The Genomic Landscape of Prostate Cancer

Abstract
:1. Introduction
2. Copy Number and Gene Expression Changes
3. Gene Fusions
3.1. Detection of ETS Gene Fusions in PCa
3.2. Detection of Non-ETS Gene Fusions in PCa
3.3. The Role of Fusion Genes in the Molecular Pathology of PCa
4. Single Base Pair Changes
4.1. Single Nucleotide Variants (SNVs)
4.1.1. The Beginning of Next-Generation Sequencing
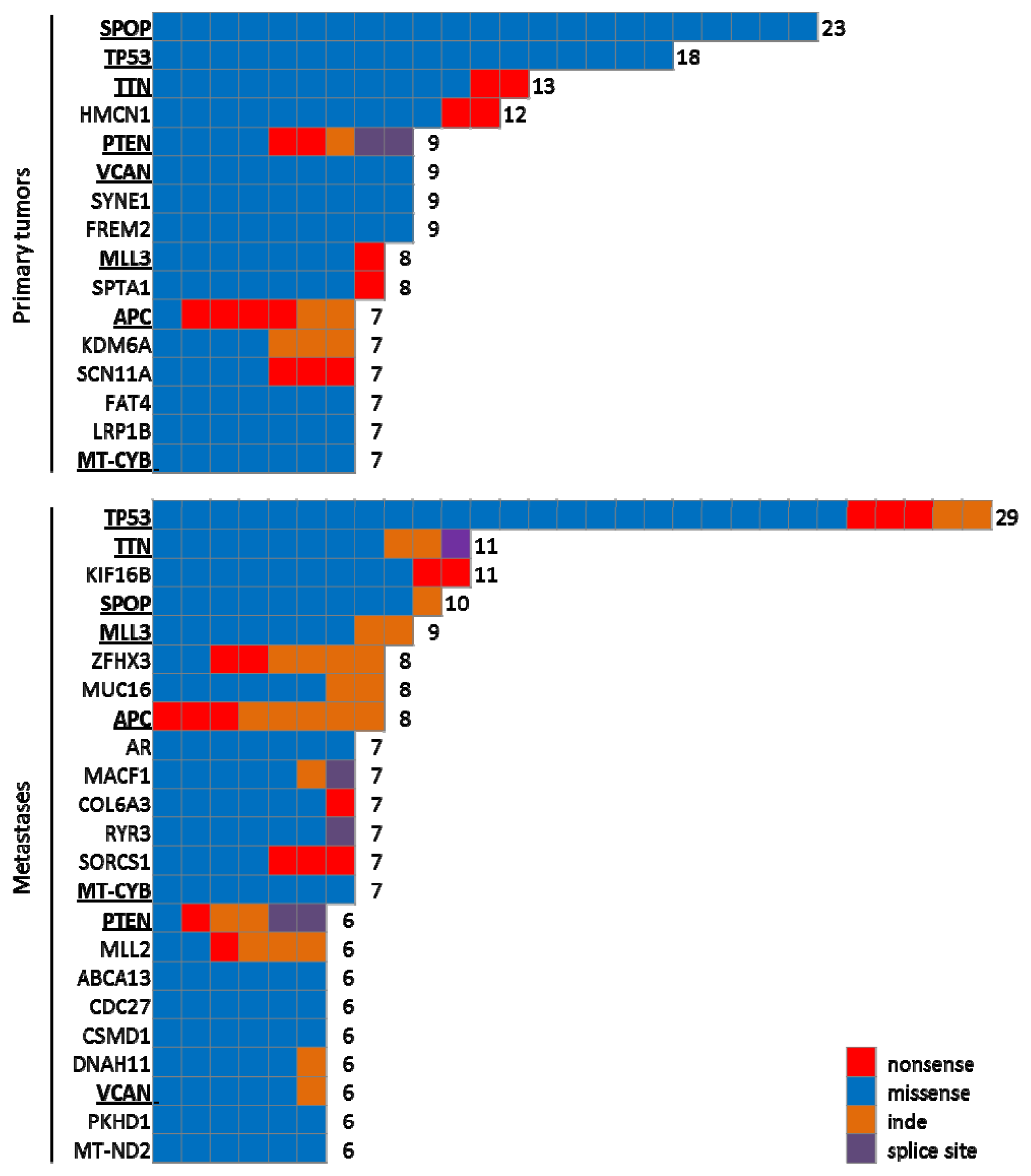
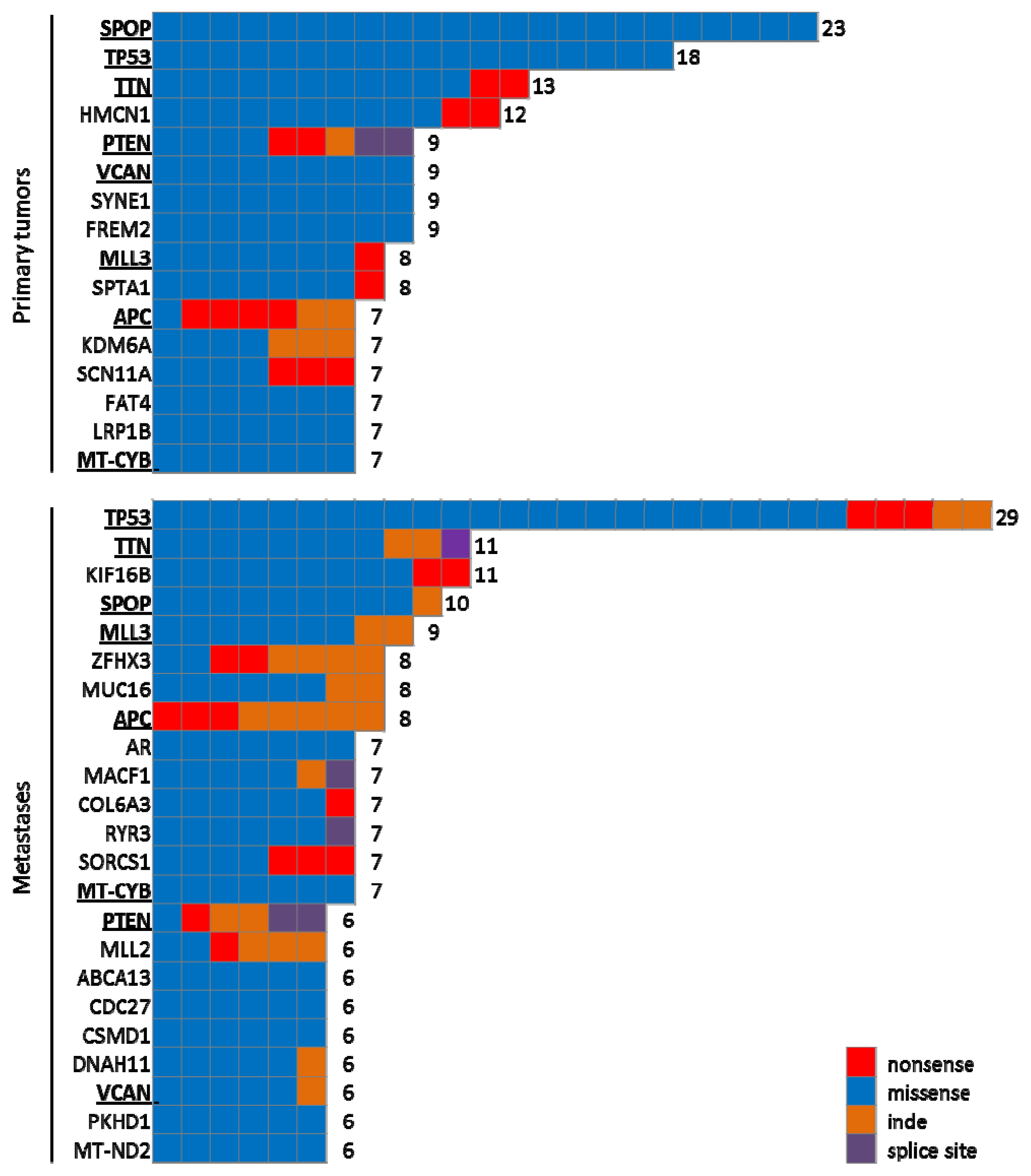
4.1.2. Large Scale Genomic Analyses
4.1.3. Future Perspectives
4.1.3.1. The Use of FFPE Samples
4.1.3.2. Prostate Cancer is a Multi-Focal Disease
4.2. Single Nucleotide Polymorphisms (SNPs)
5. DNA Methylation
6. Non-Coding RNAs
6.1. MicroRNAs
6.2. Long Non-Coding RNAs (lncRNAs)
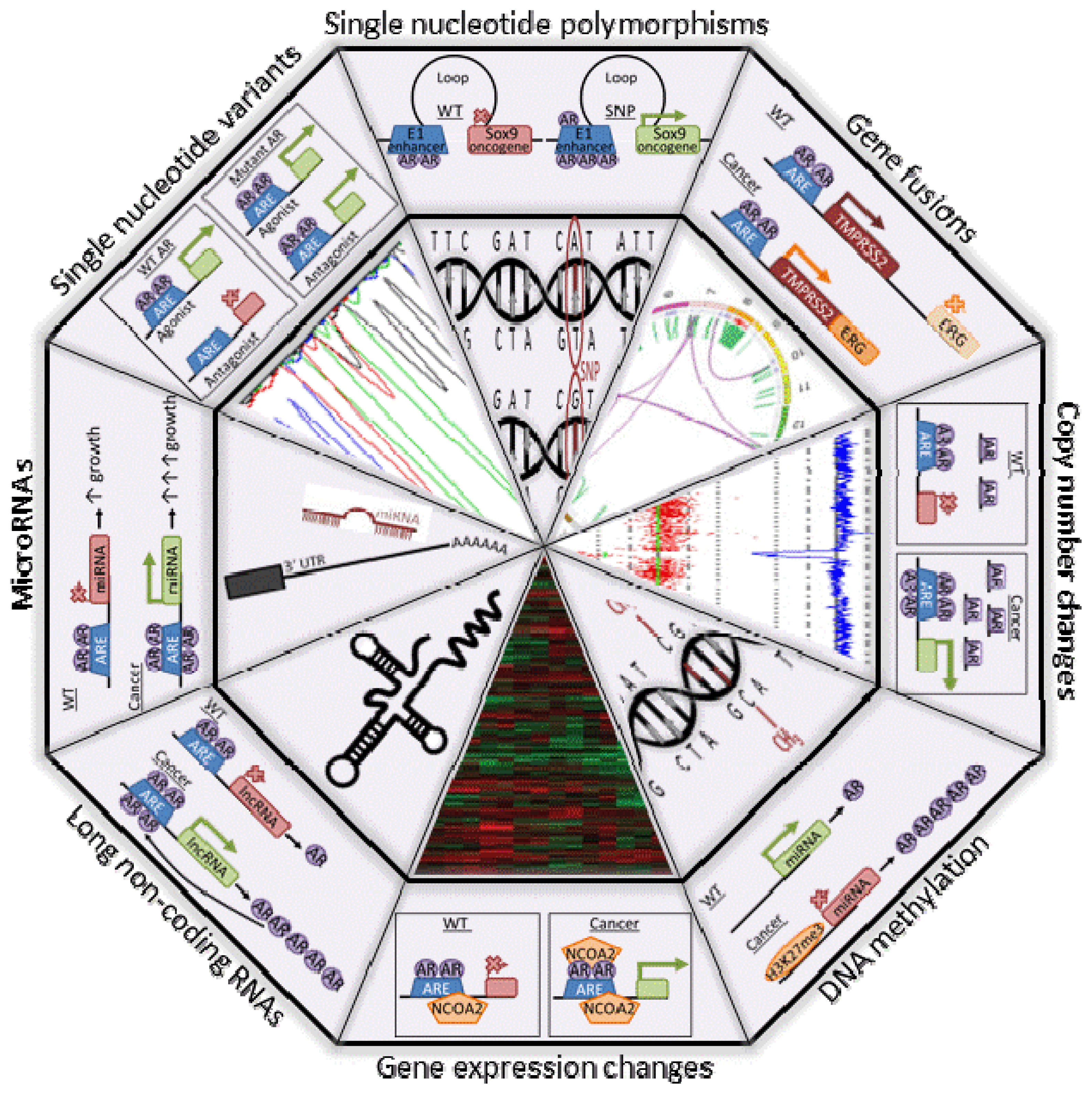
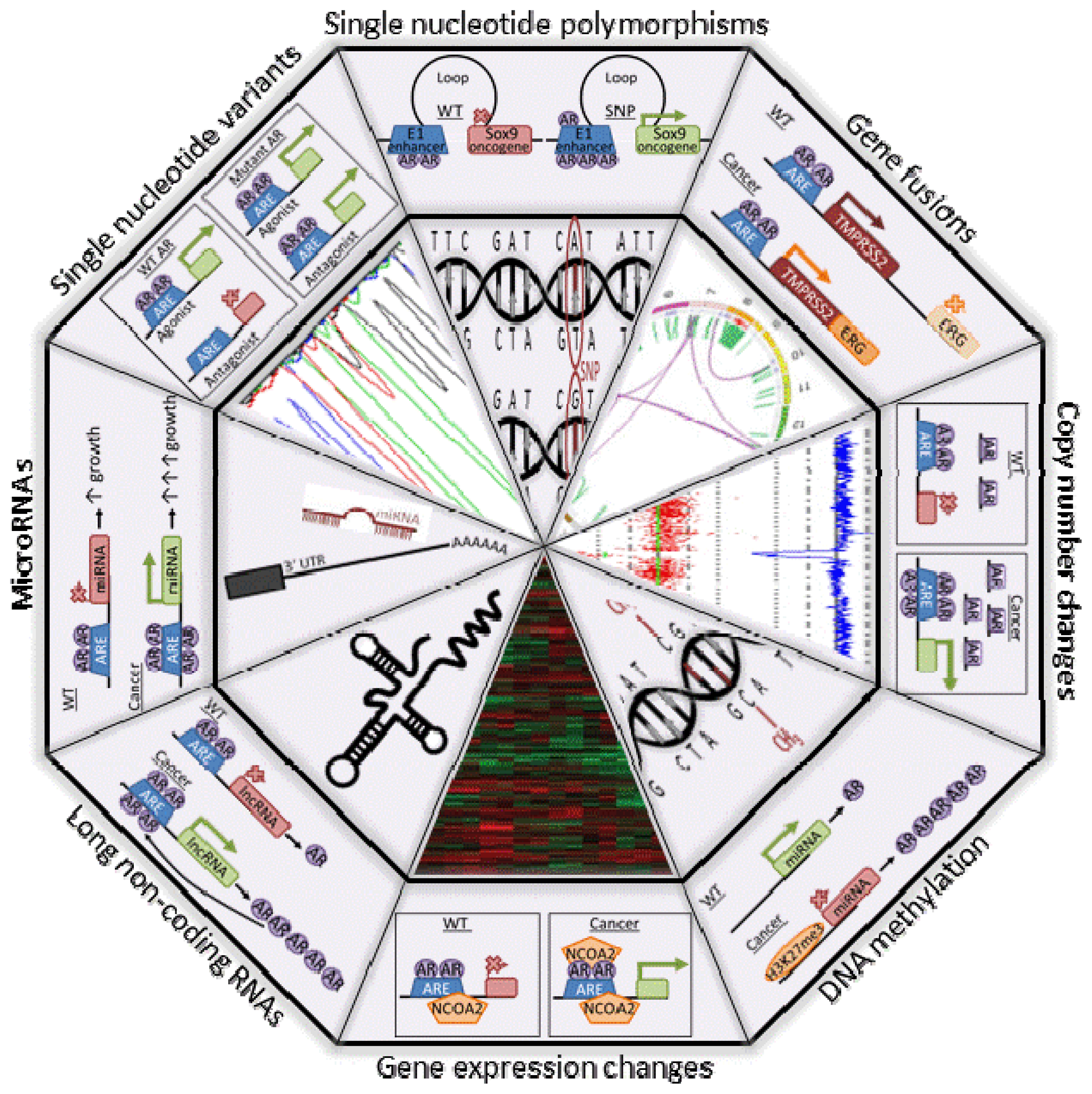
7. A Role of AR in PCa
8. Conclusions
Acknowledgments
Abbreviations
| AR | androgen receptor |
| CNA | copy number alteration |
| CRPC | castration-resistant prostate cancer |
| EMT | epithelial-to-mesenchymal transition |
| ETS | v-ets erythroblastosis virus E26 oncogene |
| FFPE | formalin fixed paraffin embedded |
| GWAS | genome-wide association study |
| indels | short insertions and deletions |
| lncRNA | long non-coding RNA |
| PCa | prostate cancer |
| PIN | prostatic intraepithelial neoplasia |
| PSA | prostate-specific antigen |
| SNP | single nucleotide polymorphism |
| SNV | single nucleotide variant |
| TMPRSS2 | transmembrane protease serine 2 |
| UTR | untranslated region |
Conflict of Interest
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar]
- Cooperberg, M.R.; Moul, J.W.; Carroll, P.R. The changing face of prostate cancer. J. Clin. Oncol 2005, 23, 8146–8151. [Google Scholar]
- Eifler, J.B.; Feng, Z.; Lin, B.M.; Partin, M.T.; Humphreys, E.B.; Han, M.; Epstein, J.I.; Walsh, P.C.; Trock, B.J.; Partin, A.W. An updated prostate cancer staging nomogram (partin tables) based on cases from 2006 to 2011. BJU Int 2013, 111, 22–29. [Google Scholar]
- Kattan, M.W.; Eastham, J.A.; Stapleton, A.M.; Wheeler, T.M.; Scardino, P.T. A preoperative nomogram for disease recurrence following radical prostatectomy for prostate cancer. J. Natl. Cancer Inst 1998, 90, 766–771. [Google Scholar]
- D’Amico, A.V.; Whittington, R.; Malkowicz, S.B.; Schultz, D.; Blank, K.; Broderick, G.A.; Tomaszewski, J.E.; Renshaw, A.A.; Kaplan, I.; Beard, C.J.; et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 1998, 280, 969–974. [Google Scholar]
- Choudhury, A.D.; Eeles, R.; Freedland, S.J.; Isaacs, W.B.; Pomerantz, M.M.; Schalken, J.A.; Tammela, T.L.; Visakorpi, T. The role of genetic markers in the management of prostate cancer. Eur. Urol 2012, 62, 577–587. [Google Scholar]
- Lonigro, R.J.; Grasso, C.S.; Robinson, D.R.; Jing, X.; Wu, Y.M.; Cao, X.; Quist, M.J.; Tomlins, S.A.; Pienta, K.J.; Chinnaiyan, A.M. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia 2011, 13, 1019–1025. [Google Scholar]
- Reynolds, M.A. Molecular alterations in prostate cancer. Cancer Lett 2008, 271, 13–24. [Google Scholar]
- Demichelis, F.; Setlur, S.R.; Beroukhim, R.; Perner, S.; Korbel, J.O.; Lafargue, C.J.; Pflueger, D.; Pina, C.; Hofer, M.D.; Sboner, A.; et al. Distinct genomic aberrations associated with ERG rearranged prostate cancer. Genes Chromosomes Cancer 2009, 48, 366–380. [Google Scholar]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [Green Version]
- Boyd, L.K.; Mao, X.; Xue, L.; Lin, D.; Chaplin, T.; Kudahetti, S.C.; Stankiewicz, E.; Yu, Y.; Beltran, L.; Shaw, G.; et al. High-resolution genome-wide copy-number analysis suggests a monoclonal origin of multifocal prostate cancer. Genes Chromosomes Cancer 2012, 51, 579–589. [Google Scholar]
- Wu, C.; Wyatt, A.W.; Lapuk, A.V.; McPherson, A.; McConeghy, B.J.; Bell, R.H.; Anderson, S.; Haegert, A.; Brahmbhatt, S.; Shukin, R.; et al. Integrated genome and transcriptome sequencing identifies a novel form of hybrid and aggressive prostate cancer. J. Pathol 2012, 227, 53–61. [Google Scholar]
- Liu, W.; Laitinen, S.; Khan, S.; Vihinen, M.; Kowalski, J.; Yu, G.; Chen, L.; Ewing, C.M.; Eisenberger, M.A.; Carducci, M.A.; et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med 2009, 15, 559–565. [Google Scholar]
- Cheng, I.; Levin, A.M.; Tai, Y.C.; Plummer, S.; Chen, G.K.; Neslund-Dudas, C.; Casey, G.; Rybicki, B.A.; Witte, J.S. Copy number alterations in prostate tumors and disease aggressiveness. Genes Chromosomes Cancer 2012, 51, 66–76. [Google Scholar]
- Rowley, J.D. Chromosome translocations: Dangerous liaisons revisited. Nat. Rev. Cancer 2001, 1, 245–250. [Google Scholar]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Smith, L.R.; Roulston, D.; Helgeson, B.E.; Cao, X.; Wei, J.T.; Rubin, M.A.; Shah, R.B.; et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res 2006, 66, 3396–3400. [Google Scholar]
- Donaldson, L.W.; Petersen, J.M.; Graves, B.J.; McIntosh, L.P. Secondary structure of the ETS domain places murine Ets-1 in the superfamily of winged helix-turn-helix DNA-binding proteins. Biochemistry 1994, 33, 13509–13516. [Google Scholar]
- Vaarala, M.H.; Porvari, K.; Kyllonen, A.; Lukkarinen, O.; Vihko, P. The TMPRSS2 gene encoding transmembrane serine protease is overexpressed in a majority of prostate cancer patients: Detection of mutated TMPRSS2 form in a case of aggressive disease. Int. J. Cancer 2001, 94, 705–710. [Google Scholar]
- Vaarala, M.H.; Porvari, K.S.; Kellokumpu, S.; Kyllonen, A.P.; Vihko, P.T. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J. Pathol 2001, 193, 134–140. [Google Scholar]
- Perner, S.; Mosquera, J.M.; Demichelis, F.; Hofer, M.D.; Paris, P.L.; Simko, J.; Collins, C.; Bismar, T.A.; Chinnaiyan, A.M.; de Marzo, A.M.; et al. TMPRSS2-ERG fusion prostate cancer: An early molecular event associated with invasion. Am. J. Surg. Pathol 2007, 31, 882–888. [Google Scholar]
- Mosquera, J.M.; Perner, S.; Demichelis, F.; Kim, R.; Hofer, M.D.; Mertz, K.D.; Paris, P.L.; Simko, J.; Collins, C.; Bismar, T.A.; et al. Morphological features of TMPRSS2-ERG gene fusion prostate cancer. J. Pathol 2007, 212, 91–101. [Google Scholar]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar]
- Pflueger, D.; Terry, S.; Sboner, A.; Habegger, L.; Esgueva, R.; Lin, P.C.; Svensson, M.A.; Kitabayashi, N.; Moss, B.J.; MacDonald, T.Y.; et al. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res 2011, 21, 56–67. [Google Scholar]
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, 97–101. [Google Scholar]
- Esgueva, R.; Perner, S.; Christopher, J.L.; Scheble, V.; Stephan, C.; Lein, M.; Fritzsche, F.R.; Dietel, M.; Kristiansen, G.; Rubin, M.A. Prevalence of TMPRSS2-ERG and SLC45A3-ERG gene fusions in a large prostatectomy cohort. Mod. Pathol 2010, 23, 539–546. [Google Scholar]
- Han, B.; Mehra, R.; Dhanasekaran, S.M.; Yu, J.; Menon, A.; Lonigro, R.J.; Wang, X.; Gong, Y.; Wang, L.; Shankar, S.; et al. A fluorescence in situ hybridization screen for E26 transformation-specific aberrations: Identification of DDX5-ETV4 fusion protein in prostate cancer. Cancer Res 2008, 68, 7629–7637. [Google Scholar]
- Helgeson, B.E.; Tomlins, S.A.; Shah, N.; Laxman, B.; Cao, Q.; Prensner, J.R.; Cao, X.; Singla, N.; Montie, J.E.; Varambally, S.; et al. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res 2008, 68, 73–80. [Google Scholar]
- Rickman, D.S.; Pflueger, D.; Moss, B.; VanDoren, V.E.; Chen, C.X.; de la Taille, A.; Kuefer, R.; Tewari, A.K.; Setlur, S.R.; Demichelis, F.; et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res 2009, 69, 2734–2738. [Google Scholar]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenhan, D.; Minner, S.; Wuttig, D.; Warnatz, H.J.; Stehr, H.; Rausch, T.; et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar]
- Maher, C.A.; Palanisamy, N.; Brenner, J.C.; Cao, X.; Kalyana-Sundaram, S.; Luo, S.; Khrebtukova, I.; Barrette, T.R.; Grasso, C.; Yu, J.; et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 12353–12358. [Google Scholar]
- Pflueger, D.; Rickman, D.S.; Sboner, A.; Perner, S.; LaFargue, C.J.; Svensson, M.A.; Moss, B.J.; Kitabayashi, N.; Pan, Y.; de la Taille, A.; et al. N-myc downstream regulated gene 1 (NDRG1) is fused to ERG in prostate cancer. Neoplasia 2009, 11, 804–811. [Google Scholar]
- Attard, G.; Clark, J.; Ambroisine, L.; Mills, I.G.; Fisher, G.; Flohr, P.; Reid, A.; Edwards, S.; Kovacs, G.; Berney, D.; et al. Heterogeneity and clinical significance of ETV1 translocations in human prostate cancer. Br. J. Cancer 2008, 99, 314–320. [Google Scholar]
- Hermans, K.G.; van der Korput, H.A.; van Marion, R.; van de Wijngaart, D.J.; Ziel-van der Made, A.; Dits, N.F.; Boormans, J.L.; van der Kwast, T.H.; van Dekken, H.; Bangma, C.H.; et al. Truncated ETV1, fused to novel tissue-specific genes, and full-length ETV1 in prostate cancer. Cancer Res 2008, 68, 7541–7549. [Google Scholar]
- Hermans, K.G.; Bressers, A.A.; van der Korput, H.A.; Dits, N.F.; Jenster, G.; Trapman, J. Two unique novel prostate-specific and androgen-regulated fusion partners of ETV4 in prostate cancer. Cancer Res 2008, 68, 3094–3098. [Google Scholar]
- Paulo, P.; Barros-Silva, J.D.; Ribeiro, F.R.; Ramalho-Carvalho, J.; Jeronimo, C.; Henrique, R.; Lind, G.E.; Skotheim, R.I.; Lothe, R.A.; Teixeira, M.R. FLI1 is a novel ETS transcription factor involved in gene fusions in prostate cancer. Gene. Chromosome. Cancer 2012, 51, 240–249. [Google Scholar]
- Lapuk, A.V.; Wu, C.; Wyatt, A.W.; McPherson, A.; McConeghy, B.J.; Brahmbhatt, S.; Mo, F.; Zoubeidi, A.; Anderson, S.; Bell, R.H.; et al. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J. Pathol 2012, 227, 286–297. [Google Scholar]
- Nhili, R.; Peixoto, P.; Depauw, S.; Flajollet, S.; Dezitter, X.; Munde, M.M.; Ismail, M.A.; Kumar, A.; Farahat, A.A.; Stephens, C.E.; et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res 2013, 41, 125–138. [Google Scholar]
- Palanisamy, N.; Ateeq, B.; Kalyana-Sundaram, S.; Pflueger, D.; Ramnarayanan, K.; Shankar, S.; Han, B.; Cao, Q.; Cao, X.; Suleman, K.; et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat. Med 2010, 16, 793–798. [Google Scholar]
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; Macdonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur. Urol 2013, 63, 920–926. [Google Scholar]
- Schaefer, G.; Mosquera, J.M.; Ramoner, R.; Park, K.; Romanel, A.; Steiner, E.; Horninger, W.; Bektic, J.; Ladurner-Rennau, M.; Rubin, M.A.; et al. Distinct ERG rearrangement prevalence in prostate cancer: Higher frequency in young age and in low PSA prostate cancer. Prostate Cancer Prostatic Dis 2013, 16, 132–138. [Google Scholar]
- Minner, S.; Enodien, M.; Sirma, H.; Luebke, A.M.; Krohn, A.; Mayer, P.S.; Simon, R.; Tennstedt, P.; Muller, J.; Scholz, L.; et al. ERG status is unrelated to PSA recurrence in radically operated prostate cancer in the absence of antihormonal therapy. Clin. Cancer Res 2011, 17, 5878–5888. [Google Scholar]
- Attard, G.; Clark, J.; Ambroisine, L.; Fisher, G.; Kovacs, G.; Flohr, P.; Berney, D.; Foster, C.S.; Fletcher, A.; Gerald, W.L.; et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 2008, 27, 253–263. [Google Scholar]
- Mehra, R.; Tomlins, S.A.; Yu, J.; Cao, X.; Wang, L.; Menon, A.; Rubin, M.A.; Pienta, K.J.; Shah, R.B.; Chinnaiyan, A.M. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res 2008, 68, 3584–3590. [Google Scholar]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; Macdonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet 2009, 41, 524–526. [Google Scholar]
- Carver, B.S.; Tran, J.; Gopalan, A.; Chen, Z.; Shaikh, S.; Carracedo, A.; Alimonti, A.; Nardella, C.; Varmeh, S.; Scardino, P.T.; et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nature Genet 2009, 41, 619–624. [Google Scholar]
- Casey, O.M.; Fang, L.; Hynes, P.G.; Abou-Kheir, W.G.; Martin, P.L.; Tillman, H.S.; Petrovics, G.; Awwad, H.O.; Ward, Y.; Lake, R.; et al. TMPRSS2-driven ERG expression in vivo increases self-renewal and maintains expression in a castration resistant subpopulation. PLoS One 2012, 7, e41668. [Google Scholar]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar]
- Goh, C.L.; Schumacher, F.R.; Easton, D.; Muir, K.; Henderson, B.; Kote-Jarai, Z.; Eeles, R.A. Genetic variants associated with predisposition to prostate cancer and potential clinical implications. J. Int. Med 2012, 271, 353–365. [Google Scholar]
- Robbins, C.M.; Tembe, W.A.; Baker, A.; Sinari, S.; Moses, T.Y.; Beckstrom-Sternberg, S.; Beckstrom-Sternberg, J.; Barrett, M.; Long, J.; Chinnaiyan, A.; et al. Copy number and targeted mutational analysis reveals novel somatic events in metastatic prostate tumors. Genome Res 2011, 21, 47–55. [Google Scholar]
- Kumar, A.; White, T.A.; MacKenzie, A.P.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Rieder, M.J.; et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar]
- Spans, L.; Atak, Z.K.; van Nieuwerburgh, F.; Deforce, D.; Lerut, E.; Aerts, S.; Claessens, F. Variations in the exome of the lncap prostate cancer cell line. Prostate 2012, 72, 1317–1327. [Google Scholar]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet 2012, 44, 685–689. [Google Scholar]
- Leach, F.S.; Velasco, A.; Hsieh, J.T.; Sagalowsky, A.I.; McConnell, J.D. The mismatch repair gene hMSH2 is mutated in the prostate cancer cell line LNCaP. J. Urol 2000, 164, 1830–1833. [Google Scholar]
- Chen, Y.; Wang, J.; Fraig, M.M.; Metcalf, J.; Turner, W.R.; Bissada, N.K.; Watson, D.K.; Schweinfest, C.W. Defects of DNA mismatch repair in human prostate cancer. Cancer Res 2001, 61, 4112–4121. [Google Scholar]
- Imamura, Y.; Sakamoto, S.; Endo, T.; Utsumi, T.; Fuse, M.; Suyama, T.; Kawamura, K.; Imamoto, T.; Yano, K.; Uzawa, K.; et al. Foxa1 promotes tumor progression in prostate cancer via the insulin-like growth factor binding protein 3 pathway. PLoS One 2012, 7, e42456. [Google Scholar]
- Sahu, B.; Laakso, M.; Ovaska, K.; Mirtti, T.; Lundin, J.; Rannikko, A.; Sankila, A.; Turunen, J.P.; Lundin, M.; Konsti, J.; et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J 2011, 30, 3962–3976. [Google Scholar]
- Makinen, N.; Mehine, M.; Tolvanen, J.; Kaasinen, E.; Li, Y.; Lehtonen, H.J.; Gentile, M.; Yan, J.; Enge, M.; Taipale, M.; et al. Med12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science 2011, 334, 252–255. [Google Scholar]
- Taatjes, D.J. The human mediator complex: A versatile, genome-wide regulator of transcription. Trends Biochem. Sci 2010, 35, 315–322. [Google Scholar]
- Majumder, P.K.; Grisanzio, C.; O’Connell, F.; Barry, M.; Brito, J.M.; Xu, Q.; Guney, I.; Berger, R.; Herman, P.; Bikoff, R.; et al. A prostatic intraepithelial neoplasia-dependent p27kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell 2008, 14, 146–155. [Google Scholar]
- Li, C.; Ao, J.; Fu, J.; Lee, D.F.; Xu, J.; Lonard, D.; O’Malley, B.W. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene 2011, 30, 4350–4364. [Google Scholar]
- Natarajan, T.G.; Kallakury, B.V.; Sheehan, C.E.; Bartlett, M.B.; Ganesan, N.; Preet, A.; Ross, J.S.; Fitzgerald, K.T. Epigenetic regulator MLL2 shows altered expression in cancer cell lines and tumors from human breast and colon. Cancer Cell Int. 2010, 10. [Google Scholar] [CrossRef]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev 2011, 25, 2158–2172. [Google Scholar]
- Lindberg, J.; Mills, I.G.; Klevebring, D.; Liu, W.; Neiman, M.; Xu, J.; Wikstrom, P.; Wiklund, P.; Wiklund, F.; Egevad, L.; et al. The mitochondrial and autosomal mutation landscapes of prostate cancer. Eur. Urol 2013, 63, 702–708. [Google Scholar]
- Lindberg, J.; Klevebring, D.; Liu, W.; Neiman, M.; Xu, J.; Wiklund, P.; Wiklund, F.; Mills, I.G.; Egevad, L.; Gronberg, H. Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. Eur. Urol 2013, 63, 347–353. [Google Scholar]
- Menon, R.; Deng, M.; Boehm, D.; Braun, M.; Fend, F.; Boehm, D.; Biskup, S.; Perner, S. Exome enrichment and SOLiD sequencing of formalin fixed paraffin embedded (FFPE) prostate cancer tissue. Int. J. Mol. Sci 2012, 13, 8933–8942. [Google Scholar]
- MacInnis, R.J.; Antoniou, A.C.; Eeles, R.A.; Severi, G.; Guy, M.; McGuffog, L.; Hall, A.L.; O’Brien, L.T.; Wilkinson, R.A.; Dearnaley, D.P.; et al. Prostate cancer segregation analyses using 4390 families from UK and Australian population-based studies. Genet. Epidemiol 2010, 34, 42–50. [Google Scholar]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer— Analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med 2000, 343, 78–85. [Google Scholar]
- Zhang, X.; Cowper-Sal lari, R.; Bailey, S.D.; Moore, J.H.; Lupien, M. Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus. Genome Res 2012, 22, 1437–1446. [Google Scholar]
- Eeles, R.A.; Olama, A.A.; Benlloch, S.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Ghoussaini, M.; Luccarini, C.; Dennis, J.; Jugurnauth-Little, S.; et al. Identification of 23 new prostate cancer susceptibility loci using the icogs custom genotyping array. Nat. Genet 2013, 45, 385–391. [Google Scholar]
- Jin, G.; Sun, J.; Isaacs, S.D.; Wiley, K.E.; Kim, S.T.; Chu, L.W.; Zhang, Z.; Zhao, H.; Zheng, S.L.; Isaacs, W.B.; et al. Human polymorphisms at long non-coding RNAs (lncRNAs) and association with prostate cancer risk. Carcinogenesis 2011, 32, 1655–1659. [Google Scholar]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar]
- Whitaker, H.C.; Kote-Jarai, Z.; Ross-Adams, H.; Warren, A.Y.; Burge, J.; George, A.; Bancroft, E.; Jhavar, S.; Leongamornlert, D.; Tymrakiewicz, M.; et al. The rs10993994 risk allele for prostate cancer results in clinically relevant changes in microseminoprotein-beta expression in tissue and urine. PLoS One 2010, 5, e13363. [Google Scholar]
- Whitaker, H.C.; Warren, A.Y.; Eeles, R.; Kote-Jarai, Z.; Neal, D.E. The potential value of microseminoprotein-β as a prostate cancer biomarker and therapeutic target. Prostate 2010, 70, 333–340. [Google Scholar]
- Cramer, S.D.; Chang, B.L.; Rao, A.; Hawkins, G.A.; Zheng, S.L.; Wade, W.N.; Cooke, R.T.; Thomas, L.N.; Bleecker, E.R.; Catalona, W.J.; et al. Association between genetic polymorphisms in the prostate-specific antigen gene promoter and serum prostate-specific antigen levels. J. Nat. Cancer Ins 2003, 95, 1044–1053. [Google Scholar]
- Severi, G.; Hayes, V.M.; Neufing, P.; Padilla, E.J.; Tilley, W.D.; Eggleton, S.A.; Morris, H.A.; English, D.R.; Southey, M.C.; Hopper, J.L.; et al. Variants in the prostate-specific antigen (PSA) gene and prostate cancer risk, survival, and circulating PSA. Cancer Epidemiol. Biomarkers Prev 2006, 15, 1142–1147. [Google Scholar]
- Kote-Jarai, Z.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Dadaev, T.; Jugurnauth-Little, S.; Ross-Adams, H.; Al Olama, A.A.; Benlloch, S.; Halim, S.; et al. Fine-mapping identifies multiple prostate cancer risk loci at 5p15, one of which associates with TERT expression. Hum. Mol. Genet. 2013. [Google Scholar] [CrossRef]
- Jin, G.; Lu, L.; Cooney, K.A.; Ray, A.M.; Zuhlke, K.A.; Lange, E.M.; Cannon-Albright, L.A.; Camp, N.J.; Teerlink, C.C.; Fitzgerald, L.M.; et al. Validation of prostate cancer risk-related loci identified from genome-wide association studies using family-based association analysis: Evidence from the international consortium for prostate cancer genetics (ICPCG). Hum. Genet 2012, 131, 1095–1103. [Google Scholar]
- Amin Al Olama, A.; Kote-Jarai, Z.; Schumacher, F.R.; Wiklund, F.; Berndt, S.I.; Benlloch, S.; Giles, G.G.; Severi, G.; Neal, D.E.; Hamdy, F.C.; et al. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum. Mol. Genet 2013, 22, 408–415. [Google Scholar]
- FitzGerald, L.M.; Kwon, E.M.; Conomos, M.P.; Kolb, S.; Holt, S.K.; Levine, D.; Feng, Z.; Ostrander, E.A.; Stanford, J.L. Genome-wide association study identifies a genetic variant associated with risk for more aggressive prostate cancer. Cancer Epidemiol. Biomark. Prev 2011, 20, 1196–1203. [Google Scholar]
- Duggan, D.; Zheng, S.L.; Knowlton, M.; Benitez, D.; Dimitrov, L.; Wiklund, F.; Robbins, C.; Isaacs, S.D.; Cheng, Y.; Li, G.; et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J. Natl. Cancer Inst 2007, 99, 1836–1844. [Google Scholar]
- Kote-Jarai, Z.; Olama, A.A.; Giles, G.G.; Severi, G.; Schleutker, J.; Weischer, M.; Campa, D.; Riboli, E.; Key, T.; Gronberg, H.; et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat. Genet 2011, 43, 785–791. [Google Scholar]
- Gudmundsson, J.; Sulem, P.; Rafnar, T.; Bergthorsson, J.T.; Manolescu, A.; Gudbjartsson, D.; Agnarsson, B.A.; Sigurdsson, A.; Benediktsdottir, K.R.; Blondal, T.; et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat. Genet 2008, 40, 281–283. [Google Scholar]
- Eeles, R.A.; Kote-Jarai, Z.; Al Olama, A.A.; Giles, G.G.; Guy, M.; Severi, G.; Muir, K.; Hopper, J.L.; Henderson, B.E.; Haiman, C.A.; et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat. Genet 2009, 41, 1116–1121. [Google Scholar]
- Schumacher, F.R.; Berndt, S.I.; Siddiq, A.; Jacobs, K.B.; Wang, Z.; Lindstrom, S.; Stevens, V.L.; Chen, C.; Mondul, A.M.; Travis, R.C.; et al. Genome-wide association study identifies new prostate cancer susceptibility loci. Hum. Mol. Genet 2011, 20, 3867–3875. [Google Scholar]
- Eeles, R.A.; Kote-Jarai, Z.; Giles, G.G.; Olama, A.A.; Guy, M.; Jugurnauth, S.K.; Mulholland, S.; Leongamornlert, D.A.; Edwards, S.M.; Morrison, J.; et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat. Genet 2008, 40, 316–321. [Google Scholar]
- Gudmundsson, J.; Sulem, P.; Gudbjartsson, D.F.; Blondal, T.; Gylfason, A.; Agnarsson, B.A.; Benediktsdottir, K.R.; Magnusdottir, D.N.; Orlygsdottir, G.; Jakobsdottir, M.; et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat. Genet 2009, 41, 1122–1126. [Google Scholar] [Green Version]
- Thomas, G.; Jacobs, K.B.; Yeager, M.; Kraft, P.; Wacholder, S.; Orr, N.; Yu, K.; Chatterjee, N.; Welch, R.; Hutchinson, A.; et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet 2008, 40, 310–315. [Google Scholar]
- Al Olama, A.A.; Kote-Jarai, Z.; Giles, G.G.; Guy, M.; Morrison, J.; Severi, G.; Leongamornlert, D.A.; Tymrakiewicz, M.; Jhavar, S.; Saunders, E.; et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat. Genet 2009, 41, 1058–1060. [Google Scholar]
- Yeager, M.; Chatterjee, N.; Ciampa, J.; Jacobs, K.B.; Gonzalez-Bosquet, J.; Hayes, R.B.; Kraft, P.; Wacholder, S.; Orr, N.; Berndt, S.; et al. Identification of a new prostate cancer susceptibility locus on chromosome 8q24. Nat. Genet 2009, 41, 1055–1057. [Google Scholar]
- Haiman, C.A.; Patterson, N.; Freedman, M.L.; Myers, S.R.; Pike, M.C.; Waliszewska, A.; Neubauer, J.; Tandon, A.; Schirmer, C.; McDonald, G.J.; et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat. Genet 2007, 39, 638–644. [Google Scholar]
- Gudmundsson, J.; Sulem, P.; Manolescu, A.; Amundadottir, L.T.; Gudbjartsson, D.; Helgason, A.; Rafnar, T.; Bergthorsson, J.T.; Agnarsson, B.A.; Baker, A.; et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat. Genet 2007, 39, 631–637. [Google Scholar]
- Yeager, M.; Orr, N.; Hayes, R.B.; Jacobs, K.B.; Kraft, P.; Wacholder, S.; Minichiello, M.J.; Fearnhead, P.; Yu, K.; Chatterjee, N.; et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat. Genet 2007, 39, 645–649. [Google Scholar]
- Amundadottir, L.T.; Sulem, P.; Gudmundsson, J.; Helgason, A.; Baker, A.; Agnarsson, B.A.; Sigurdsson, A.; Benediktsdottir, K.R.; Cazier, J.B.; Sainz, J.; et al. A common variant associated with prostate cancer in european and african populations. Nat. Genet 2006, 38, 652–658. [Google Scholar]
- Chung, C.C.; Ciampa, J.; Yeager, M.; Jacobs, K.B.; Berndt, S.I.; Hayes, R.B.; Gonzalez-Bosquet, J.; Kraft, P.; Wacholder, S.; Orr, N.; et al. Fine mapping of a region of chromosome 11q13 reveals multiple independent loci associated with risk of prostate cancer. Hum. Mol. Genet 2011, 20, 2869–2878. [Google Scholar]
- Sun, J.; Zheng, S.L.; Wiklund, F.; Isaacs, S.D.; Purcell, L.D.; Gao, Z.; Hsu, F.C.; Kim, S.T.; Liu, W.; Zhu, Y.; et al. Evidence for two independent prostate cancer risk-associated loci in the HNF1B gene at 17q12. Nat. Genet 2008, 40, 1153–1155. [Google Scholar]
- Gudmundsson, J.; Sulem, P.; Steinthorsdottir, V.; Bergthorsson, J.T.; Thorleifsson, G.; Manolescu, A.; Rafnar, T.; Gudbjartsson, D.; Agnarsson, B.A.; Baker, A.; et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat. Genet 2007, 39, 977–983. [Google Scholar]
- Majumdar, S.; Buckles, E.; Estrada, J.; Koochekpour, S. Aberrant DNA methylation and prostate cancer. Curr. Genomics 2011, 12, 486–505. [Google Scholar]
- Yegnasubramanian, S.; Haffner, M.C.; Zhang, Y.; Gurel, B.; Cornish, T.C.; Wu, Z.; Irizarry, R.A.; Morgan, J.; Hicks, J.; DeWeese, T.L.; et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res 2008, 68, 8954–8967. [Google Scholar]
- Lee, W.H.; Morton, R.A.; Epstein, J.I.; Brooks, J.D.; Campbell, P.A.; Bova, G.S.; Hsieh, W.S.; Isaacs, W.B.; Nelson, W.G. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc. Natl. Acad. .Sci. USA 1994, 91, 11733–11737. [Google Scholar]
- Shi, X.B.; Xue, L.; Ma, A.H.; Tepper, C.G.; Gandour-Edwards, R.; Kung, H.J.; Devere White, R.W. Tumor suppressive miR-124 targets androgen receptor and inhibits proliferation of prostate cancer cells. Oncogene 2012. [Google Scholar] [CrossRef]
- Kron, K.; Pethe, V.; Briollais, L.; Sadikovic, B.; Ozcelik, H.; Sunderji, A.; Venkateswaran, V.; Pinthus, J.; Fleshner, N.; van der Kwast, T.; et al. Discovery of novel hypermethylated genes in prostate cancer using genomic CpG island microarrays. PLoS One 2009, 4, e4830. [Google Scholar]
- Kim, S.J.; Kelly, W.K.; Fu, A.; Haines, K.; Hoffman, A.; Zheng, T.; Zhu, Y. Genome-wide methylation analysis identifies involvement of TNF-α mediated cancer pathways in prostate cancer. Cancer Lett 2011, 302, 47–53. [Google Scholar]
- Kim, Y.J.; Yoon, H.Y.; Kim, S.K.; Kim, Y.W.; Kim, E.J.; Kim, I.Y.; Kim, W.J. EFEMP1 as a novel DNA methylation marker for prostate cancer: Array-based DNA methylation and expression profiling. Clin. Cancer Res 2011, 17, 4523–4530. [Google Scholar]
- Kobayashi, Y.; Absher, D.M.; Gulzar, Z.G.; Young, S.R.; McKenney, J.K.; Peehl, D.M.; Brooks, J.D.; Myers, R.M.; Sherlock, G. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res 2011, 21, 1017–1027. [Google Scholar]
- Kim, J.H.; Dhanasekaran, S.M.; Prensner, J.R.; Cao, X.; Robinson, D.; Kalyana-Sundaram, S.; Huang, C.; Shankar, S.; Jing, X.; Iyer, M.; et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res 2011, 21, 1028–1041. [Google Scholar]
- Kim, J.W.; Kim, S.T.; Turner, A.R.; Young, T.; Smith, S.; Liu, W.; Lindberg, J.; Egevad, L.; Gronberg, H.; Isaacs, W.B.; et al. Identification of new differentially methylated genes that have potential functional consequences in prostate cancer. PLoS One 2012, 7, e48455. [Google Scholar]
- Aryee, M.J.; Liu, W.; Engelmann, J.C.; Nuhn, P.; Gurel, M.; Haffner, M.C.; Esopi, D.; Irizarry, R.A.; Getzenberg, R.H.; Nelson, W.G.; et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Borno, S.T.; Fischer, A.; Kerick, M.; Falth, M.; Laible, M.; Brase, J.C.; Kuner, R.; Dahl, A.; Grimm, C.; Sayanjali, B.; et al. Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative prostate cancers implicate an EZH2-dependent mechanism with miR-26a hypermethylation. Cancer Discov 2012, 2, 1024–1035. [Google Scholar]
- Schwartzman, J.; Mongoue-Tchokote, S.; Gibbs, A.; Gao, L.; Corless, C.L.; Jin, J.; Zarour, L.; Higano, C.; True, L.D.; Vessella, R.L.; et al. A DNA methylation microarray-based study identifies ERG as a gene commonly methylated in prostate cancer. Epigenetics 2011, 6, 1248–1256. [Google Scholar]
- Agell, L.; Hernandez, S.; Nonell, L.; Lorenzo, M.; Puigdecanet, E.; de Muga, S.; Juanpere, N.; Bermudo, R.; Fernandez, P.L.; Lorente, J.A.; et al. A 12-gene expression signature is associated with aggressive histological in prostate cancer: SEC14L1 and TCEB1 genes are potential markers of progression. Am. J. Pathol 2012, 181, 1585–1594. [Google Scholar]
- Bismar, T.A.; Demichelis, F.; Riva, A.; Kim, R.; Varambally, S.; He, L.; Kutok, J.; Aster, J.C.; Tang, J.; Kuefer, R.; et al. Defining aggressive prostate cancer using a 12-gene model. Neoplasia 2006, 8, 59–68. [Google Scholar]
- Sun, R.; Fu, X.; Li, Y.; Xie, Y.; Mao, Y. Global gene expression analysis reveals reduced abundance of putative microRNA targets in human prostate tumours. BMC Genomics 2009, 10, e93. [Google Scholar]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar]
- Carlsson, J.; Davidsson, S.; Helenius, G.; Karlsson, M.; Lubovac, Z.; Andren, O.; Olsson, B.; Klinga-Levan, K. A miRNA expression signature that separates between normal and malignant prostate tissues. Cancer Cell Int 2011, 11, e14. [Google Scholar]
- Fuse, M.; Kojima, S.; Enokida, H.; Chiyomaru, T.; Yoshino, H.; Nohata, N.; Kinoshita, T.; Sakamoto, S.; Naya, Y.; Nakagawa, M.; et al. Tumor suppressive microRNAs (miR-222 and miR-31) regulate molecular pathways based on microRNA expression signature in prostate cancer. J. Hum. Genet 2012, 57, 691–699. [Google Scholar]
- O’Kelly, F.; Marignol, L.; Meunier, A.; Lynch, T.H.; Perry, A.S.; Hollywood, D. MicroRNAs as putative mediators of treatment response in prostate cancer. Nat. Rev. Urol 2012, 9, 397–407. [Google Scholar]
- Ostling, P.; Leivonen, S.K.; Aakula, A.; Kohonen, P.; Makela, R.; Hagman, Z.; Edsjo, A.; Kangaspeska, S.; Edgren, H.; Nicorici, D.; et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res 2011, 71, 1956–1967. [Google Scholar]
- Shi, X.B.; Xue, L.; Yang, J.; Ma, A.H.; Zhao, J.; Xu, M.; Tepper, C.G.; Evans, C.P.; Kung, H.J.; deVere White, R.W. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 19983–19988. [Google Scholar]
- Ribas, J.; Ni, X.; Haffner, M.; Wentzel, E.A.; Salmasi, A.H.; Chowdhury, W.H.; Kudrolli, T.A.; Yegnasubramanian, S.; Luo, J.; Rodriguez, R.; et al. miR-21: An androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res 2009, 69, 7165–7169. [Google Scholar]
- Waltering, K.K.; Porkka, K.P.; Jalava, S.E.; Urbanucci, A.; Kohonen, P.J.; Latonen, L.M.; Kallioniemi, O.P.; Jenster, G.; Visakorpi, T. Androgen regulation of micro-RNAs in prostate cancer. Prostate 2011, 71, 604–614. [Google Scholar]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature Cell Biol 2008, 10, 593–601. [Google Scholar]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem 2008, 283, 14910–14914. [Google Scholar]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008, 22, 894–907. [Google Scholar]
- Tucci, P.; Agostini, M.; Grespi, F.; Markert, E.K.; Terrinoni, A.; Vousden, K.H.; Muller, P.A.; Dotsch, V.; Kehrloesser, S.; Sayan, B.S.; et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 15312–15317. [Google Scholar]
- Kong, D.; Li, Y.; Wang, Z.; Banerjee, S.; Ahmad, A.; Kim, H.R.; Sarkar, F.H. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells 2009, 27, 1712–1721. [Google Scholar]
- Peng, X.; Guo, W.; Liu, T.; Wang, X.; Tu, X.; Xiong, D.; Chen, S.; Lai, Y.; Du, H.; Chen, G.; et al. Identification of miRs-143 and -145 that is associated with bone metastasis of prostate cancer and involved in the regulation of EMT. PLoS One 2011, 6, e20341. [Google Scholar]
- Li, T.; Li, D.; Sha, J.; Sun, P.; Huang, Y. MicroRNA-21 directly targets marcks and promotes apoptosis resistance and invasion in prostate cancer cells. Biochem. Biophys. Res. Commun 2009, 383, 280–285. [Google Scholar]
- Lee, Y.S.; Kim, H.K.; Chung, S.; Kim, K.S.; Dutta, A. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J. Biol. Chem 2005, 280, 16635–16641. [Google Scholar]
- Spahn, M.; Kneitz, S.; Scholz, C.J.; Stenger, N.; Rudiger, T.; Strobel, P.; Riedmiller, H.; Kneitz, B. Expression of microRNA-221 is progressively reduced in aggressive prostate cancer and metastasis and predicts clinical recurrence. Int. J. Cancer. 2010, 127, 394–403. [Google Scholar]
- Selth, L.A.; Townley, S.; Gillis, J.L.; Ochnik, A.M.; Murti, K.; Macfarlane, R.J.; Chi, K.N.; Marshall, V.R.; Tilley, W.D.; Butler, L.M. Discovery of circulating micrornas associated with human prostate cancer using a mouse model of disease. Int. J. Cancer 2012, 131, 652–661. [Google Scholar]
- Bussemakers, M.J.; van Bokhoven, A.; Verhaegh, G.W.; Smit, F.P.; Karthaus, H.F.; Schalken, J.A.; Debruyne, F.M.; Ru, N.; Isaacs, W.B. Dd3: A new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res 1999, 59, 5975–5979. [Google Scholar]
- Wang, R.; Chinnaiyan, A.M.; Dunn, R.L.; Wojno, K.J.; Wei, J.T. Rational approach to implementation of prostate cancer antigen 3 into clinical care. Cancer 2009, 115, 3879–3886. [Google Scholar]
- Srikantan, V.; Zou, Z.; Petrovics, G.; Xu, L.; Augustus, M.; Davis, L.; Livezey, J.R.; Connell, T.; Sesterhenn, I.A.; Yoshino, K.; et al. Pcgem1, a prostate-specific gene, is overexpressed in prostate cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 12216–12221. [Google Scholar]
- Chung, S.; Nakagawa, H.; Uemura, M.; Piao, L.; Ashikawa, K.; Hosono, N.; Takata, R.; Akamatsu, S.; Kawaguchi, T.; Morizono, T.; et al. Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci 2011, 102, 245–252. [Google Scholar]
- Cui, Z.; Ren, S.; Lu, J.; Wang, F.; Xu, W.; Sun, Y.; Wei, M.; Chen, J.; Gao, X.; Xu, C.; et al. The prostate cancer-up-regulated long noncoding RNA PlncRNA-1 modulates apoptosis and proliferation through reciprocal regulation of androgen receptor. Urol. Oncol. 2012. [Google Scholar] [CrossRef]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D.; et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol 2011, 29, 742–749. [Google Scholar]
- Balk, S.P.; Knudsen, K.E. Ar, the cell cycle, and prostate cancer. Nucl. Recept. Signal 2008, 6, e001. [Google Scholar]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar]

{kind=link}
{kind=link}
| 5′ partner | 3′ partner | Reference | 5′ partner | 3′ partner | Reference |
|---|---|---|---|---|---|
| TMPRSS2 | ERG | [18] | TMPRSS2 | ETV1 | [18] |
| HERPUD1 | ERG | [33] | SLC45A3 | ETV1 | [25] |
| SLC45A3 | ERG | [29] | C15orf21 | ETV1 | [25] |
| NDRG1 | ERG | [34] | HNRPA2B1 | ETV1 | [25] |
| FKBP5 | ERG | [26] | FLJ35294 | ETV1 | [29] |
| TMPRSS2 | ETV4 | [19] | ACSL3 | ETV1 | [35] |
| DDX5 | ETV4 | [29] | EST14 | ETV1 | [36] |
| CANT1 | ETV4 | [37] | HERVK17 | ETV1 | [36] |
| KLK2 | ETV4 | [37] | HERVK22q11.23 | ETV1 | [25] |
| TMPRSS2 | ETV5 | [30] | FOXP1 | ETV1 | [36] |
| SLC45A3 | ETV5 | [30] | KLK2 | ETV1 | [26] |
| SLC45A3 | FLI1 | [38] | FUBP1 | ETV1 | [39] |
| SLC45A3 | ELK4 | [27] | SNURF | ETV1 | [32] |
| Nearest Known Gene Within 100 kb | Chromosomal Locus | SNP | Region | References | OR a |
|---|---|---|---|---|---|
| KCNN3 | 1q23 | rs1218582 | Intronic | [75] | 1.03–1.09 |
| MDM4 | 1q32 | rs4245739 | Exonic/Coding | [75] | 0.88–0.95 |
| GGCX | 2p11 | rs10187424 | Intergenic | [76] | 1.06–1.19 |
| EHBP1 | 2p15 | rs721048 | Intronic | [77] | 1.15 |
| THADA | 2p21 | rs1465618 | Intronic | [78] | 1.16–1.20 |
| TAF1B:GRHL1 | 2p25 | rs11902236 | Intronic | [75] | 1.03–1.10 |
| ITGA6 | 2q31 | rs12621278 | Intronic | [78] | 1.32–1.47 |
| MLPH | 2q37 | rs2292884 | Intronic | [79] | 1.14 |
| FARP2 | 2q37 | rs3771570 | Intronic | [75] | 1.08–1.17 |
| VGLL3 | 3p12 | rs2660753 | Intergenic | [80] | 1.11–1.48 |
| SIDT1 | 3q13 | rs7611694 | Intronic | [75] | 0.88–0.93 |
| EEFSEC | 3q21 | rs10934853 | Intronic | [81] | 1.12 |
| ZBTB38 | 3q23 | rs6763931 | Intronic | [79] | 1.04–1.18 |
| CLDN11 | 3q26 | rs10936632 | Intergenic | [76] | 1.08–1.28 |
| AFM, RASSF6 | 4q13 | rs1894292 | Intronic | [75] | 0.89–0.94 |
| PDLIM5 | 4q22 | rs12500426 | Intronic | [78] | 1.14–1.17 |
| PDLIM5 | 4q22 | rs17021918 | Intronic | [78] | 1.12–1.25 |
| TET2 | 4q24 | rs7679673 | Intergenic | [78] | 1.15–1.37 |
| FGF10 | 5p12 | rs2121875 | Intronic | [76] | 1.05–1.11 |
| TERT | 5p15 | rs2242652 | Intronic | [79] | 1.15–1.39 |
| FAM44B (BOD1) | 5q35 | rs6869841 | Intergenic | [75] | 1.04–1.11 |
| CCHCR1 | 6p21 | rs130067 | Exonic/Coding | [79] | 1.05–1.20 |
| NOTCH4 | 6p21 | rs3096702 | Intergenic | [75] | 1.04–1.10 |
| ARMC2, SESN1 | 6q21 | rs2273669 | Intronic | [75] | 1.03–1.11 |
| SLC22A3 | 6q25 | rs9364554 | Intronic | [80] | 1.17–1.26 |
| RSG17 | 6q25 | rs1933488 | Intronic | [75] | 0.87–0.92 |
| JAZF1 | 7p15 | rs10486567 | Intronic | [82] | 1.12–1.35 |
| SP8 | 7p21 | rs12155172 | Intergenic | [75] | 1.07–1.15 |
| LMTK2 | 7q21 | rs6465657 | Intronic | [80] | 1.03–1.19 |
| SLC25A37 | 8p21 | rs2928679 | Intergenic | [78] | 1.16–1.26 |
| NKX3-1 | 8p21 | rs1512268 | Intergenic | [78] | 1.13–1.28 |
| EBF2 | 8p21 | rs11135910 | Intronic | [75] | 1.07–1.16 |
| None | 8q24 | rs10086908 | Intergenic | [83] | 1.14–1.25 |
| None | 8q24 | rs7841060 | Intergenic | [84] | 1.19 |
| None | 8q24 | rs13254738 | Intergenic | [85] | 1.11 |
| None | 8q24 | rs16901979 | Intergenic | [86] | 1.66 |
| None | 8q24 | rs16902094 | Intergenic | [81] | 1.21 |
| None | 8q24 | rs445114 | Intergenic | [81] | 1.14 |
| None | 8q24 | rs620861 | Intergenic | [83,84] | 1.11–1.28 |
| None | 8q24 | rs6983267 | Intergenic | [82,83,85,87] | 1.13–1.42 |
| None | 8q24 | rs7000448 | Intergenic | [85] | 1.14 |
| None | 8q24 | rs1447295 | Intergenic | [86–88] | 1.29–1.72 |
| MSMB | 10q11 | rs10993994 | Intergenic | [80] | 1.15–1.42 |
| TRIM8 | 10q24 | rs3850699 | Intronic | [75] | 0.89–0.94 |
| CTBP2 | 10q26 | rs4962416 | Intronic | [82] | 1.17–1.20 |
| TH | 11p15 | rs7127900 | Intergenic | [78] | 1.29–1.40 |
| MYEOV | 11q13 | rs11228565 | Intergenic | [81] | 1.23 |
| MYEOV | 11q13 | rs7931342 | Intergenic | [80] | 1.19–1.25 |
| MYEOV | 11q13 | rs10896449 | Intergenic | [89] | 1.09–1.20 |
| MYEOV | 11q13 | rs12793759 | Intergenic | [89] | 1.04–1.18 |
| MYEOV | 11q13 | rs10896438 | Intergenic | [89] | 1.02–1.12 |
| MMP7 | 11q22 | rs11568818 | Intergenic | [75] | 0.88–0.94 |
| KRT8 | 12q13 | rs902774 | Intergenic | [79] | 1.17 |
| TUBA1C | 12q13 | rs10875943 | Intergenic | [76] | 1.02–1.18 |
| TBX5 | 12q24 | rs1270884 | Intergenic | [75] | 1.04–1.10 |
| FERMT2 | 14q22 | rs8008270 | Intronic | [75] | 0.86–0.93 |
| RAD51L1 | 14q24 | rs7141529 | Intronic | [75] | 1.06–1.12 |
| VPS53, FAM57A | 17p13 | rs684232 | Intergenic | [75] | 1.07–1.14 |
| HNF1B | 17q12 | rs11649743 | Intronic | [90] | 1.28 |
| HNF1B | 17q12 | rs4430796 | Intronic | [90,91] | 1.16–1.38 |
| HOXB13 | 17q21 | rs11650494 | Intergenic | [75] | 1.09–1.22 |
| None | 17q24 | rs1859962 | Intergenic | [91] | 1.20 |
| SALL3 | 18q23 | rs7241993 | Intergenic | [75] | 0.89–0.95 |
| PPP1R14A | 19q13 | rs8102476 | Intergenic | [81] | 1.12 |
| KLK3 | 19q13 | rs2735839 | Intergenic | [80] | 1.25–1.72 |
| GATAS, CABLES2 | 20q13 | rs2427345 | Intergenic | [75] | 0.91–0.97 |
| ZGPAT | 20q13 | rs6062509 | Intronic | [75] | 0.86–0.92 |
| BIK | 22q13 | rs5759167 | Intergenic | [78] | 1.14–1.20 |
| NUDT11 | Xp11 | rs5945619 | Intergenic | [80] | 1.19–1.46 |
| SHROOM2 | Xp22 | rs2405942 | Intronic | [75] | 0.83–0.92 |
| AR | Xq12 | rs5919432 | Intergenic | [79] | 1.06–1.14 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Spans, L.; Clinckemalie, L.; Helsen, C.; Vanderschueren, D.; Boonen, S.; Lerut, E.; Joniau, S.; Claessens, F. The Genomic Landscape of Prostate Cancer. Int. J. Mol. Sci. 2013, 14, 10822-10851. https://doi.org/10.3390/ijms140610822
Spans L, Clinckemalie L, Helsen C, Vanderschueren D, Boonen S, Lerut E, Joniau S, Claessens F. The Genomic Landscape of Prostate Cancer. International Journal of Molecular Sciences. 2013; 14(6):10822-10851. https://doi.org/10.3390/ijms140610822
Chicago/Turabian StyleSpans, Lien, Liesbeth Clinckemalie, Christine Helsen, Dirk Vanderschueren, Steven Boonen, Evelyne Lerut, Steven Joniau, and Frank Claessens. 2013. "The Genomic Landscape of Prostate Cancer" International Journal of Molecular Sciences 14, no. 6: 10822-10851. https://doi.org/10.3390/ijms140610822