Targeting Alternative Sites on the Androgen Receptor to Treat Castration-Resistant Prostate Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
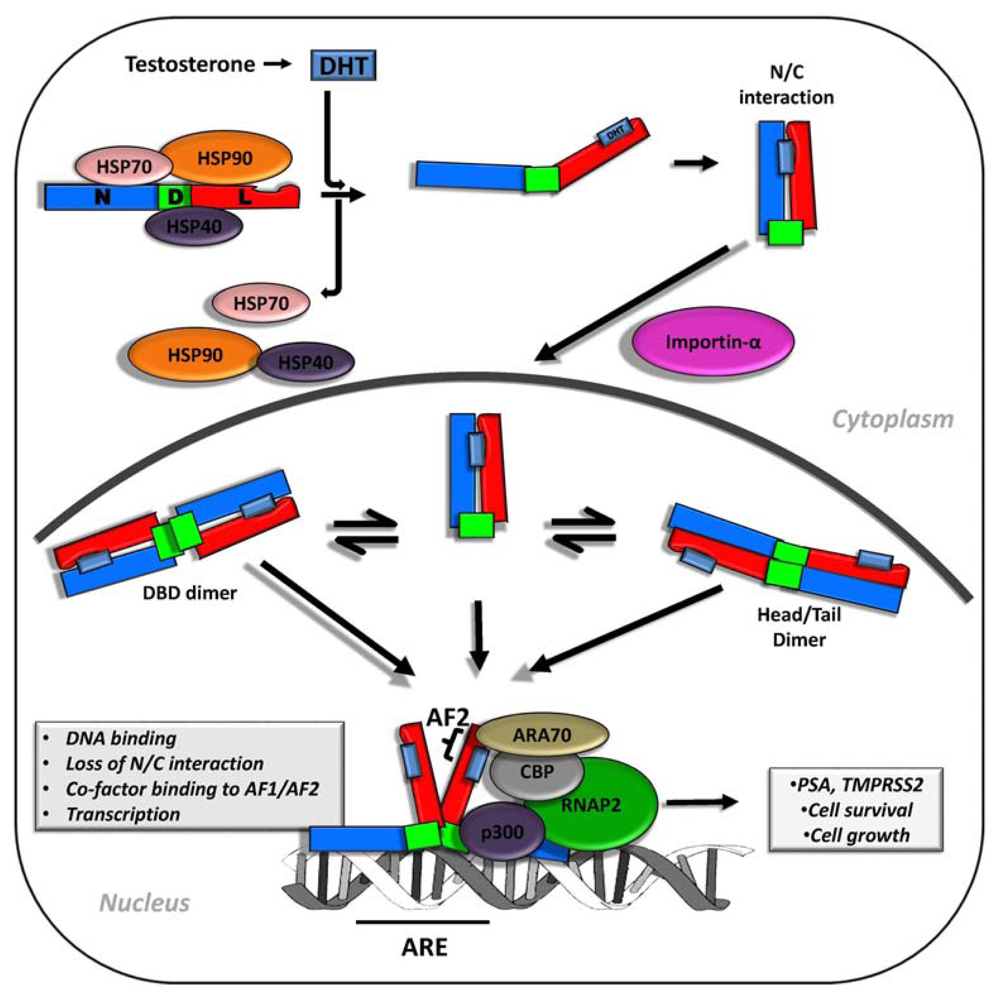
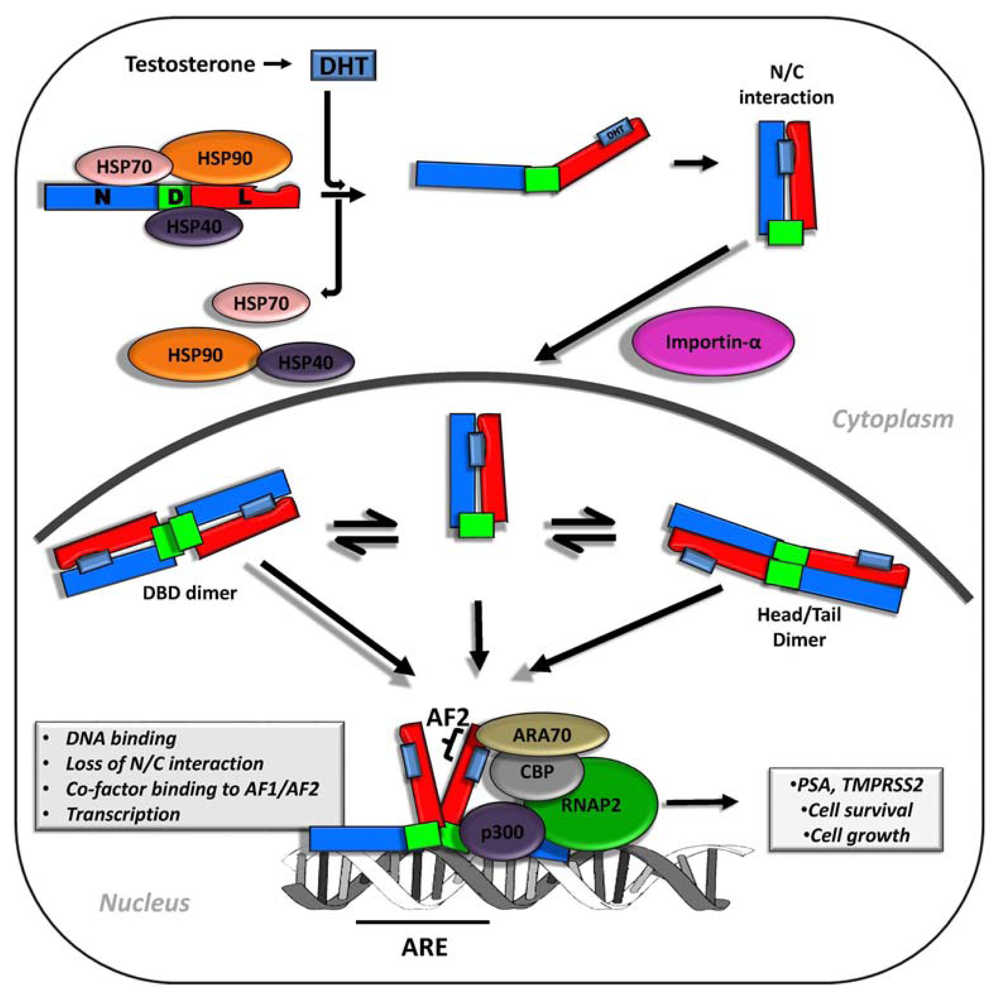
2. Androgen Signaling and AR Function in Early and Advanced Prostate Cancer
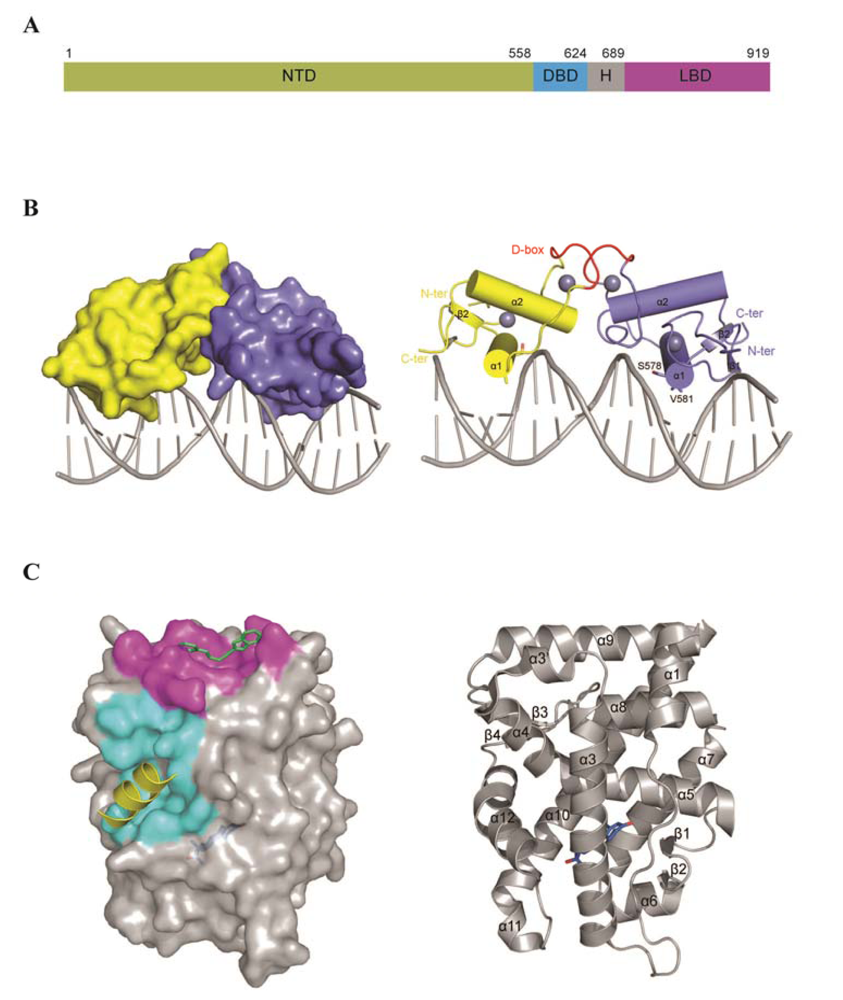
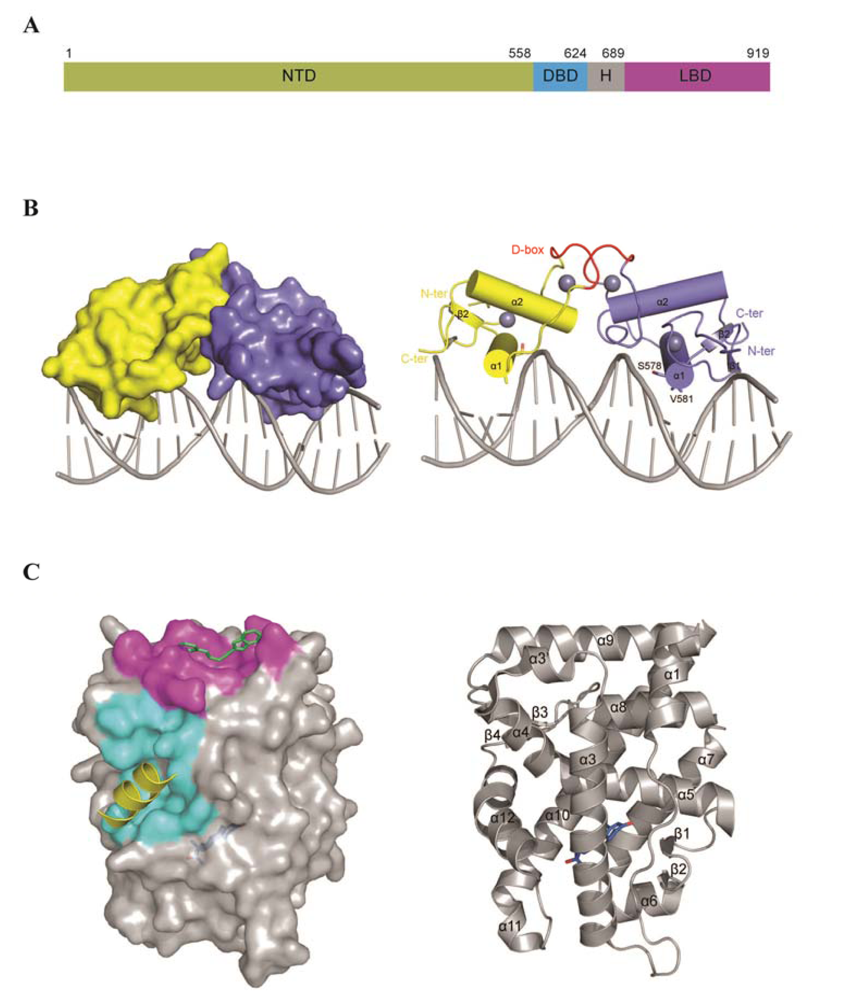
3. AR Structure and Domain Organization
3.1. N-Terminus Domain (NTD)
3.2. The DNA Binding Domain (DBD)
3.3. Hinge Region
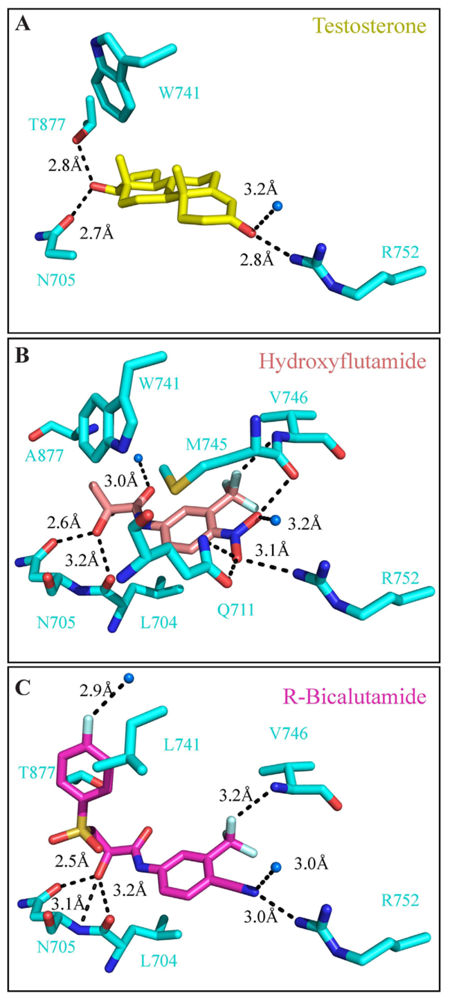
3.4. Ligand Binding Domain (LBD)
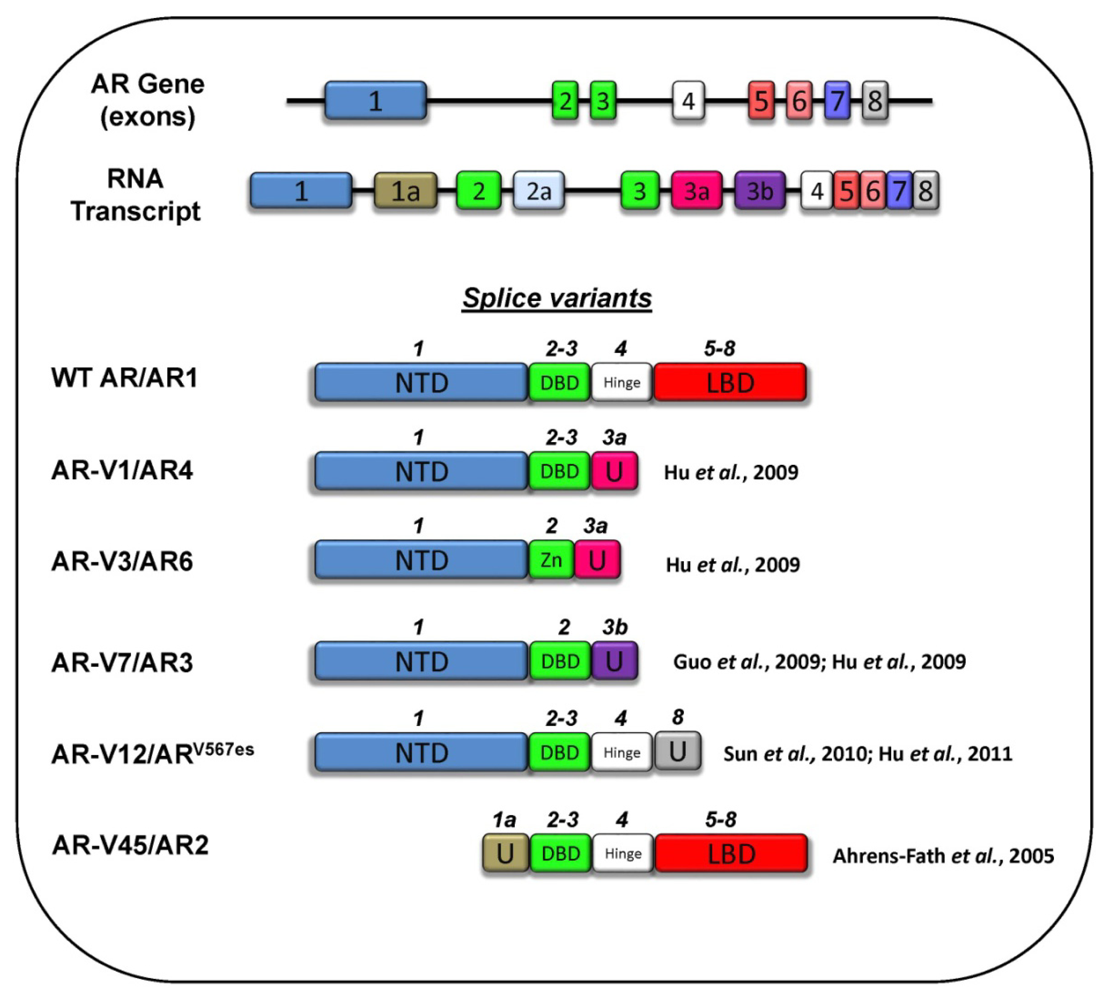
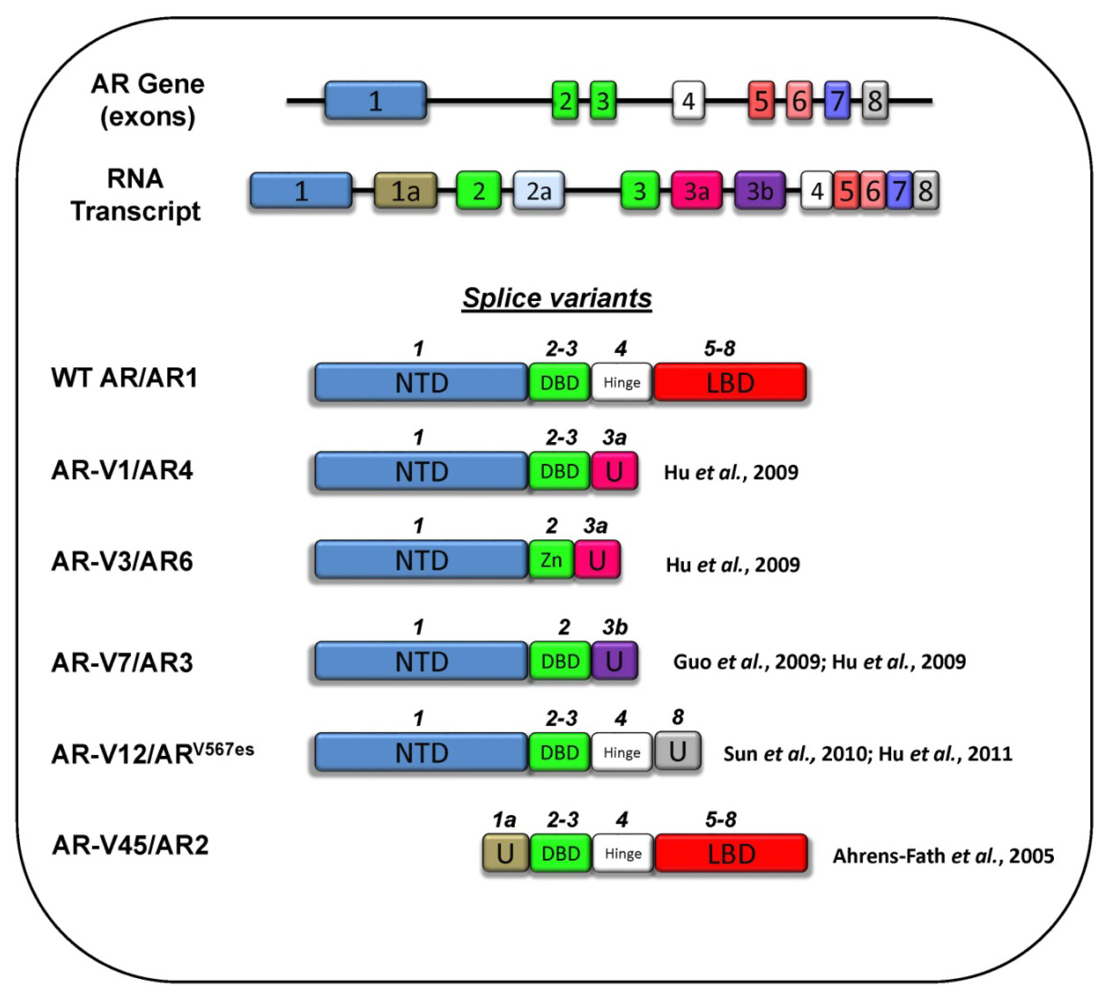
3.5 Splice Variants
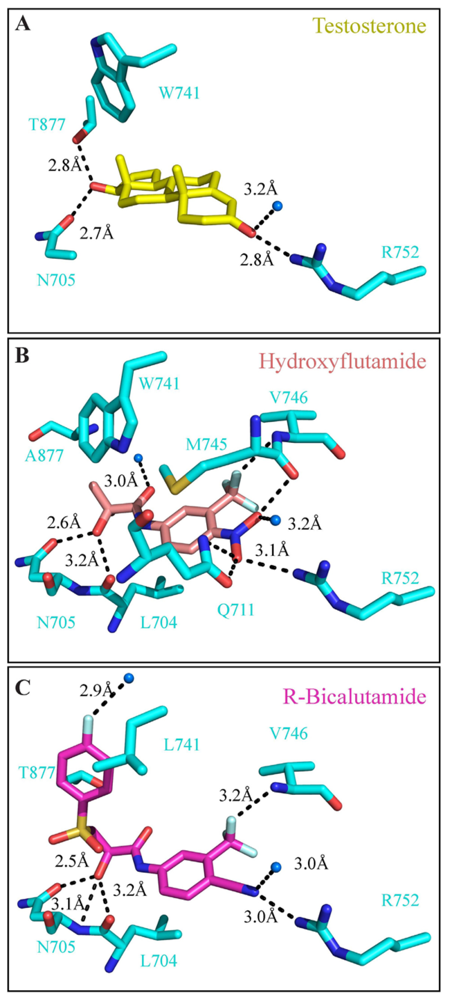
4. Targeting the LBD
5. Targeting the N-Terminal Domain
6. Targeting the DNA Binding Domain
7. Future Outlook
Acknowledgements
Conflict of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin 2013, 63, 11–30. [Google Scholar]
- Green, S.M.; Mostaghel, E.A.; Nelson, P.S. Androgen action and metabolism in prostate cancer. Mol. Cell. Endocrinol 2012, 360, 3–13. [Google Scholar]
- Sampson, N.; Neuwirt, H.; Puhr, M.; Klocker, H.; Eder, I.E. In vitro model systems to study androgen receptor signaling in prostate cancer. Endocr. Relat. Cancer 2013. [Google Scholar] [CrossRef]
- Hiipakka, R.A.; Liao, S. Molecular mechanism of androgen action. Trends Endocrinol. Metab 1998, 9, 317–324. [Google Scholar]
- Jenster, G.; van der Korput, H.A.; van Vroonhoven, C.; van der Kwast, T.H.; Trapman, J.; Brinkmann, A.O. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol. Endocrinol 1991, 5, 1396–1404. [Google Scholar]
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of nomenclature of nuclear receptors. Pharmacol. Rev 2006, 58, 685–704. [Google Scholar]
- Eick, G.N.; Thornton, J.W. Evolution of steroid receptors from an estrogen-sensitive ancestral receptor. Mol. Cell. Endocrinol 2011, 334, 31–38. [Google Scholar]
- Lamont, K.R.; Tindall, D.J. Androgen regulation of gene expression. Adv. Cancer Res 2010, 107, 137–162. [Google Scholar]
- Saad, F.; Miller, K. Treatment options in castration-resistant prostate cancer: Current therapies and emerging docetaxel-based regimens. Urol. Oncol. 2013. [Google Scholar] [CrossRef]
- Lack, N.A.; Axerio-Cilies, P.; Tavassoli, P.; Han, F.Q.; Chan, K.H.; Feau, C.; LeBlanc, E.; Guns, E.T.; Guy, R.K.; Rennie, P.S.; et al. Targeting the binding function 3 (BF3) site of the human androgen receptor through virtual screening. J. Med. Chem 2011, 54, 8563–8573. [Google Scholar]
- Shen, H.C.; Shanmugasundaram, K.; Simon, N.I.; Cai, C.; Wang, H.; Chen, S.; Balk, S.P.; Rigby, A.C. In silico discovery of androgen receptor antagonists with activity in castration resistant prostate cancer. Mol. Endocrinol 2012, 26, 1836–1846. [Google Scholar]
- Song, C.H.; Yang, S.H.; Park, E.; Cho, S.H.; Gong, E.Y.; Khadka, D.B.; Cho, W.J.; Lee, K. Structure-based virtual screening and identification of a novel androgen receptor antagonist. J. Biol. Chem 2012, 287, 30769–30780. [Google Scholar]
- Munuganti, R.S.; Leblanc, E.; Axerio-Cilies, P.; Labriere, C.; Frewin, K.; Singh, K.; Hassona, M.D.; Lack, N.A.; Li, H.; Ban, F.; et al. Targeting the binding function 3 (BF3) site of the androgen receptor through virtual screening. 2. Development of 2-((2-phenoxyethyl) thio)-1H-benzimidazole derivatives. J. Med. Chem 2013, 56, 1136–1148. [Google Scholar]
- Yang, F.; Nickols, N.G.; Li, B.C.; Marinov, G.K.; Said, J.W.; Dervan, P.B. Antitumor activity of a pyrrole-imidazole polyamide. Proc. Natl. Acad. Sci. USA 2013, 110, 1863–1868. [Google Scholar]
- Voet, A.; Helsen, C.; Zhang, K.Y.; Claessens, F. The discovery of novel human androgen receptor antagonist chemotypes using a combined pharmacophore screening procedure. Chem. Med. Chem 2013, 8, 644–651. [Google Scholar]
- Caboni, L.; Lloyd, D.G. Beyond the ligand-binding pocket: Targeting alternate sites in nuclear receptors. Med. Res. Rev. 2012. [Google Scholar] [CrossRef]
- Mohler, J.L.; Gregory, C.W.; Ford, O.H., 3rd; Kim, D.; Weaver, C.M.; Petrusz, P.; Wilson, E.M.; French, F.S. The androgen axis in recurrent prostate cancer. Clin. Cancer Res 2004, 10, 440–448. [Google Scholar]
- Azzouni, F.; Mohler, J. Biology of castration-recurrent prostate cancer. Urol. Clin 2012, 39, 435–452. [Google Scholar]
- Prescott, J.; Coetzee, G.A. Molecular chaperones throughout the life cycle of the androgen receptor. Cancer Lett 2006, 231, 12–19. [Google Scholar]
- Eichholz, A.; Ferraldeschi, R.; Attard, G.; de Bono, J.S. Putting the brakes on continued androgen receptor signaling in castration-resistant prostate cancer. Mol. Cell. Endocrinol 2012, 360, 68–75. [Google Scholar]
- Centenera, M.M.; Fitzpatrick, A.K.; Tilley, W.D.; Butler, L.M. Hsp90: Still a viable target in prostate cancer. Biochim. Biophys. Acta 2013, 1835, 211–218. [Google Scholar]
- Cutress, M.L.; Whitaker, H.C.; Mills, I.G.; Stewart, M.; Neal, D.E. Structural basis for the nuclear import of the human androgen receptor. J. Cell Sci 2008, 121, 957–968. [Google Scholar]
- Kaku, N.; Matsuda, K.; Tsujimura, A.; Kawata, M. Characterization of nuclear import of the domain-specific androgen receptor in association with the importin alpha/beta and Ran-guanosine 5′-triphosphate systems. Endocrinology 2008, 149, 3960–3969. [Google Scholar]
- Van Royen, M.E.; van Cappellen, W.A.; de Vos, C.; Houtsmuller, A.B.; Trapman, J. Stepwise androgen receptor dimerization. J. Cell Sci 2012, 125, 1970–1979. [Google Scholar]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev 2004, 25, 276–308. [Google Scholar]
- Van der Sluis, T.M.; van Moorselaar, R.J.; Meuleman, E.J.; Ter Haar, R.W.; Bui, H.N.; Heijboer, A.C.; Vis, A.N. Relationship between body mass index and serum testosterone Concentration in patients receiving luteinizing hormone-releasing hormone agonist therapy for prostate cancer. Urology 2013, 81, 1005–1009. [Google Scholar]
- Tammela, T.L. Endocrine prevention and treatment of prostate cancer. Mol. Cell. Endocrinol 2012, 360, 59–67. [Google Scholar]
- Liu, S.V.; Liu, S.; Pinski, J. Luteinizing hormone-releasing hormone receptor targeted agents for prostate cancer. Expert Opin. Investig. Drugs 2011, 20, 769–778. [Google Scholar]
- Friedlander, T.W.; Ryan, C.J. Targeting the androgen receptor. Urol. Clin 2012, 39, 453–464. [Google Scholar]
- Sadar, M.D. Advances in small molecule inhibitors of androgen receptor for the treatment of advanced prostate cancer. World J. Urol 2012, 30, 311–318. [Google Scholar]
- Ferraldeschi, R.; de Bono, J. Agents that target androgen synthesis in castration-resistant prostate cancer. Cancer J 2013, 19, 34–42. [Google Scholar]
- Lim, A.C.; Attard, G. Improved therapeutic targeting of the androgen receptor: Rational drug design improves survival in castration-resistant prostate cancer. Curr. Drug Targets 2013, 14, 408–419. [Google Scholar]
- Pont, A.; Williams, P.L.; Azhar, S.; Reitz, R.E.; Bochra, C.; Smith, E.R.; Stevens, D.A. Ketoconazole blocks testosterone synthesis. Arch. Intern. Med 1982, 142, 2137–2140. [Google Scholar]
- Lamberts, S.W.; Bons, E.G.; Bruining, H.A.; de Jong, F.H. Differential effects of the imidazole derivatives etomidate, ketoconazole and miconazole and of metyrapone on the secretion of cortisol and its precursors by human adrenocortical cells. J. Pharmacol. Exp. Ther 1987, 240, 259–264. [Google Scholar]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol 2012, 13, 983–992. [Google Scholar]
- Zhu, H.; Garcia, J.A. Targeting the adrenal gland in castration-resistant prostate cancer: A case for orteronel, a selective CYP-17 17,20-lyase inhibitor. Curr. Oncol. Rep 2013, 15, 105–112. [Google Scholar]
- Bruno, R.D.; Gover, T.D.; Burger, A.M.; Brodie, A.M.; Njar, V.C. 17alpha-Hydroxylase/17,20 lyase inhibitor VN/124–1 inhibits growth of androgen-independent prostate cancer cells via induction of the endoplasmic reticulum stress response. Mol. Cancer Ther 2008, 7, 2828–2836. [Google Scholar]
- Ferraldeschi, R.; Sharifi, N.; Auchus, R.J.; Attard, G. Molecular pathways: Inhibiting steroid biosynthesis in prostate cancer. Clin. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar]
- Schweizer, M.T.; Antonarakis, E.S. Abiraterone and other novel androgen-directed strategies for the treatment of prostate cancer: A new era of hormonal therapies is born. Ther. Adv. Urol 2012, 4, 167–178. [Google Scholar]
- Rathkopf, D.; Scher, H.I. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J 2013, 19, 43–49. [Google Scholar]
- Liao, X.; Tang, S.; Thrasher, J.B.; Griebling, T.L.; Li, B. Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Mol. Cancer. Ther 2005, 4, 505–515. [Google Scholar]
- Cheng, H.; Snoek, R.; Ghaidi, F.; Cox, M.E.; Rennie, P.S. Short hairpin RNA knockdown of the androgen receptor attenuates ligand-independent activation and delays tumor progression. Cancer Res 2006, 66, 10613–10620. [Google Scholar]
- Snoek, R.; Cheng, H.; Margiotti, K.; Wafa, L.A.; Wong, C.A.; Wong, E.C.; Fazli, L.; Nelson, C.C.; Gleave, M.E.; Rennie, P.S. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin. Cancer Res 2009, 15, 39–47. [Google Scholar]
- Lee, J.B.; Zhang, K.; Tam, Y.Y.; Tam, Y.K.; Belliveau, N.M.; Sung, V.Y.; Lin, P.J.; LeBlanc, E.; Ciufolini, M.A.; Rennie, P.S.; et al. Lipid nanoparticle sirna systems for silencing the androgen receptor in human prostate cancer in vivo. Int. J. Cancer 2012, 131, E781–790. [Google Scholar]
- Sternberg, C.N.; Petrylak, D.P.; Sartor, O.; Witjes, J.A.; Demkow, T.; Ferrero, J.M.; Eymard, J.C.; Falcon, S.; Calabro, F.; James, N.; et al. Multinational, double-blind, phase iii study of prednisone and either satraplatin or placebo in patients with castrate-refractory prostate cancer progressing after prior chemotherapy: The sparc trial. J. Clin. Oncol 2009, 27, 5431–5438. [Google Scholar]
- Molina, A.; Belldegrun, A. Novel therapeutic strategies for castration resistant prostate cancer: Inhibition of persistent androgen production and androgen receptor mediated signaling. J. Urol 2011, 185, 787–794. [Google Scholar]
- Cheng, H.H.; Lin, D.W.; Yu, E.Y. Advanced clinical states in prostate cancer. Urol. Clin 2012, 39, 561–571. [Google Scholar]
- Loriot, Y.; Zoubeidi, A.; Gleave, M.E. Targeted therapies in metastatic castration-resistant prostate cancer: Beyond the androgen receptor. Urol. Clin 2012, 39, 517–531. [Google Scholar]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med 2004, 10, 33–39. [Google Scholar]
- Taplin, M.E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med 1995, 332, 1393–1398. [Google Scholar]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 2006, 66, 2815–2825. [Google Scholar]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res 2008, 68, 4447–4454. [Google Scholar]
- Nyquist, M.D.; Dehm, S.M. Interplay between genomic alterations and androgen receptor signaling during prostate cancer development and progression. Horm. Cancer 2013, 4, 61–69. [Google Scholar]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar]
- Brand, L.J.; Dehm, S.M. Androgen receptor gene rearrangements: New perspectives on prostate cancer progression. Curr. Drug Targets 2013, 14, 441–449. [Google Scholar]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–196. [Google Scholar]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res 2013, 73, 483–489. [Google Scholar]
- Cao, B.; Liu, X.; Li, J.; Liu, S.; Qi, Y.; Xiong, Z.; Zhang, A.; Wiese, T.; Fu, X.; Gu, J.; et al. 20(S)-protopanaxadiol-aglycone downregulation of the full-length and splice variants of androgen receptor. Int. J. Cancer 2013, 132, 1277–1287. [Google Scholar]
- Lamont, K.R.; Tindall, D.J. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer. Mol. Endocrinol 2011, 25, 897–907. [Google Scholar]
- Rice, P.; Longden, I.; Bleasby, A. Emboss: The european molecular biology open software suite. Trends Genet 2000, 16, 276–277. [Google Scholar]
- Robins, D.M.; Albertelli, M.A.; O’Mahony, O.A. Androgen receptor variants and prostate cancer in humanized AR mice. J. Steroid Biochem 2008, 108, 230–236. [Google Scholar]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in x-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar]
- Choong, C.S.; Kemppainen, J.A.; Wilson, E.M. Evolution of the primate androgen receptor: A structural basis for disease. J. Mol. Evol 1998, 47, 334–342. [Google Scholar]
- Davies, P.; Watt, K.; Kelly, S.M.; Clark, C.; Price, N.C.; McEwan, I.J. Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J. Mol. Endocrinol 2008, 41, 301–314. [Google Scholar]
- Betney, R.; McEwan, I.J. Role of conserved hydrophobic amino acids in androgen receptor AF-1 function. J. Mol. Endocrinol 2003, 31, 427–439. [Google Scholar]
- Jenster, G.; van der Korput, H.A.; Trapman, J.; Brinkmann, A.O. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J. Biol. Chem 1995, 270, 7341–7346. [Google Scholar]
- Reid, J.; Betney, R.; Watt, K.; McEwan, I.J. The androgen receptor transactivation domain: The interplay between protein conformation and protein-protein interactions. Biochem. Soc. Trans 2003, 31, 1042–1046. [Google Scholar]
- He, B.; Kemppainen, J.A.; Wilson, E.M. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem 2000, 275, 22986–22994. [Google Scholar]
- He, B.; Wilson, E.M. The NH(2)-terminal and carboxyl-terminal interaction in the human androgen receptor. Mol. Genet. Metab 2002, 75, 293–298. [Google Scholar]
- McEwan, I.J. Molecular mechanisms of androgen receptor-mediated gene regulation: Structure-function analysis of the AF-1 domain. Endocr. Relat. Cancer 2004, 11, 281–293. [Google Scholar]
- McEwan, I.J. Intrinsic disorder in the androgen receptor: Identification, characterisation and drugability. Mol. Biosyst 2012, 8, 82–90. [Google Scholar]
- McEwan, I.J.; Gustafsson, J. Interaction of the human androgen receptor transactivation function with the general transcription factor TFIIF. Proc. Natl. Acad. Sci. USA 1997, 94, 8485–8490. [Google Scholar]
- Reid, J.; Murray, I.; Watt, K.; Betney, R.; McEwan, I.J. The androgen receptor interacts with multiple regions of the large subunit of general transcription factor TFIIF. J. Biol. Chem 2002, 277, 41247–41253. [Google Scholar]
- Lee, D.K.; Duan, H.O.; Chang, C. From androgen receptor to the general transcription factor tfiih. Identification of cdk activating kinase (CAK) as an androgen receptor NH(2)-terminal associated coactivator. J. Biol. Chem 2000, 275, 9308–9313. [Google Scholar]
- Fronsdal, K.; Engedal, N.; Slagsvold, T.; Saatcioglu, F. CREB binding protein is a coactivator for the androgen receptor and mediates cross-talk with AP-1. J. Biol. Chem 1998, 273, 31853–31859. [Google Scholar]
- Aarnisalo, P.; Palvimo, J.J.; Janne, O.A. CREB-binding protein in androgen receptor-mediated signaling. Proc. Natl. Acad. Sci. USA 1998, 95, 2122–2127. [Google Scholar]
- Dotzlaw, H.; Moehren, U.; Mink, S.; Cato, A.C.; Iniguez Lluhi, J.A.; Baniahmad, A. The amino terminus of the human AR is target for corepressor action and antihormone agonism. Mol. Endocrinol 2002, 16, 661–673. [Google Scholar]
- Umesono, K.; Evans, R.M. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell 1989, 57, 1139–1146. [Google Scholar]
- Claessens, F.; Alen, P.; Devos, A.; Peeters, B.; Verhoeven, G.; Rombauts, W. The androgen-specific probasin response element 2 interacts differentially with androgen and glucocorticoid receptors. J. Biol. Chem 1996, 271, 19013–19016. [Google Scholar]
- Schoenmakers, E.; Alen, P.; Verrijdt, G.; Peeters, B.; Verhoeven, G.; Rombauts, W.; Claessens, F. Differential DNA binding by the androgen and glucocorticoid receptors involves the second Zn-finger and a C-terminal extension of the DNA-binding domains. Biochem. J 1999, 341, 515–521. [Google Scholar]
- Denayer, S.; Helsen, C.; Thorrez, L.; Haelens, A.; Claessens, F. The rules of DNA recognition by the androgen receptor. Mol. Endocrinol 2010, 24, 898–913. [Google Scholar]
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T. Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. USA 2004, 101, 4758–4763. [Google Scholar]
- Zhou, Z.X.; Sar, M.; Simental, J.A.; Lane, M.V.; Wilson, E.M. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. J. Biol. Chem 1994, 269, 13115–13123. [Google Scholar]
- Jenster, G.; Trapman, J.; Brinkmann, A.O. Nuclear import of the human androgen receptor. Biochem. J 1993, 293, 761–768. [Google Scholar]
- Clinckemalie, L.; Vanderschueren, D.; Boonen, S.; Claessens, F. The hinge region in androgen receptor control. Mol. Cell. Endocrinol 2012, 358, 1–8. [Google Scholar]
- Simental, J.A.; Sar, M.; Lane, M.V.; French, F.S.; Wilson, E.M. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J. Biol. Chem 1991, 266, 510–518. [Google Scholar]
- Pereira de Jesus-Tran, K.; Cote, P.L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci 2006, 15, 987–999. [Google Scholar]
- He, B.; Gampe, R.T., Jr; Kole, A.J.; Hnat, A.T.; Stanley, T.B.; An, G.; Stewart, E.L.; Kalman, R.I.; Minges, J.T.; Wilson, E.M. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol. Cell 2004, 16, 425–438. [Google Scholar]
- Gao, W.; Bohl, C.E.; Dalton, J.T. Chemistry and structural biology of androgen receptor. Chem. Rev 2005, 105, 3352–3370. [Google Scholar]
- Zhou, X.E.; Suino-Powell, K.M.; Li, J.; He, Y.; Mackeigan, J.P.; Melcher, K.; Yong, E.L.; Xu, H.E. Identification of SRC3/AIB1 as a preferred coactivator for hormone-activated androgen receptor. J. Biol. Chem 2010, 285, 9161–9171. [Google Scholar]
- Wilson, E.M. Analysis of interdomain interactions of the androgen receptor. Methods Mol. Biol 2011, 776, 113–129. [Google Scholar]
- Estebanez-Perpina, E.; Arnold, L.A.; Nguyen, P.; Rodrigues, E.D.; Mar, E.; Bateman, R.; Pallai, P.; Shokat, K.M.; Baxter, J.D.; Guy, R.K.; et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. USA 2007, 104, 16074–16079. [Google Scholar]
- Grosdidier, S.; Carbo, L.R.; Buzon, V.; Brooke, G.; Nguyen, P.; Baxter, J.D.; Bevan, C.; Webb, P.; Estebanez-Perpina, E.; Fernandez-Recio, J. Allosteric conversation in the androgen receptor ligand-binding domain surfaces. Mol. Endocrinol 2012, 26, 1078–1090. [Google Scholar]
- De Leon, J.T.; Iwai, A.; Feau, C.; Garcia, Y.; Balsiger, H.A.; Storer, C.L.; Suro, R.M.; Garza, K.M.; Lee, S.; Kim, Y.S.; et al. Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11878–11883. [Google Scholar]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat 2012, 33, 887–894. [Google Scholar]
- Guo, Z.; Qiu, Y. A new trick of an old molecule: Androgen receptor splice variants taking the stage?! Int. J. Biol. Sci 2011, 7, 815–822. [Google Scholar]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J 2005, 272, 74–84. [Google Scholar]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; Kung, H.J.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 2009, 69, 2305–2313. [Google Scholar]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest 2010, 120, 2715–2730. [Google Scholar]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 2009, 69, 16–22. [Google Scholar]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 2008, 68, 5469–5477. [Google Scholar]
- Suzuki, H.; Sato, N.; Watabe, Y.; Masai, M.; Seino, S.; Shimazaki, J. Androgen receptor gene mutations in human prostate cancer. J. Steroid Biochem 1993, 46, 759–765. [Google Scholar]
- Bohl, C.E.; Gao, W.; Miller, D.D.; Bell, C.E.; Dalton, J.T. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 6201–6206. [Google Scholar]
- Bohl, C.E.; Miller, D.D.; Chen, J.; Bell, C.E.; Dalton, J.T. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem 2005, 280, 37747–37754. [Google Scholar]
- Salvati, M.E.; Balog, A.; Shan, W.; Wei, D.D.; Pickering, D.; Attar, R.M.; Geng, J.; Rizzo, C.A.; Gottardis, M.M.; Weinmann, R.; et al. Structure based approach to the design of bicyclic-1H-isoindole-1,3(2H)-dione based androgen receptor antagonists. Bioorg. Med. Chem. Lett 2005, 15, 271–276. [Google Scholar]
- Duke, C.B.; Jones, A.; Bohl, C.E.; Dalton, J.T.; Miller, D.D. Unexpected binding orientation of bulky-B-ring anti-androgens and implications for future drug targets. J. Med. Chem 2011, 54, 3973–3976. [Google Scholar]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar]
- Kauppi, B.; Jakob, C.; Farnegardh, M.; Yang, J.; Ahola, H.; Alarcon, M.; Calles, K.; Engstrom, O.; Harlan, J.; Muchmore, S.; et al. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: Ru-486 induces a transconformation that leads to active antagonism. J. Biol. Chem 2003, 278, 22748–22754. [Google Scholar]
- Pal, S.K.; Stein, C.A.; Sartor, O. Enzalutamide for the treatment of prostate cancer. Expert Opin. Pharmacother 2013, 14, 679–685. [Google Scholar]
- Scher, H.I.; Beer, T.M.; Higano, C.S.; Anand, A.; Taplin, M.E.; Efstathiou, E.; Rathkopf, D.; Shelkey, J.; Yu, E.Y.; Alumkal, J.; et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1–2 study. Lancet 2010, 375, 1437–1446. [Google Scholar]
- Nique, F.; Hebbe, S.; Peixoto, C.; Annoot, D.; Lefrancois, J.M.; Duval, E.; Michoux, L.; Triballeau, N.; Lemoullec, J.M.; Mollat, P.; et al. Discovery of diarylhydantoins as new selective androgen receptor modulators. J. Med. Chem 2012, 55, 8225–8235. [Google Scholar]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res 2012, 72, 1494–1503. [Google Scholar]
- Yang, S.H.; Song, C.H.; van, H.T.; Park, E.; Khadka, D.B.; Gong, E.Y.; Lee, K.; Cho, W.J. SAR based design of nicotinamides as a novel class of androgen receptor antagonists for prostate cancer. J. Med. Chem 2013, 56, 3414–3418. [Google Scholar]
- Helsen, C.; Marchand, A.; Chaltin, P.; Munck, S.; Voet, A.; Verstuyf, A.; Claessens, F. Identification and characterization of MEL-3, a novel ar antagonist that suppresses prostate cancer cell growth. Mol. Cancer Ther 2012, 11, 1257–1268. [Google Scholar]
- Buzon, V.; Carbo, L.R.; Estruch, S.B.; Fletterick, R.J.; Estebanez-Perpina, E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric control. Mol. Cell. Endocrinol 2012, 348, 394–402. [Google Scholar]
- Axerio-Cilies, P.; Lack, N.A.; Nayana, M.R.; Chan, K.H.; Yeung, A.; Leblanc, E.; Guns, E.S.; Rennie, P.S.; Cherkasov, A. Inhibitors of androgen receptor activation function-2 (AF2) site identified through virtual screening. J. Med. Chem 2011, 54, 6197–6205. [Google Scholar]
- Sadar, M.D. Small molecule inhibitors targeting the “achilles’ heel” of androgen receptor activity. Cancer Res 2011, 71, 1208–1213. [Google Scholar]
- Sadar, M.D.; Williams, D.E.; Mawji, N.R.; Patrick, B.O.; Wikanta, T.; Chasanah, E.; Irianto, H.E.; Soest, R.V.; Andersen, R.J. Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. That inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org. Lett 2008, 10, 4947–4950. [Google Scholar]
- Meimetis, L.G.; Williams, D.E.; Mawji, N.R.; Banuelos, C.A.; Lal, A.A.; Park, J.J.; Tien, A.H.; Fernandez, J.G.; de Voogd, N.J.; Sadar, M.D.; et al. Niphatenones, glycerol ethers from the sponge niphates digitalis block androgen receptor transcriptional activity in prostate cancer cells: Structure elucidation, synthesis, and biological activity. J. Med. Chem 2012, 55, 503–514. [Google Scholar]
- Quayle, S.N.; Mawji, N.R.; Wang, J.; Sadar, M.D. Androgen receptor decoy molecules block the growth of prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 1331–1336. [Google Scholar]
- Van de Wijngaart, D.J.; Dubbink, H.J.; van Royen, M.E.; Trapman, J.; Jenster, G. Androgen receptor coregulators: Recruitment via the coactivator binding groove. Mol. Cell. Endocrinol 2012, 352, 57–69. [Google Scholar]
- Van de Wijngaart, D.J.; van Royen, M.E.; Hersmus, R.; Pike, A.C.; Houtsmuller, A.B.; Jenster, G.; Trapman, J.; Dubbink, H.J. Novel FXXFF and FXXMF motifs in androgen receptor cofactors mediate high affinity and specific interactions with the ligand-binding domain. J. Biol. Chem 2006, 281, 19407–19416. [Google Scholar]
- Van Royen, M.E.; Cunha, S.M.; Brink, M.C.; Mattern, K.A.; Nigg, A.L.; Dubbink, H.J.; Verschure, P.J.; Trapman, J.; Houtsmuller, A.B. Compartmentalization of androgen receptor protein-protein interactions in living cells. J. Cell Biol 2007, 177, 63–72. [Google Scholar]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar]
- Cherian, M.T.; Wilson, E.M.; Shapiro, D.J. A competitive inhibitor that reduces recruitment of androgen receptor to androgen-responsive genes. J. Biol. Chem 2012, 287, 23368–23380. [Google Scholar]
- Chenoweth, D.M.; Dervan, P.B. Structural basis for cyclic Py-Im polyamide allosteric inhibition of nuclear receptor binding. J. Am. Chem. Soc 2010, 132, 14521–14529. [Google Scholar]
- Nickols, N.G.; Dervan, P.B. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc. Natl. Acad. Sci. USA 2007, 104, 10418–10423. [Google Scholar]
- Chenoweth, D.M.; Dervan, P.B. Allosteric modulation of DNA by small molecules. Proc. Natl. Acad. Sci. USA 2009, 106, 13175–13179. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lallous, N.; Dalal, K.; Cherkasov, A.; Rennie, P.S. Targeting Alternative Sites on the Androgen Receptor to Treat Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2013, 14, 12496-12519. https://doi.org/10.3390/ijms140612496
Lallous N, Dalal K, Cherkasov A, Rennie PS. Targeting Alternative Sites on the Androgen Receptor to Treat Castration-Resistant Prostate Cancer. International Journal of Molecular Sciences. 2013; 14(6):12496-12519. https://doi.org/10.3390/ijms140612496
Chicago/Turabian StyleLallous, Nada, Kush Dalal, Artem Cherkasov, and Paul S. Rennie. 2013. "Targeting Alternative Sites on the Androgen Receptor to Treat Castration-Resistant Prostate Cancer" International Journal of Molecular Sciences 14, no. 6: 12496-12519. https://doi.org/10.3390/ijms140612496