting, L.; Sun, G.; Lawson, G.; Kato, M.; Natarajan, R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol 2012, 23, 458–469. [Google Scholar]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar]
- Krützfeldt, J.; Kuwajima, S.; Braich, R.; Rajeev, K.G.; Pena, J.; Tuschl, T.; Manoharan, M.; Stoffel, M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res 2007, 35, 2885–2892. [Google Scholar]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar]
- Lorenzen, J.M.; Thum, T. Circulating and urinary microRNAs in kidney disease. Clin. J. Am. Soc. Nephrol 2012, 7, 1528–1533. [Google Scholar]



© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hou, J.; Zhao, D. MicroRNA Regulation in Renal Pathophysiology. Int. J. Mol. Sci. 2013, 14, 13078-13092. https://doi.org/10.3390/ijms140713078
Hou J, Zhao D. MicroRNA Regulation in Renal Pathophysiology. International Journal of Molecular Sciences. 2013; 14(7):13078-13092. https://doi.org/10.3390/ijms140713078
Chicago/Turabian StyleHou, Jianghui, and Dan Zhao. 2013. "MicroRNA Regulation in Renal Pathophysiology" International Journal of Molecular Sciences 14, no. 7: 13078-13092. https://doi.org/10.3390/ijms140713078
APA StyleHou, J., & Zhao, D. (2013). MicroRNA Regulation in Renal Pathophysiology. International Journal of Molecular Sciences, 14(7), 13078-13092. https://doi.org/10.3390/ijms140713078