Analysis of Transcriptional Regulation of the Human miR-17-92 Cluster; Evidence for Involvement of Pim-1

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
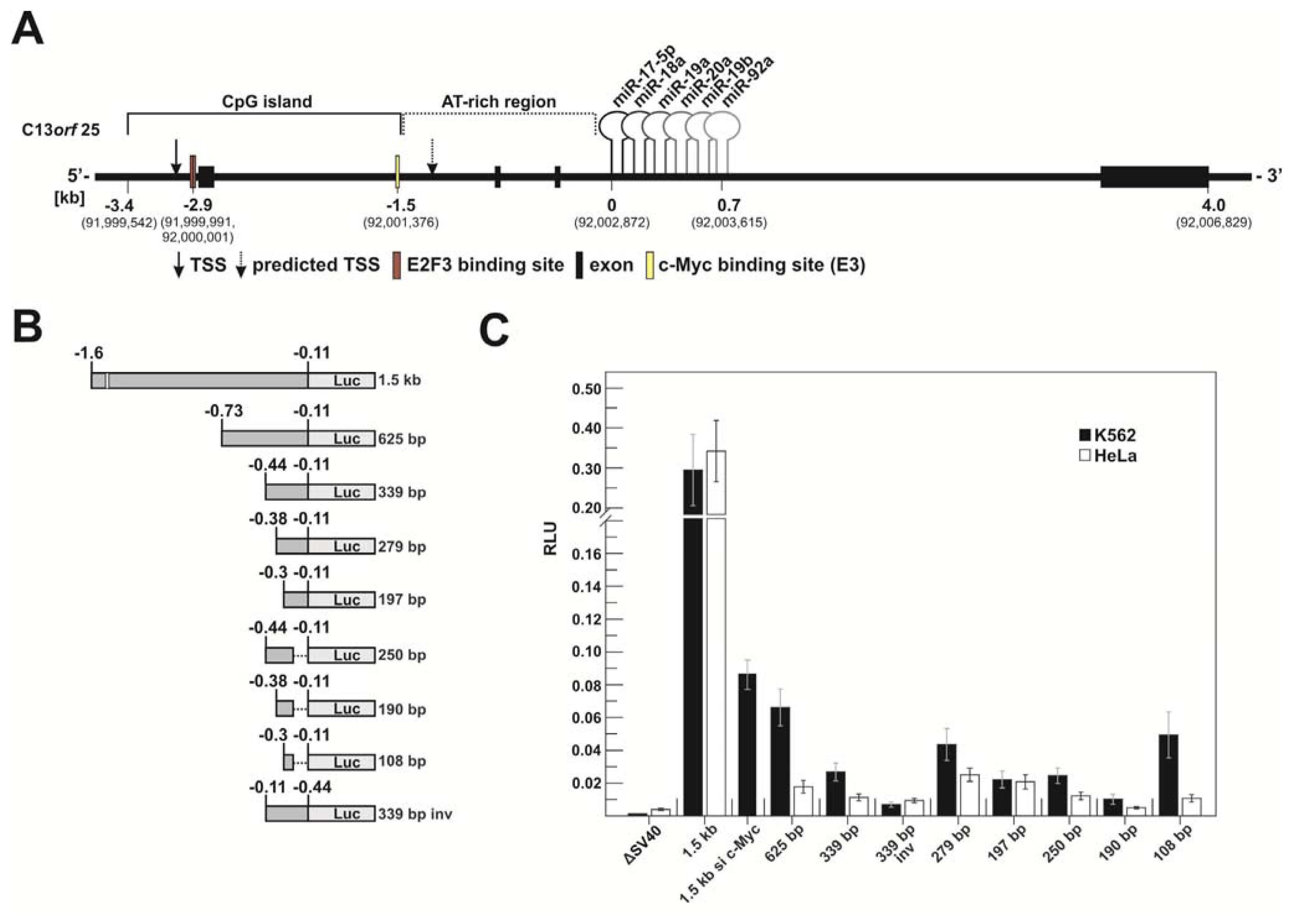
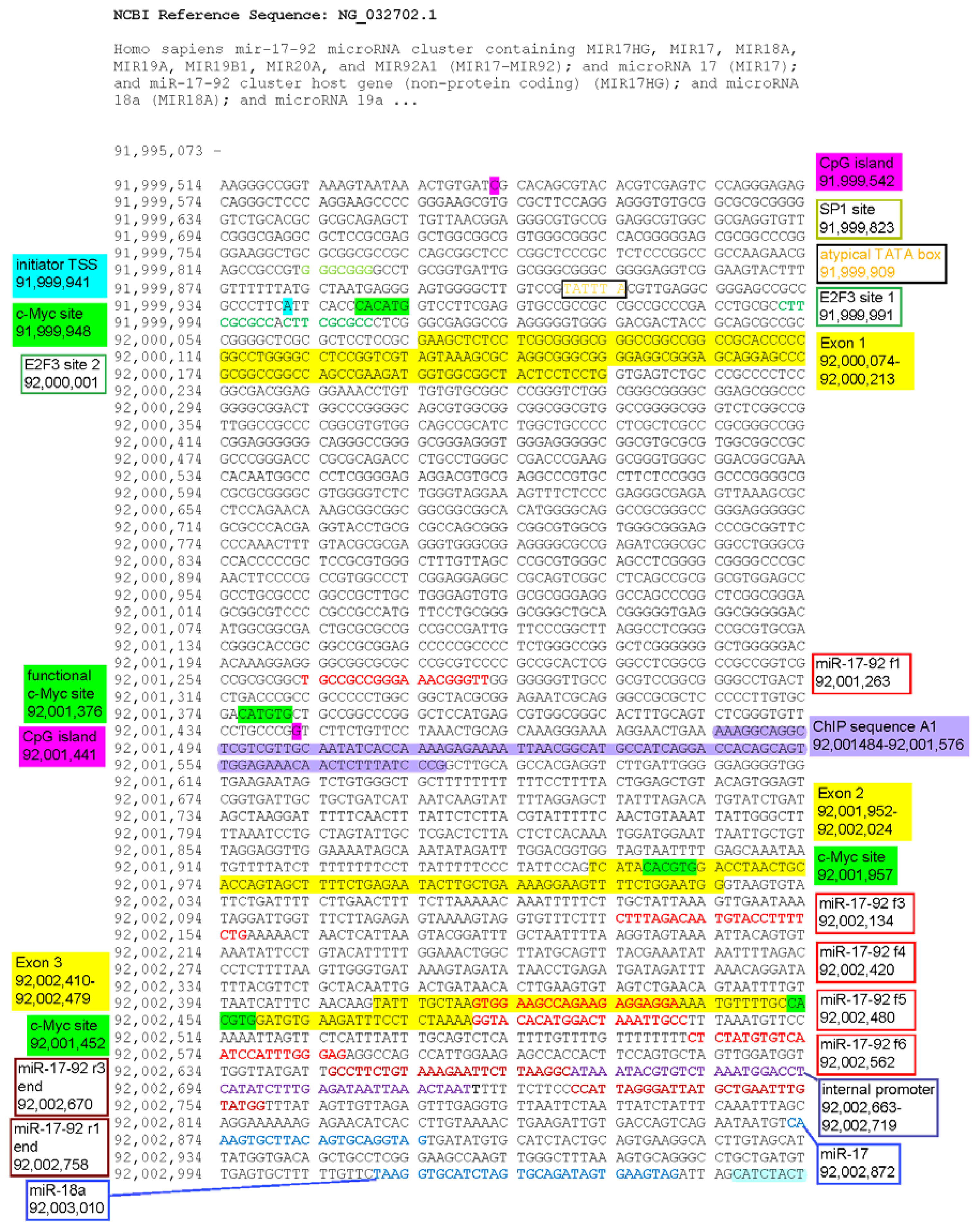
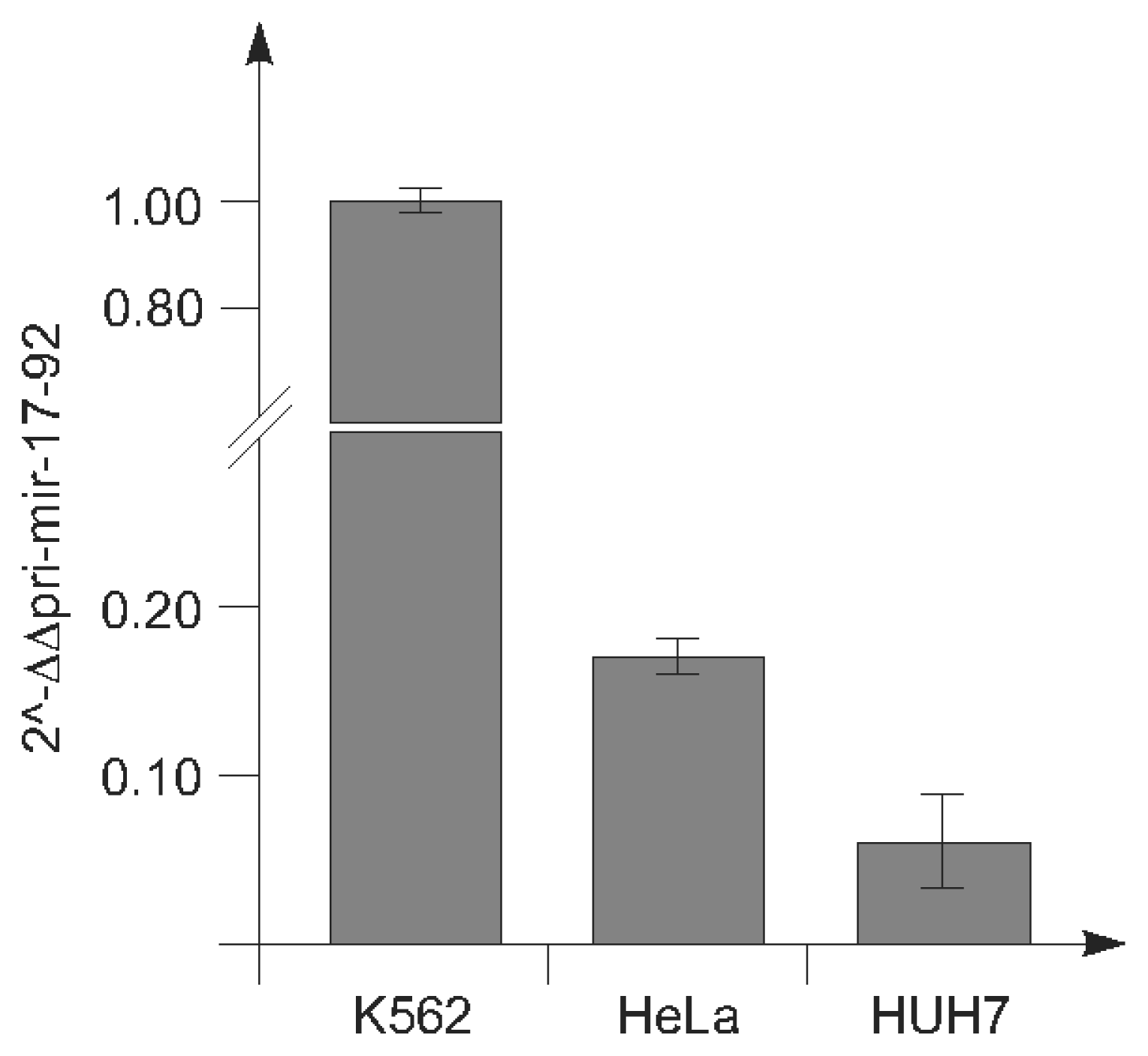
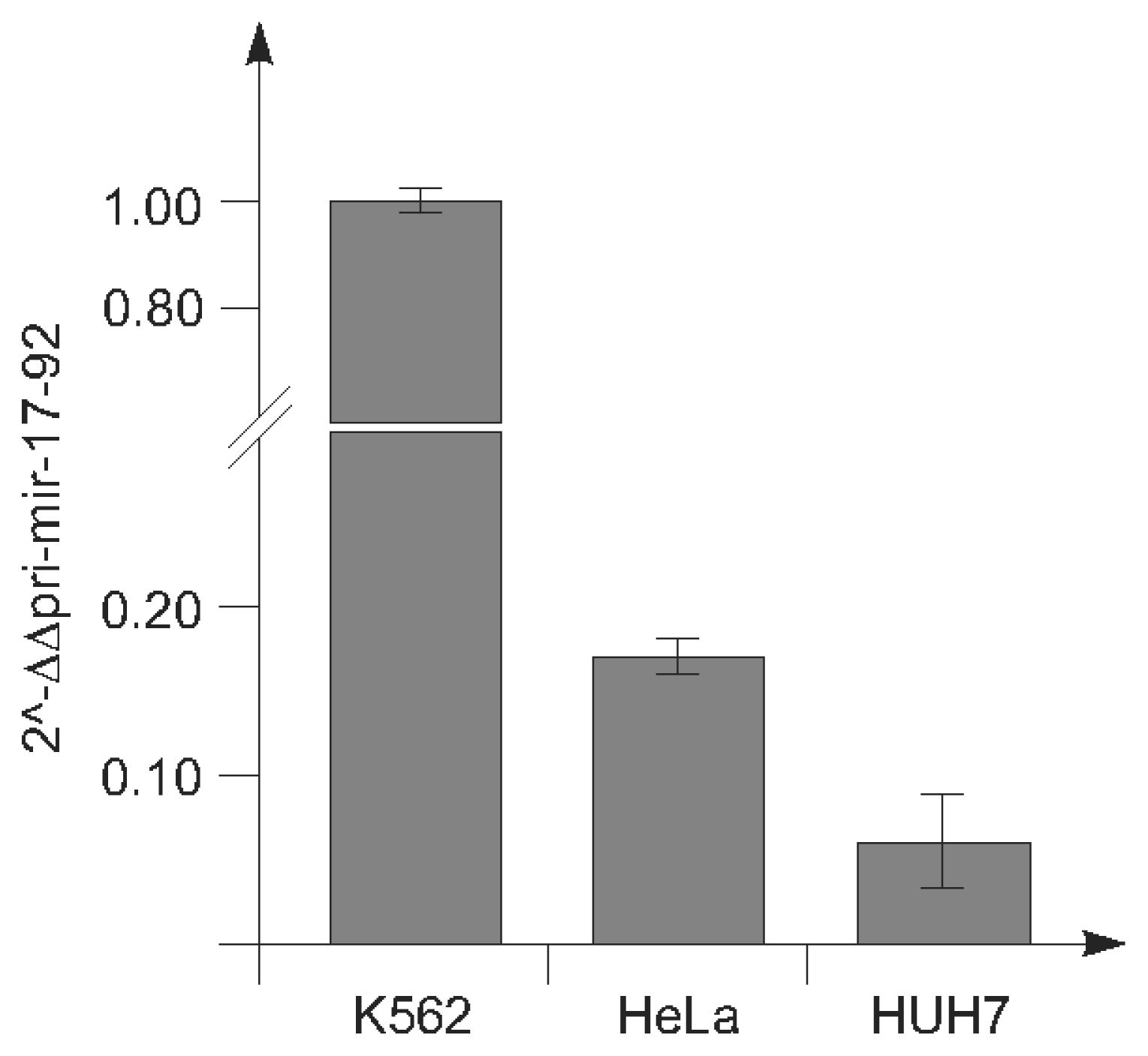
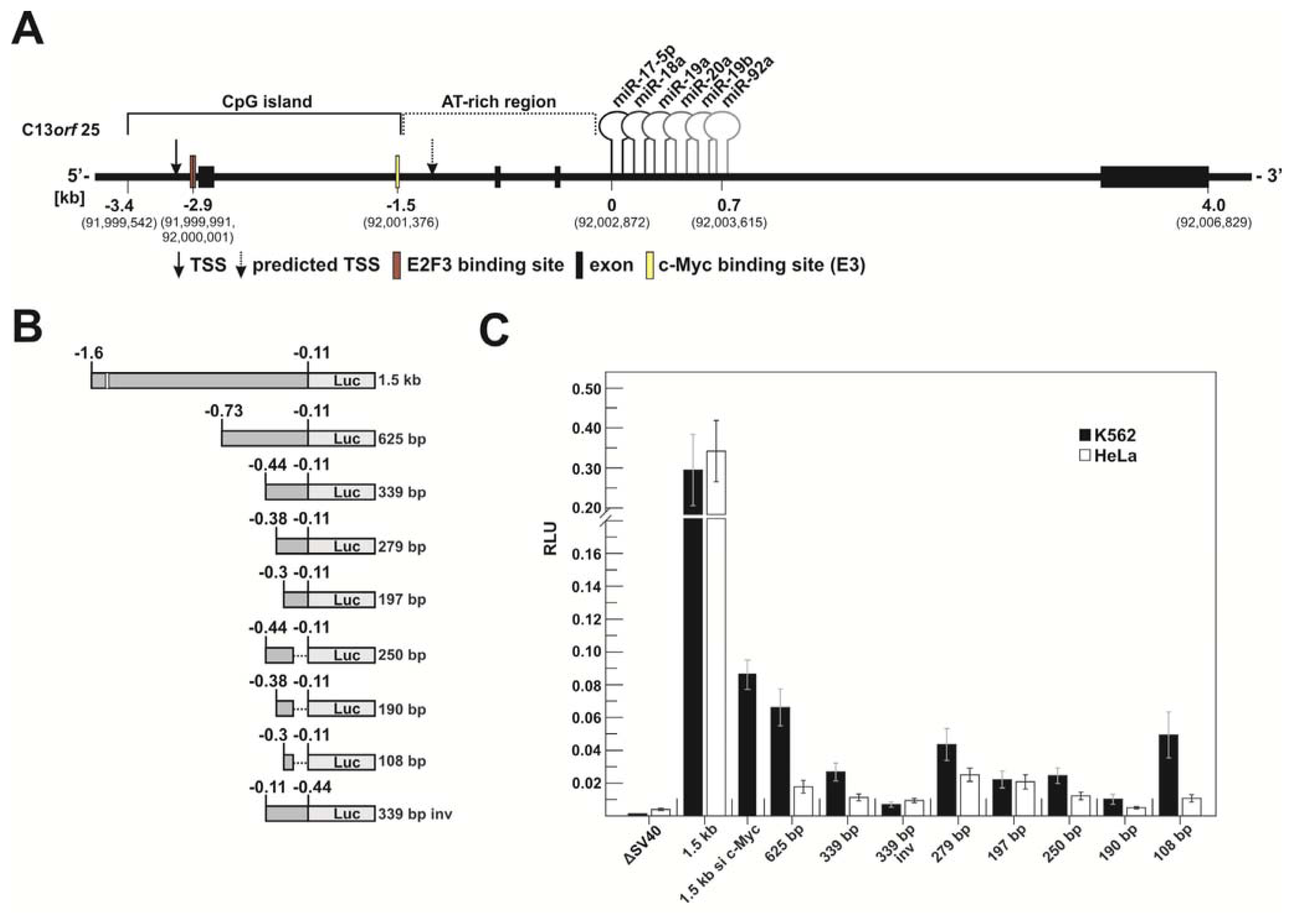
2.1.1. c-Myc-Dependent Intronic Transcriptional Activity within the Human miR-17-92 Locus
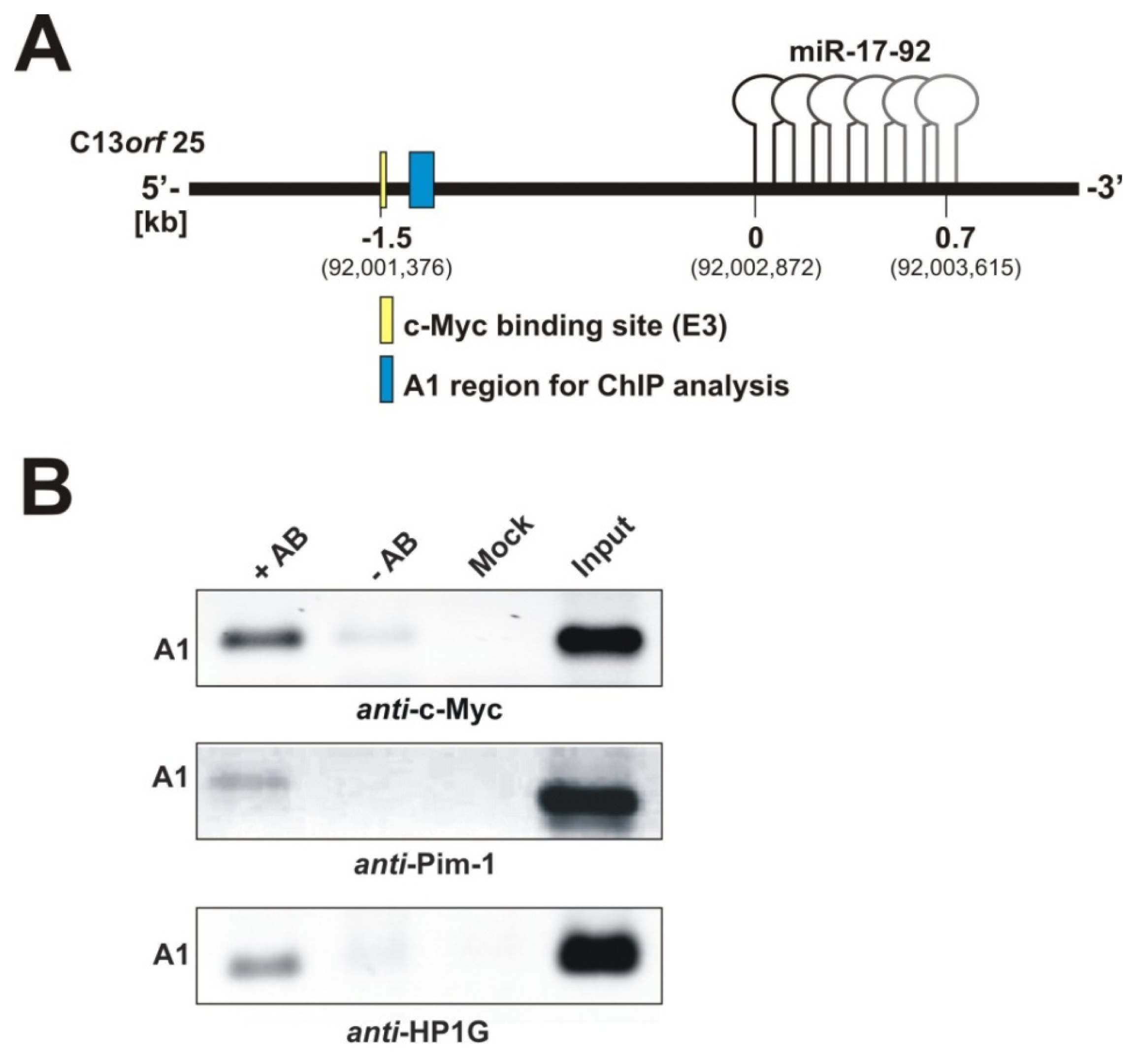
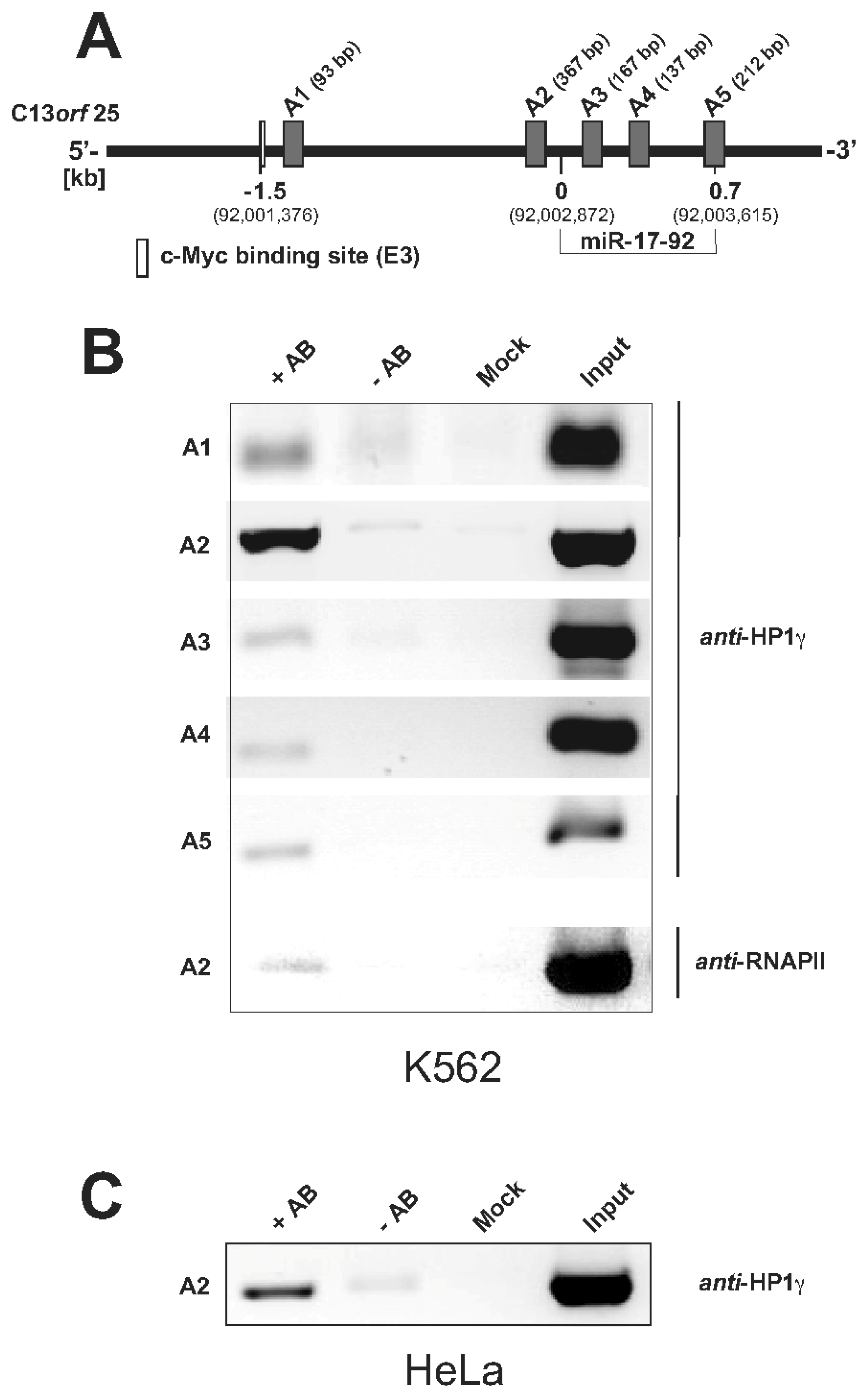
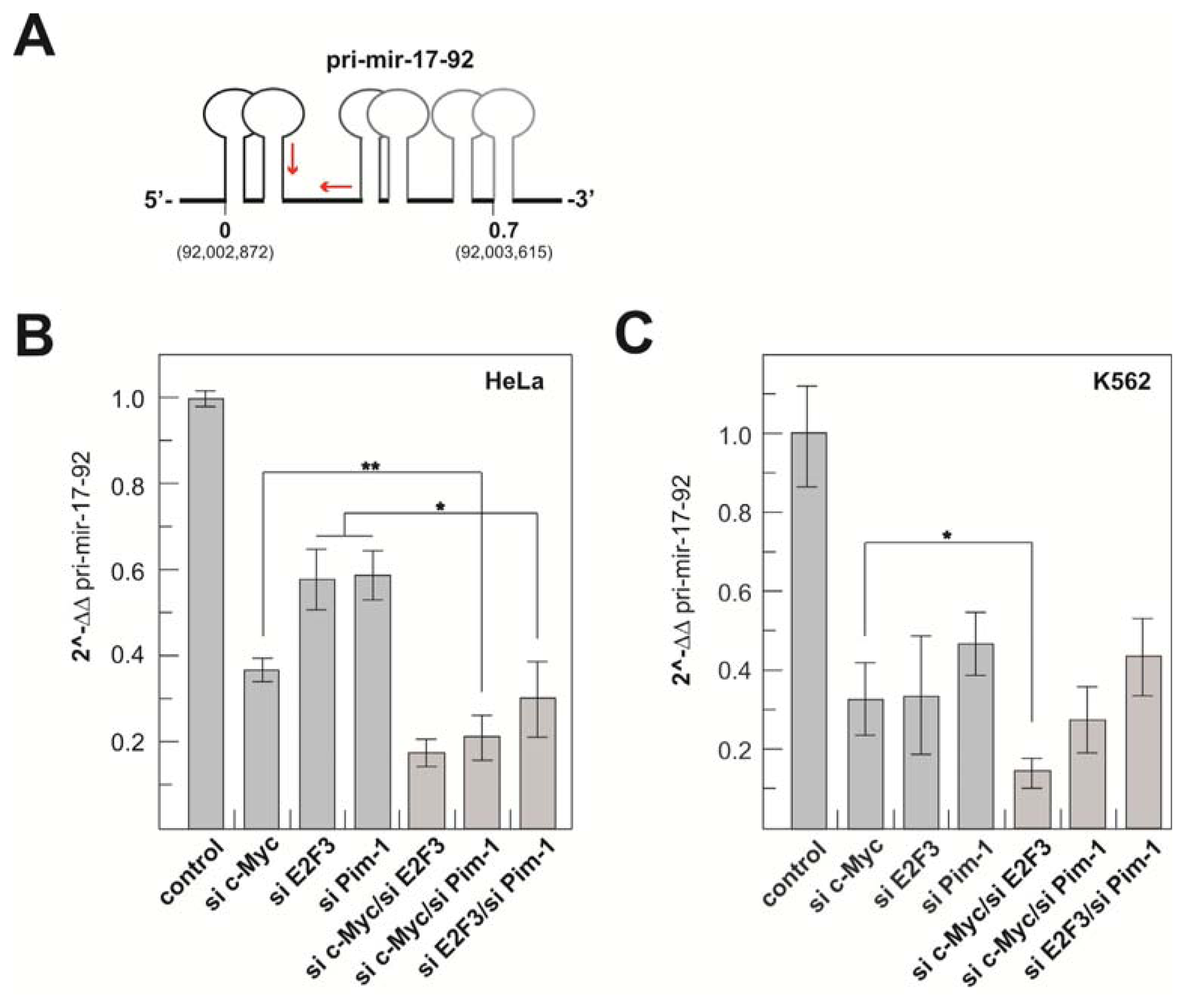
2.1.2. Pim-1 and HP1γ Are Associated with the Intronic c-Myc Binding Site
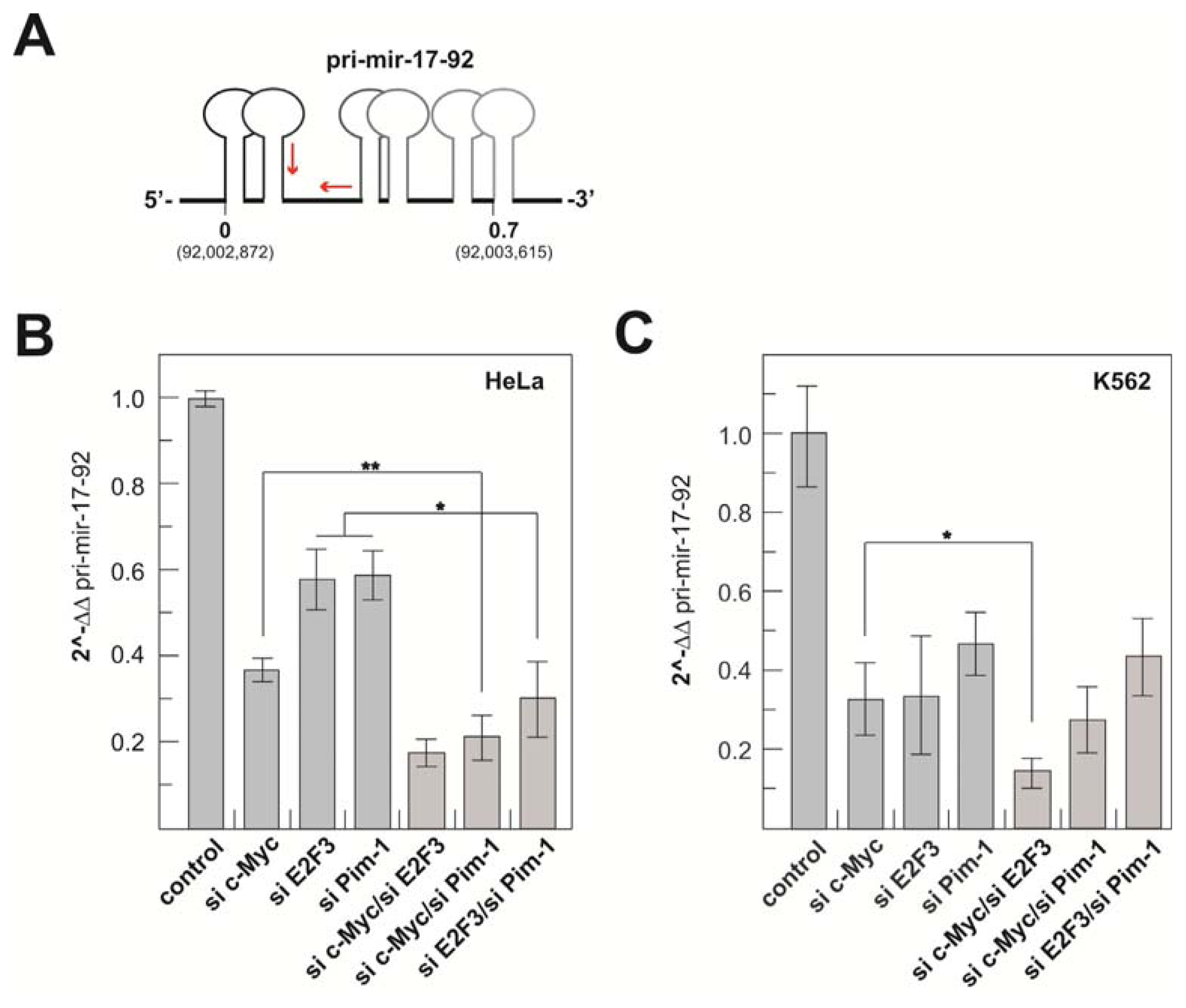
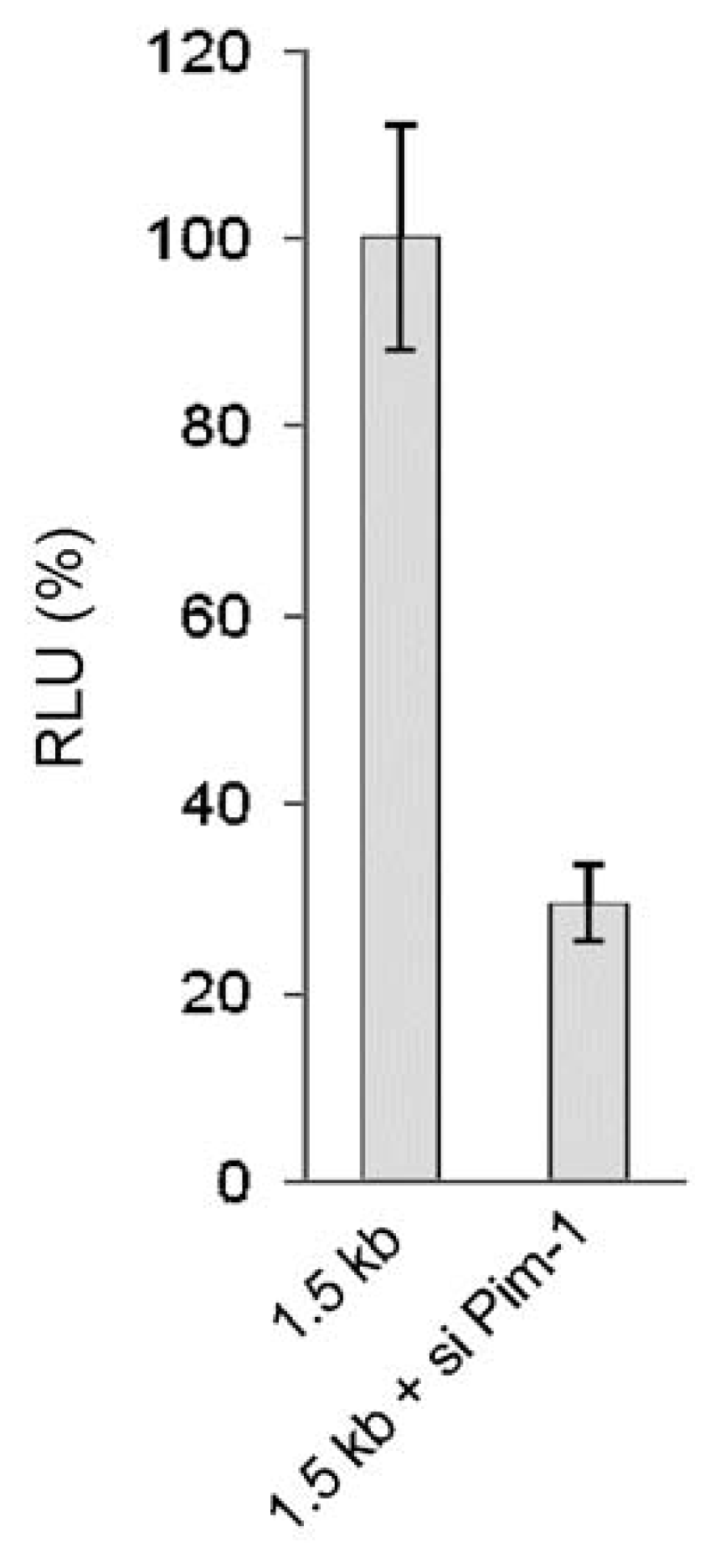
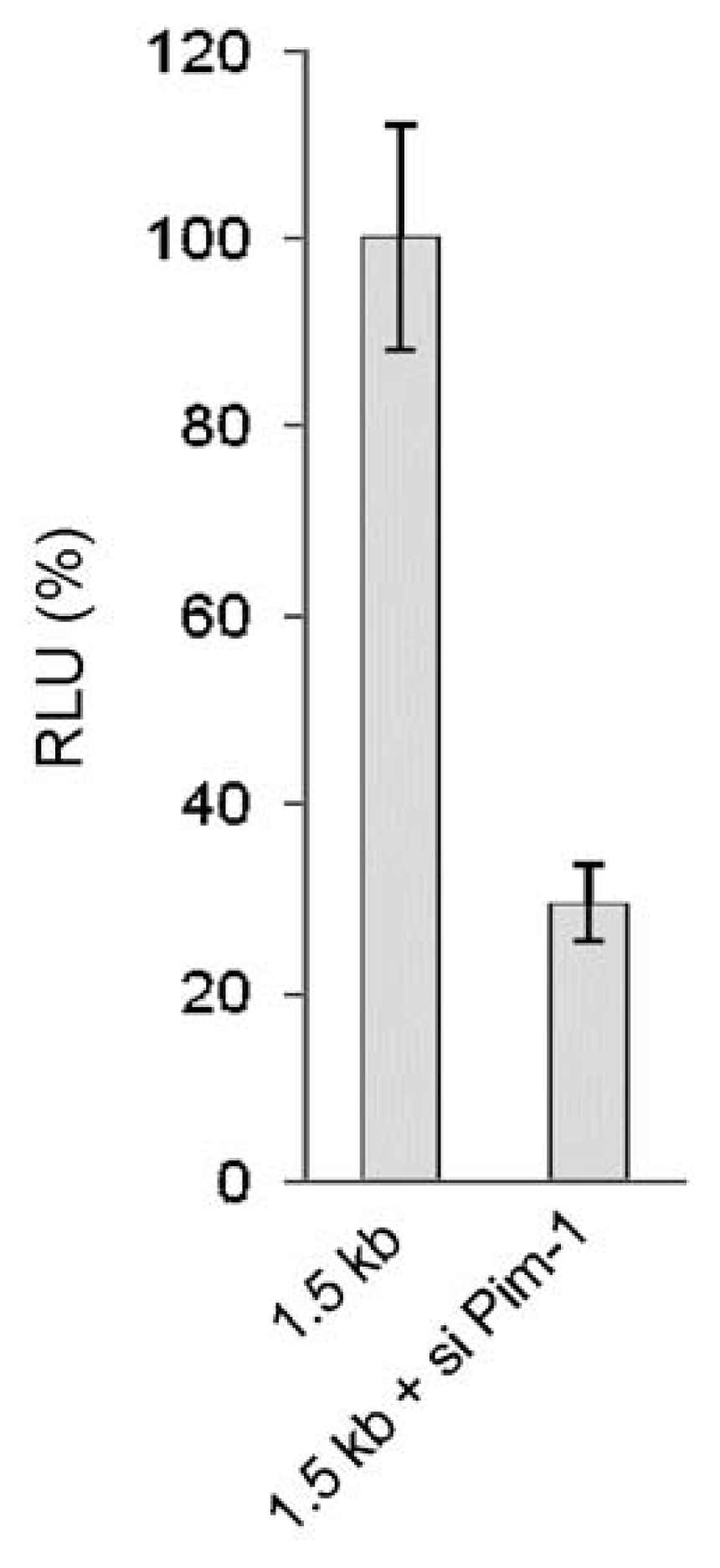
2.1.3. Transcriptional Activity of the Human miR-17-92 Cluster Depends on c-Myc and Pim-1
2.2. Discussion
3. Experimental Section
3.1. Oligonucleotides
3.2. Antibodies
3.3. Plasmid Construction and Seed Mutagenesis
3.4. Transfection Procedures and Luciferase Reporter Assays
3.5. Chromatin Immunoprecipitation
4. Conclusions
Supplementary Figures
Figure S1
Supplementary Table

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| K562 | c-Myc KD | E2F3 KD | Pim-1 KD | |||
|---|---|---|---|---|---|---|
| 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | |
| 0.13 | 77 | 0.21 | 95 | 1.23 | 72 | |
| 0.02 | 87 | 0.16 | 98 | 0.16 | 69 | |
| 0.56 | 59 | 0.60 | 19 | 1.39 | 55 | |
| 0.75 | 74 | 0.76 | 59 | |||
| 0.33 | 75 | 0.22 | 49 | |||
| 0.14 | 99 | 2.38 | 78 | |||
| 0.20 | 97 | 1.84 | 87 | |||
| 0.55 | 80 | 0.80 | 88 | |||
| 0.27 | 85 | |||||
| 0.40 | 89 | |||||
| 0.52 | 67 | |||||
| 0.69 | 65 | |||||
| 0.37 | 87 | |||||
| Mean | 0.33 | 81 | 0.34 | 89 | 0.47 | 73 |
| SEM | 0.09 | 5.0 | 0.15 | 8.0 | 0.08 | 5.0 |
| K562 | c-Myc/E2F3 KD | c-Myc/Pim-1 KD | Pim-1/E2F3 KD | |||
|---|---|---|---|---|---|---|
| 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | |
| 0.22 | 82/75 | 0.45 | 72/55 | 0.69 | 88/86 | |
| 0.23 | 86/72 | 0.19 | 90/54 | 0.56 | 68/73 | |
| 0.15 | 81/48 | 0.19 | 95/48 | 0.19 | 95/99 | |
| 0.08 | 99/99 | 0.52 | 87/81 | |||
| 0.04 | 99/99 | 0.23 | 91/93 | |||
| Mean | 0.15 | 89/79 | 0.28 | 86/52 | 0.44 | 86/86 |
| SEM | 0.04 | 4.0/9.6 | 0.08 | 7.0/2.2 | 0.09 | 4.7/4.5 |
| HeLa | c-Myc KD | E2F3 KD | Pim-1 KD | |||
|---|---|---|---|---|---|---|
| 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | |
| 0.37 | 70 | 0.72 | 51 | 0.50 | 47 | |
| 0.29 | 82 | 0.66 | 78 | 0.34 | 72 | |
| 0.32 | 61 | 0.40 | 76 | 0.71 | 63 | |
| 0.41 | 99 | 0.53 | 73 | 0.58 | 80 | |
| 0.48 | 55 | 0.64 | 73 | 0.54 | 87 | |
| 0.37 | 64 | 0.78 | 72 | |||
| 0.70 | 76 | |||||
| Mean | 0.37 | 65 | 0.59 | 70 | 0.59 | 71 |
| SEM | 0.03 | 6.7 | 0.07 | 6.3 | 0.06 | 4.9 |
| HeLa | c-Myc/E2F3 KD | c-Myc/Pim-1 KD | Pim-1/E2F3 KD | |||
|---|---|---|---|---|---|---|
| 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | 2^-Δ ΔCT pri-mir-17-92 | KD efficiency | |
| 0.24 | 79/64 | 0.24 | 79/84 | 0.49 | 81/96 | |
| 0.13 | 91/81 | 0.25 | 77/61 | 0.33 | 91/85 | |
| 0.11 | 93/86 | 0.15 | 86/58 | 0.10 | 97/94 | |
| 0.12 | 90/91 | 0.12 | 95/91 | 0.26 | 92/94 | |
| 0.23 | 87/74 | 0.41 | 84/73 | |||
| 0.26 | 83/66 | 0.10 | 97/95 | |||
| Mean | 0.18 | 87/77 | 0.21 | 86/77 | 0.30 | 90/92 |
| SEM | 0.03 | 2.2/4.4 | 0.053 | 3.3/6.0 | 0.08 | 3.3/2.5 |
Acknowledgments
Appendix
Cell Culture
Chromatin Immunoprecipitation (ChIP)
- A1 forward 5′-AAA GGC AGG CTC GTC GTT G
- A1 reverse 5′-CGG GAT AAA GAG TTG TTT CTC CAA
- A2 forward 5′-ACA TGG ACT AAA TTG CCT TTA AAT G
- A2 reverse 5′-AAT CTT CAG TTT TAC AAG GTG ATG
- A3 forward 5′-ACT GCA GTG AAG GCA CTT GT
- A3 reverse 5′-TGC CAG AAG GAG CAC TTA GG
- A4 forward 5′-CCA ATA ATT CAA GCC AAG CAA
- A4 reverse 5′-AAA TAG CAG GCC ACC ATC AG
- A5 forward 5′-GCC CAA TCA AAC TGT CCT GT
- A5 reverse 5′-CGG GAC AAG TGC AAT ACC AT
Transfection of Reporter Constructs
Transfection of siRNAs
RNA Preparation and Quantitative Real-Time PCR
- Pri-miR-17-92: forward primer 5′-CAT CTA CTG CCC TAA GTG CTC CTT and reverse primer 5′-GCT TGG CTT GAA TTA TTG GAT GA;
- 5S rRNA: 5′-TCT CGT CTG ATC TCG GAA GC and 5′-AGC CTA CAG CAC CCG GTA TT;
- c-Myc mRNA: forward primer 5′-CCT TGC AGC TGC TTA GAC and reverse primer 5′-GAG TCG TAG TCG AGG TCA T;
- E2F3 mRNA: forward primer 5′-GAG ACT GAA ACA CAC AGT CC and reverse primer 5′-CCT GAG TTG GTT GAA GCC;
- Pim-1 mRNA: forward primer 5′-ATC AGG GGC CAG GTT TTC T and reverse primer 5′-GGG CCA AGC ACC ATC TAA T;
- Actin mRNA: forward primer 5′-CCA ACC GCG AGA AGA TGA and reverse primer 5′-CCA GAG GCG TAC AGG GAT AG.
Luciferase Reporter Assays
Plasmid Construction and Seed Mutagenesis
- pGL3 1.5 kb: 5′-ATA TAG ATC TTG CCG CCG GGA AAC GGG TT and reverse primer R1 5′-ATA TAA GCT TCC ATA CAA ATT CAG CAT AAT CCC TAA TGG;
- pGL3 625 bp: 5′-ATA TAG ATC TCT TTA GAC AAT GTA CCT TTT CTG and reverse primer R1 (see above);
- pGL3 339 bp: 5′-ATA TAG ATC TGT GGA AGC CAG AAG AGG AGG A and reverse primer R1 (see above);
- pGL3 279 bp: 5′-ATA TAG ATC TGG TAC ACA TGG ACT AAA TTG CC and reverse primer R1 (see above);
- pGL3 197 bp: 5′-ATA TAG ATC TCT CTA TGT GTC AAT CCA TTT GGG AG and reverse primer R1 (see above);
- pGL3 250 bp: 5′-ATA TAG ATC TGT GGA AGC CAG AAG AGG AGG A and reverse primer R3 5′-ATA TAA GCT TGC CTT AAG AAT TCT TTA CAG AAG GC;
- pGL3 190 bp: 5′-ATA TAG ATC TGG TAC ACA TGG ACT AAA TTG CC and reverse primer R3 (see above);
- pGL3 108 bp: 5′-ATA TAG ATC TCT CTA TGT GTC AAT CCA TTT GGG AG and reverse primer R3 (see above);
- pGL3 339 inv: 5′-ATA TAA GCT TGT GGA AGC CAG AAG AGG AGG A and 5′-ATA TAG ATC TCC ATA CAA ATT CAG CAT AAT CCC TAA TGG.
Bioinformatical Promoter Analysis
Statistical Analysis
Conflict of Interest
References
- Iorio, M.V.; Croce, C.M. MicroRNAs in cancer: Small molecules with a huge impact. J. Clin. Oncol 2009, 27, 5848–5856. [Google Scholar]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet 2010, 11, 597–610. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 2003, 17, 3011–3016. [Google Scholar]
- Lee, Y.; Jeon, K.; Lee, J.-T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J 2002, 21, 4663–4670. [Google Scholar]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar]
- Ota, A.; Tagawa, H.; Karnan, S.; Tsuzuki, S.; Karpas, A.; Kira, S.; Yoshida, Y.; Seto, M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31–q32 amplification in malignant lymphoma. Cancer Res 2004, 64, 3087–3095. [Google Scholar]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar]
- Tanzer, A.; Stadler, P.F. Molecular evolution of a microRNA cluster. J. Mol. Biol 2004, 339, 327–335. [Google Scholar]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar]
- Olive, V.; Bennett, M.J.; Walker, J.C.; Ma, C.; Jiang, I.; Cordon-Cardo, C.; Li, Q.-J.; Lowe, S.W.; Hannon, G.J.; He, L. miR-19 is a key oncogenic component of mir-17–92. Genes Dev 2009, 23, 2839–2849. [Google Scholar]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar]
- Mu, P.; Han, Y.-C.; Betel, D.; Yao, E.; Squatrito, M.; Ogrodowski, P.; de Stanchina, E.; D’Andrea, A.; Sander, C.; Ventura, A. Genetic dissection of the miR-17–92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev 2009, 23, 2806–2811. [Google Scholar]
- Ozsolak, F.; Poling, L.L.; Wang, Z.; Liu, H.; Liu, X.S.; Roeder, R.G.; Zhang, X.; Song, J.S.; Fisher, D.E. Chromatin structure analyses identify miRNA promoters. Genes Dev 2008, 22, 3172–3183. [Google Scholar]
- Woods, K.; Thomson, J.M.; Hammond, S.M. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J. Biol. Chem 2007, 282, 2130–2134. [Google Scholar]
- Sylvestre, Y.; de Guire, V.; Querido, E.; Mukhopadhyay, U.K.; Bourdeau, V.; Major, F.; Ferbeyre, G.; Chartrand, P. An E2F/miR-20a autoregulatory feedback loop. J. Biol. Chem 2007, 282, 2135–2143. [Google Scholar]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar]
- Ji, M.; Rao, E.; Ramachandrareddy, H.; Shen, Y.; Jiang, C.; Chen, J.; Hu, Y.; Rizzino, A.; Chan, W.C.; Fu, K.; et al. The miR-17–92 microRNA cluster is regulated by multiple mechanisms in B-cell malignancies. Am. J. Pathol 2011, 179, 1645–1656. [Google Scholar]
- Pospisil, V.; Vargova, K.; Kokavec, J.; Rybarova, J.; Savvulidi, F.; Jonasova, A.; Necas, E.; Zavadil, J.; Laslo, P.; Stopka, T. Epigenetic silencing of the oncogenic miR-17–92 cluster during PU.1-directed macrophage differentiation. EMBO J 2011, 30, 4450–4464. [Google Scholar]
- Koike, N.; Maita, H.; Taira, T.; Ariga, H.; Iguchi-Ariga, S.M.M. Identification of heterochromatin protein 1 (HP1) as a phosphorylation target by Pim-1 kinase and the effect of phosphorylation on the transcriptional repression function of HP1. FEBS Lett 2000, 467, 17–21. [Google Scholar]
- Nawijn, M.C.; Alendar, A.; Berns, A. For better or for worse: The role of Pim oncogenes in tumorigenesis. Nat. Rev. Cancer 2011, 11, 23–34. [Google Scholar]
- Zippo, A.; de Robertis, A.; Serafini, R.; Oliviero, S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat. Cell Biol 2007, 9, 932–944. [Google Scholar]
- UCSC Genome Bioinformatics. Available online: http://genome.ucsc.edu (assessed on 1 February 2009).
- Thomas, M.; Lange-Grünweller, K.; Weirauch, U.; Gutsch, D.; Aigner, A.; Grünweller, A.; Hartmann, R.K. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene 2012, 31, 918–928. [Google Scholar]
- Venturini, L.; Battmer, K.; Castoldi, M.; Schultheis, B.; Hochhaus, A.; Muckenthaler, M.U.; Ganser, A.; Eder, M.; Scherr, M. Expression of the miR-17–92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood 2007, 109, 4399–4405. [Google Scholar]
- Kwon, S.H.; Workman, J.L. HP1c casts light on dark matter. Cell Cycle 2011, 15, 625–630. [Google Scholar]
- Vakoc, C.R.; Mandat, S.A.; Olenchock, B.A.; Blobel, G.A. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell 2005, 19, 381–391. [Google Scholar]
- Minc, E.; Allory, Y.; Worman, H.J.; Courvalin, J.; Buendia, B. Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma 1999, 108, 220–234. [Google Scholar]
- Mateescu, B.; Bourachot, B.; Rachez, C.; Ogryzko, V.; Muchardt, C. Regulation of an inducible promoter by an HP1beta-HP1gamma switch. EMBO Rep 2008, 9, 267–272. [Google Scholar]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar]
- Adams, M.R.; Sears, R.; Nuckolls, F.; Leone, G.; Nevins, J.R. Complex transcriptional regulatory mechanisms control expression of the E2F3 locus. Mol. Cell Biol 2000, 20, 3633–3639. [Google Scholar]
- Thalmeier, K.; Synovzik, H.; Mertz, R.; Winnacker, E.L.; Lipp, M. Nuclear factor E2F mediates basic transcription and trans-activation by E1a of the human MYC promoter. Genes Dev 1989, 3, 527–536. [Google Scholar]
- Wong, J.V.; Dong, P.; Nevins, J.R.; Mathey-Prevot, B.; You, L. Network calisthenics: Control of E2F dynamics in cell cycle entry. Cell Cycle 2011, 15, 3086–3094. [Google Scholar]
- Grünweller, A.; Wyszko, E.; Bieber, B.; Jahnel, R.; Erdmann, V.A.; Kurreck, J. Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res 2003, 31, 3185–3193. [Google Scholar]
- Gozani Lab. Available online: http://www.stanford.edu/group/gozani (assessed on 1 June 2005).
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Prot 2008, 3, 1101–1108. [Google Scholar]
- Reese, M.G. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem 2001, 26, 51–56. [Google Scholar]
- Ohler, U. Identification of core promoter modules in Drosophila and their application in accurate transcription start site prediction. Nucleic Acids Res 2006, 34, 5943–5950. [Google Scholar]
- Knudsen, S. Promoter2.0: For the recognition of PolII promoter sequences. Bioinformatics 1999, 15, 356–361. [Google Scholar]
- Rangannan, V.; Bansal, M. Relative stability of DNA as a generic criterion for promoter prediction: whole genome annotation of microbial genomes with varying nucleotide base composition. Mol. BioSys 2009, 5, 1758–1769. [Google Scholar]
- Zhang, M.Q. Identification of human gene core promoters in silico. Genome Res 1998, 8, 319–326. [Google Scholar]
- The R Development Core Team, R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012.

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Thomas, M.; Lange-Grünweller, K.; Hartmann, D.; Golde, L.; Schlereth, J.; Streng, D.; Aigner, A.; Grünweller, A.; Hartmann, R.K. Analysis of Transcriptional Regulation of the Human miR-17-92 Cluster; Evidence for Involvement of Pim-1. Int. J. Mol. Sci. 2013, 14, 12273-12296. https://doi.org/10.3390/ijms140612273
Thomas M, Lange-Grünweller K, Hartmann D, Golde L, Schlereth J, Streng D, Aigner A, Grünweller A, Hartmann RK. Analysis of Transcriptional Regulation of the Human miR-17-92 Cluster; Evidence for Involvement of Pim-1. International Journal of Molecular Sciences. 2013; 14(6):12273-12296. https://doi.org/10.3390/ijms140612273
Chicago/Turabian StyleThomas, Maren, Kerstin Lange-Grünweller, Dorothee Hartmann, Lara Golde, Julia Schlereth, Dennis Streng, Achim Aigner, Arnold Grünweller, and Roland K. Hartmann. 2013. "Analysis of Transcriptional Regulation of the Human miR-17-92 Cluster; Evidence for Involvement of Pim-1" International Journal of Molecular Sciences 14, no. 6: 12273-12296. https://doi.org/10.3390/ijms140612273