Remotely Triggered Scaffolds for Controlled Release of Pharmaceuticals


Abstract
:1. Introduction
2. Results and Discussion
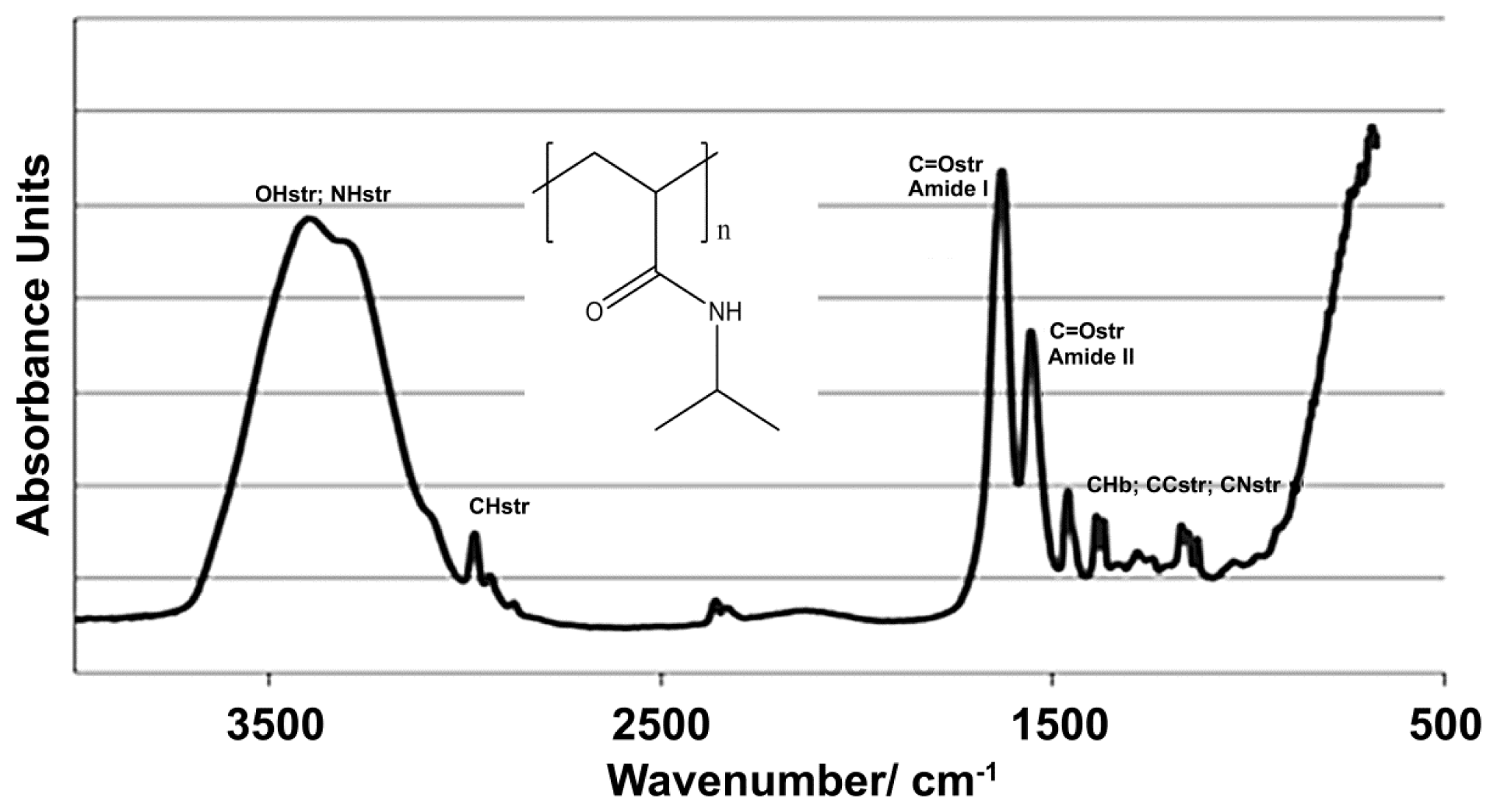
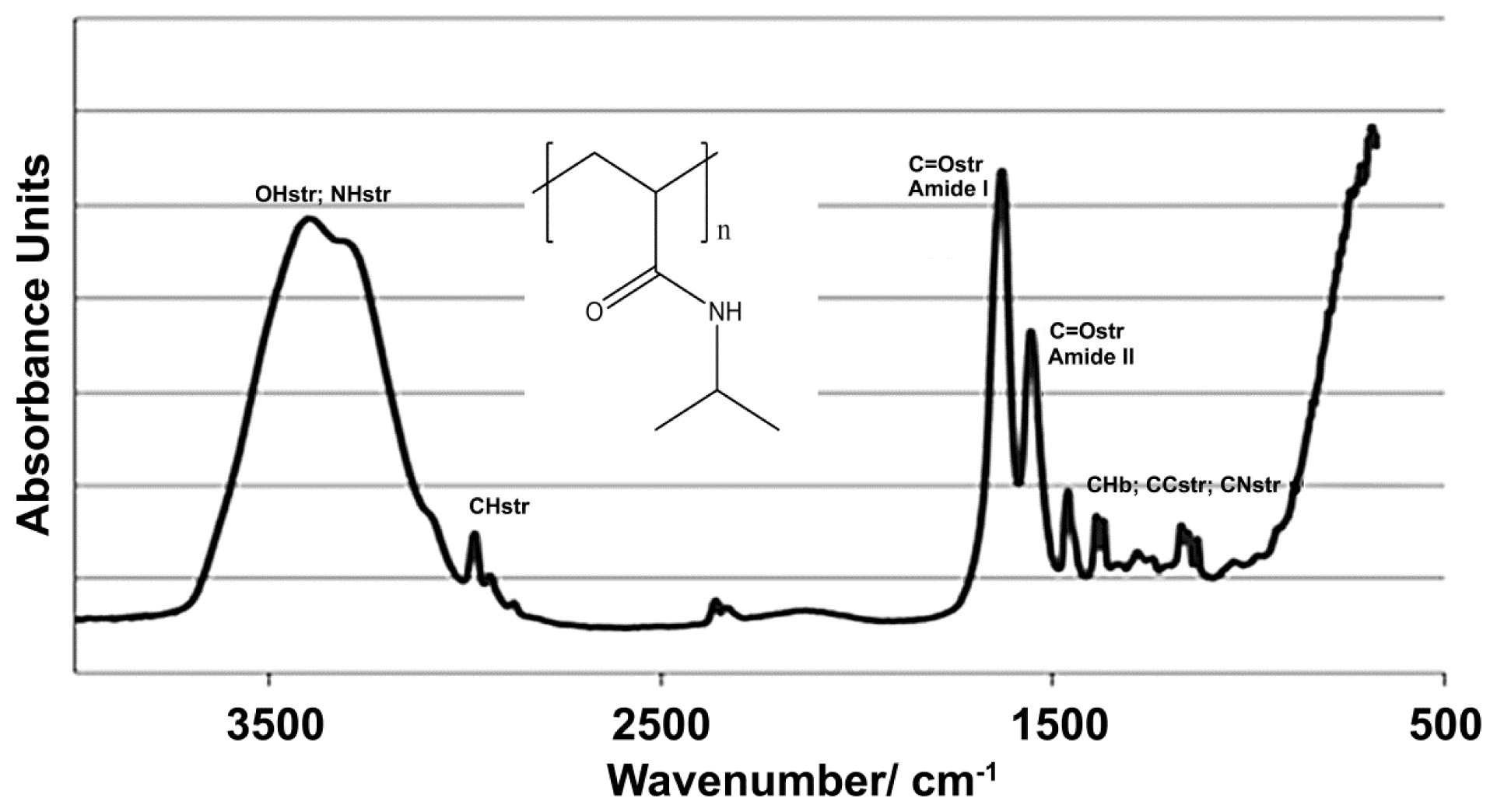
2.1. NiPAM Scaffold Charcteristics
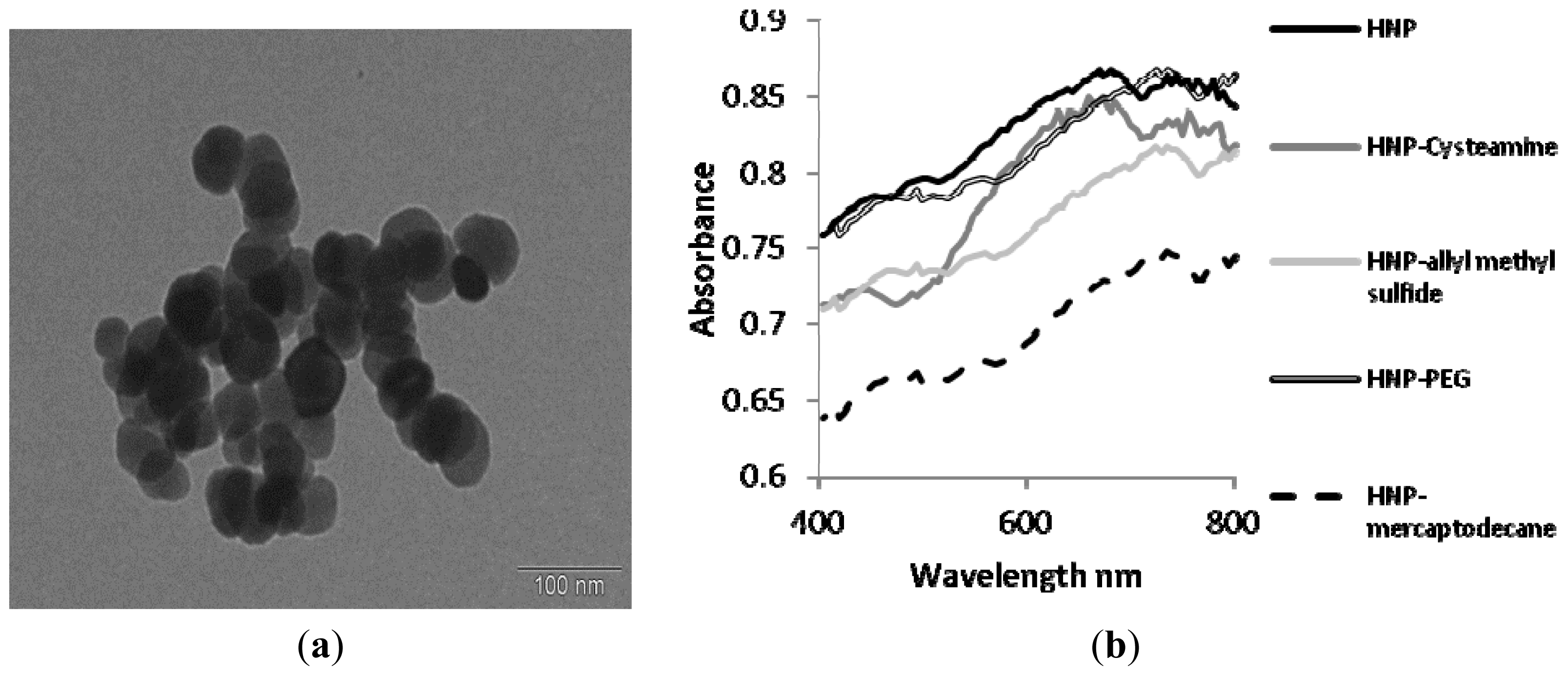
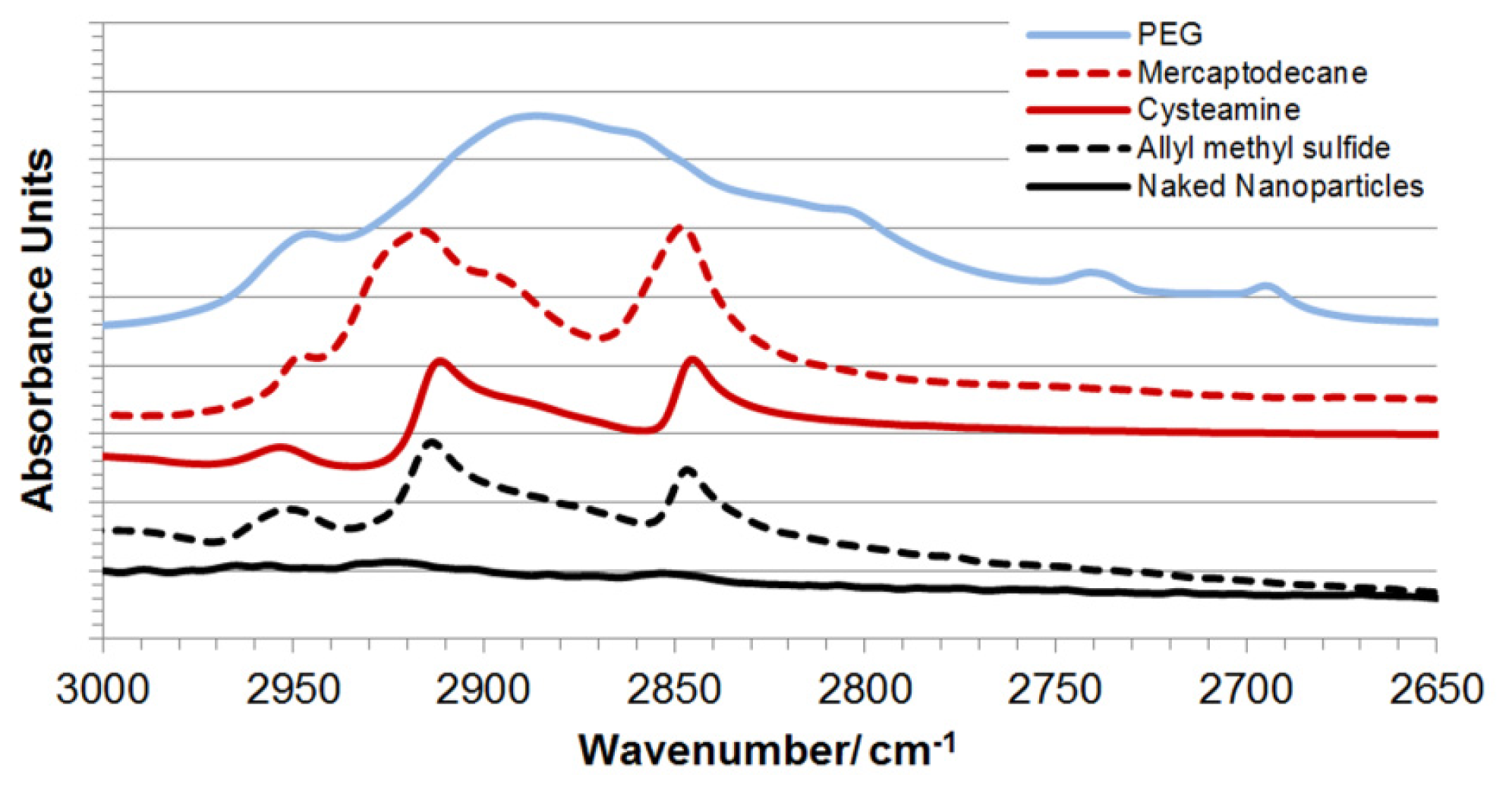
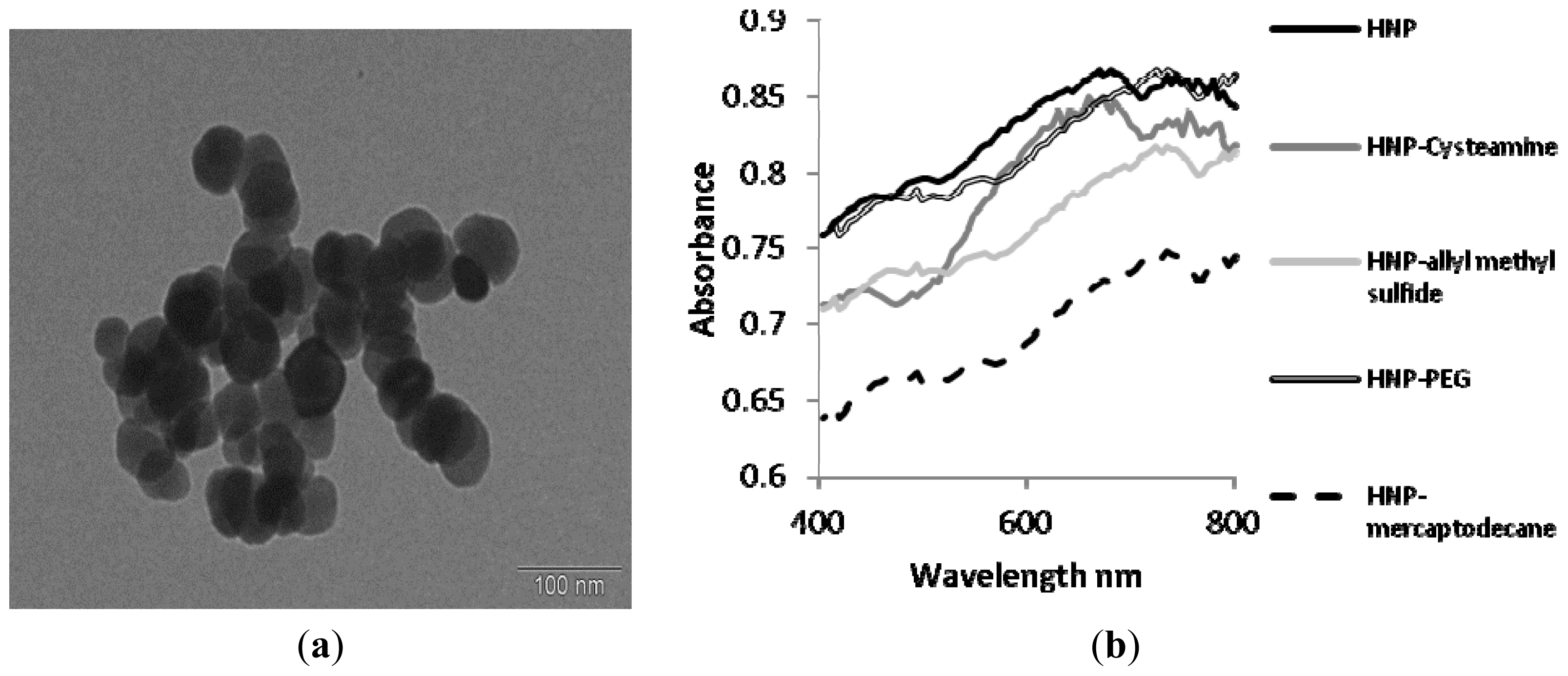
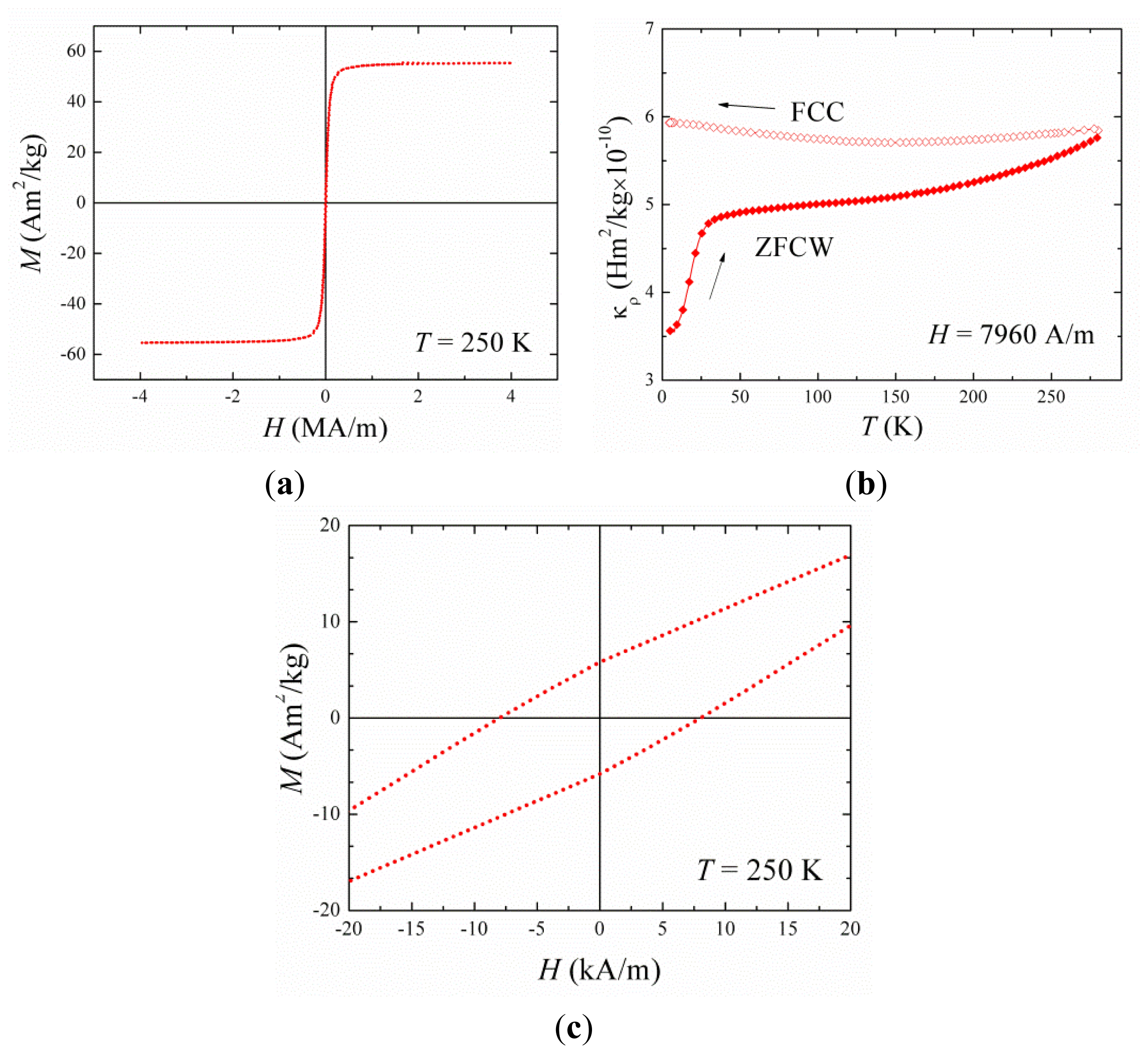
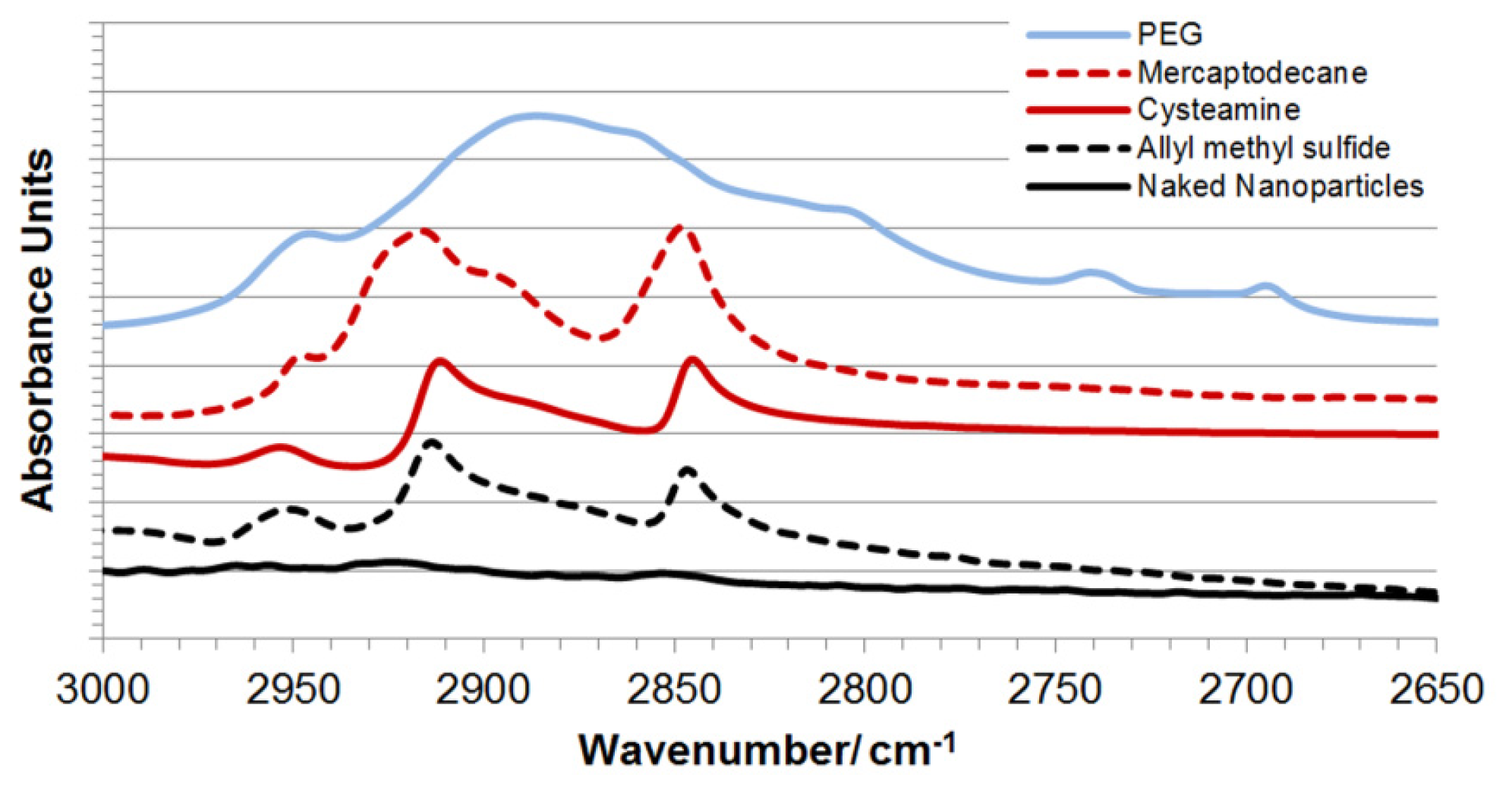
2.2. Synthesis and Characterization of Hybrid Nanoparticles
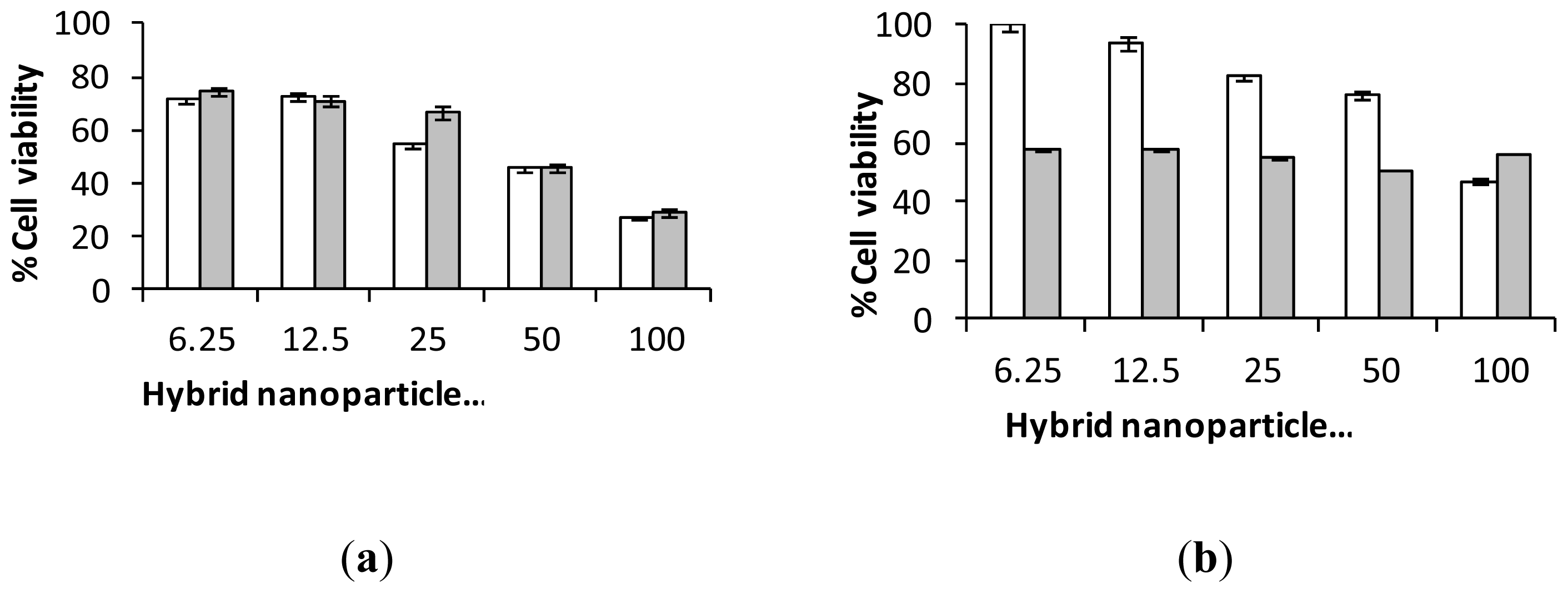
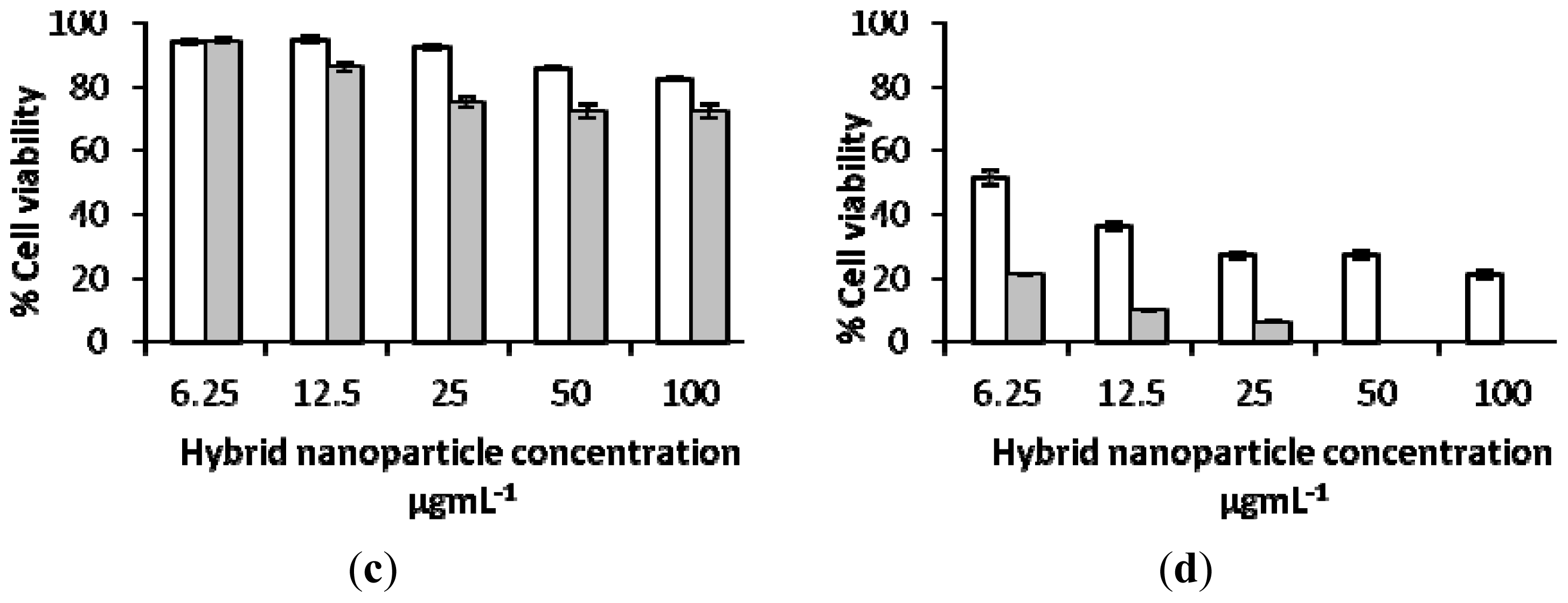
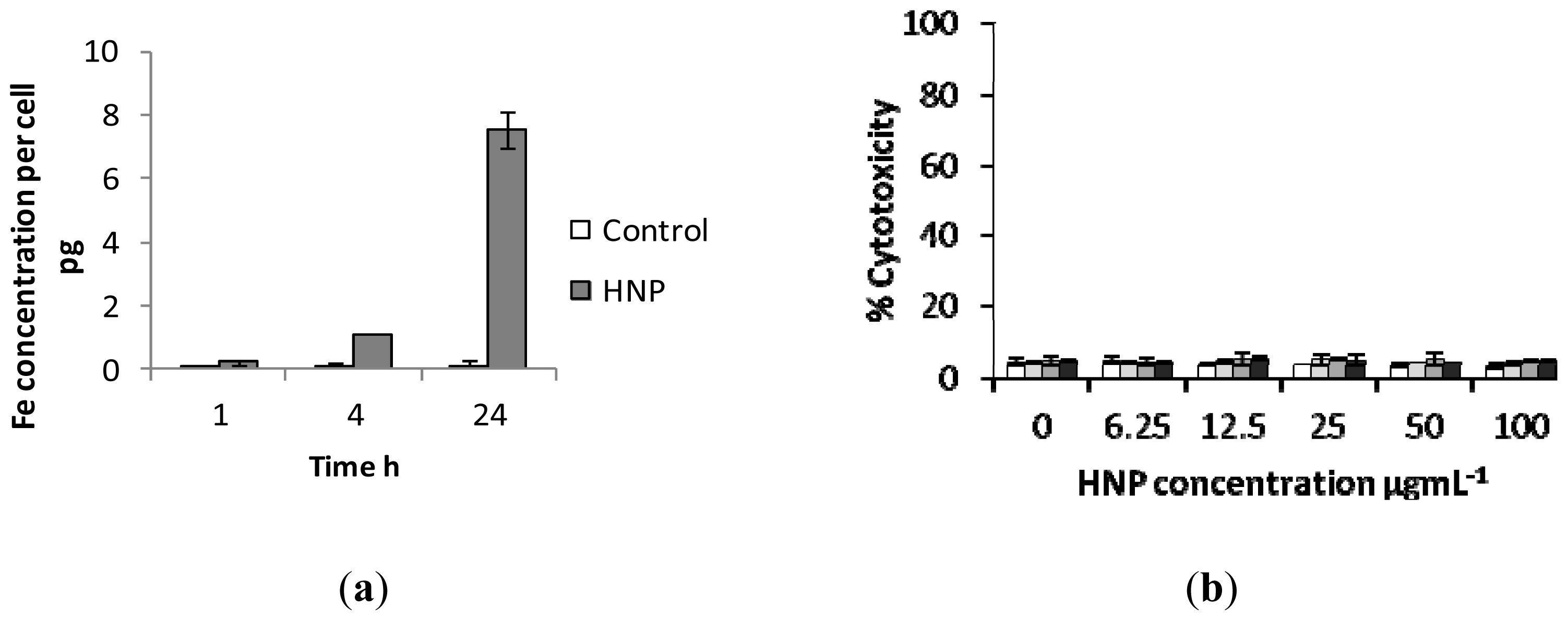
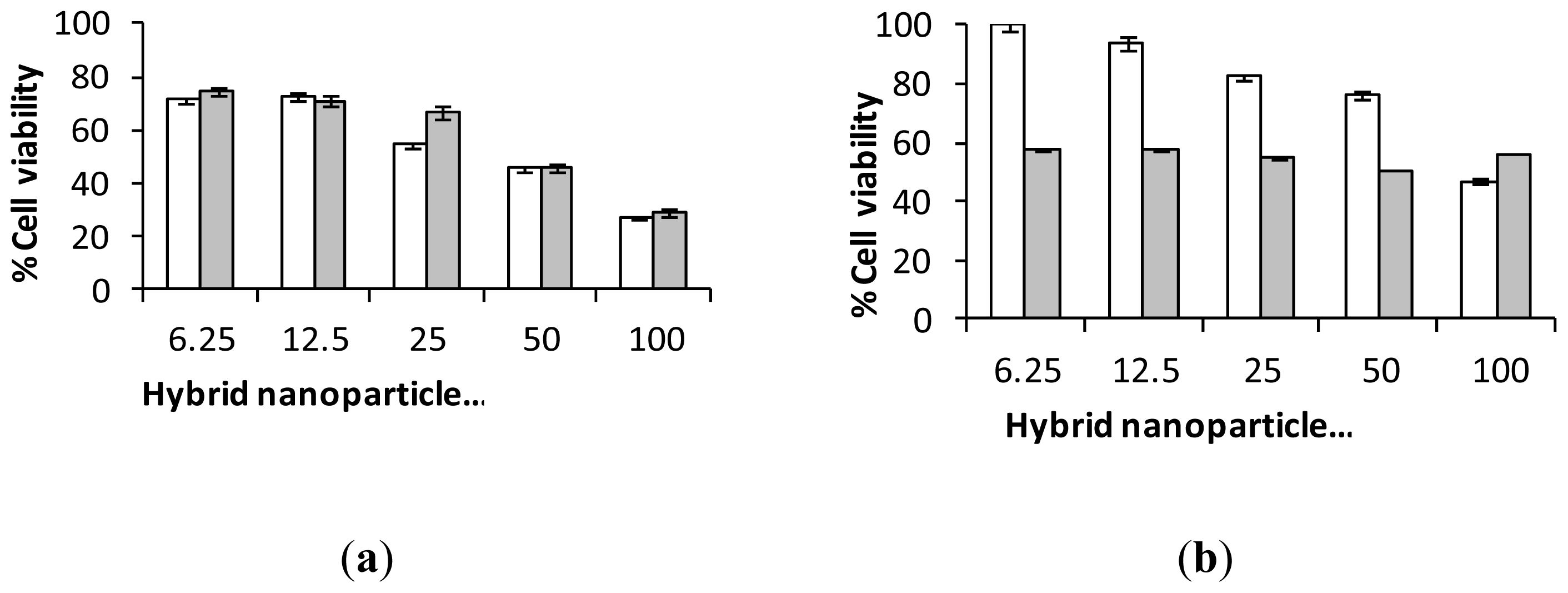
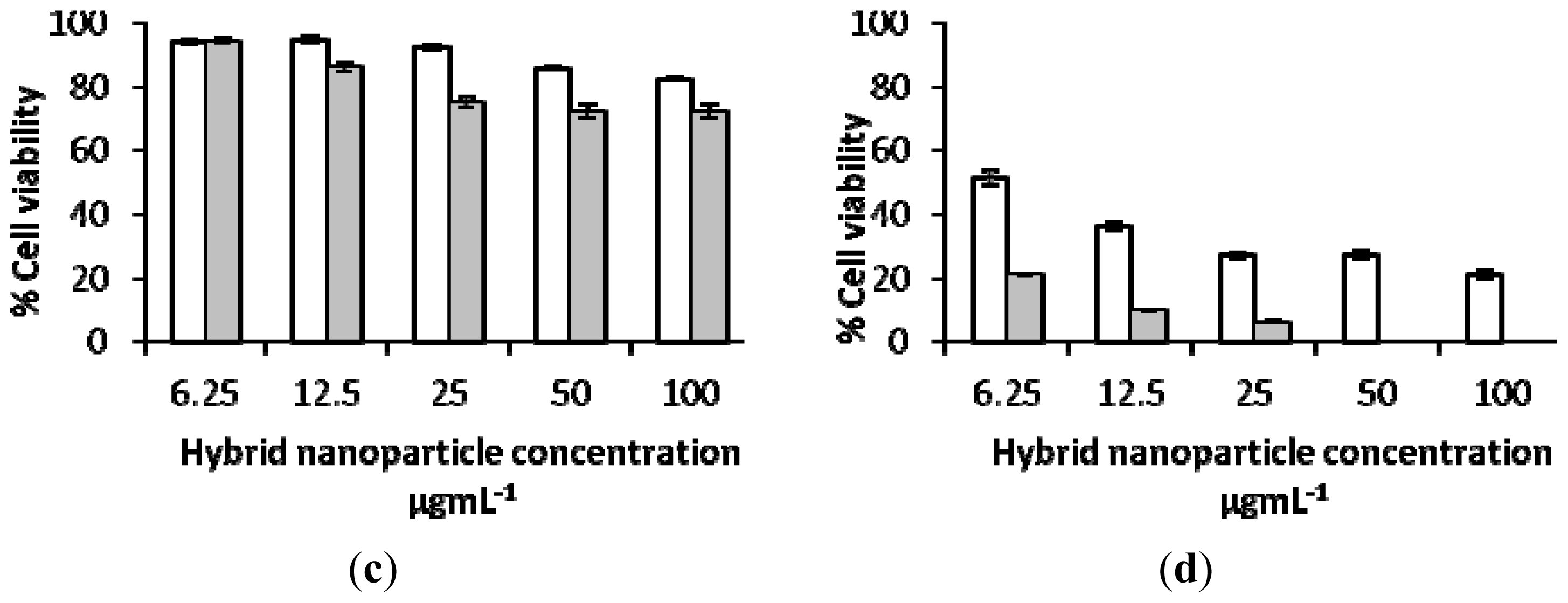
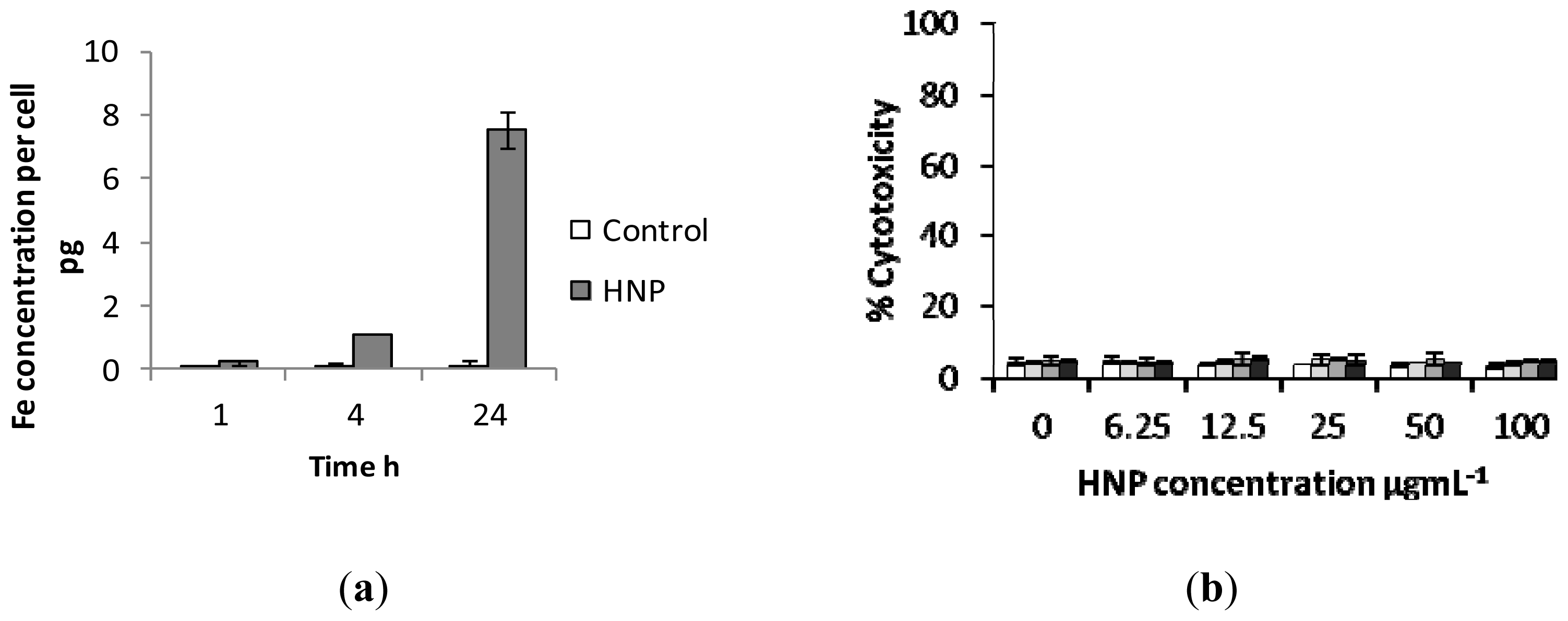
2.3. Biological Characterization of Polymer Coated Hybrid Nanoparticles
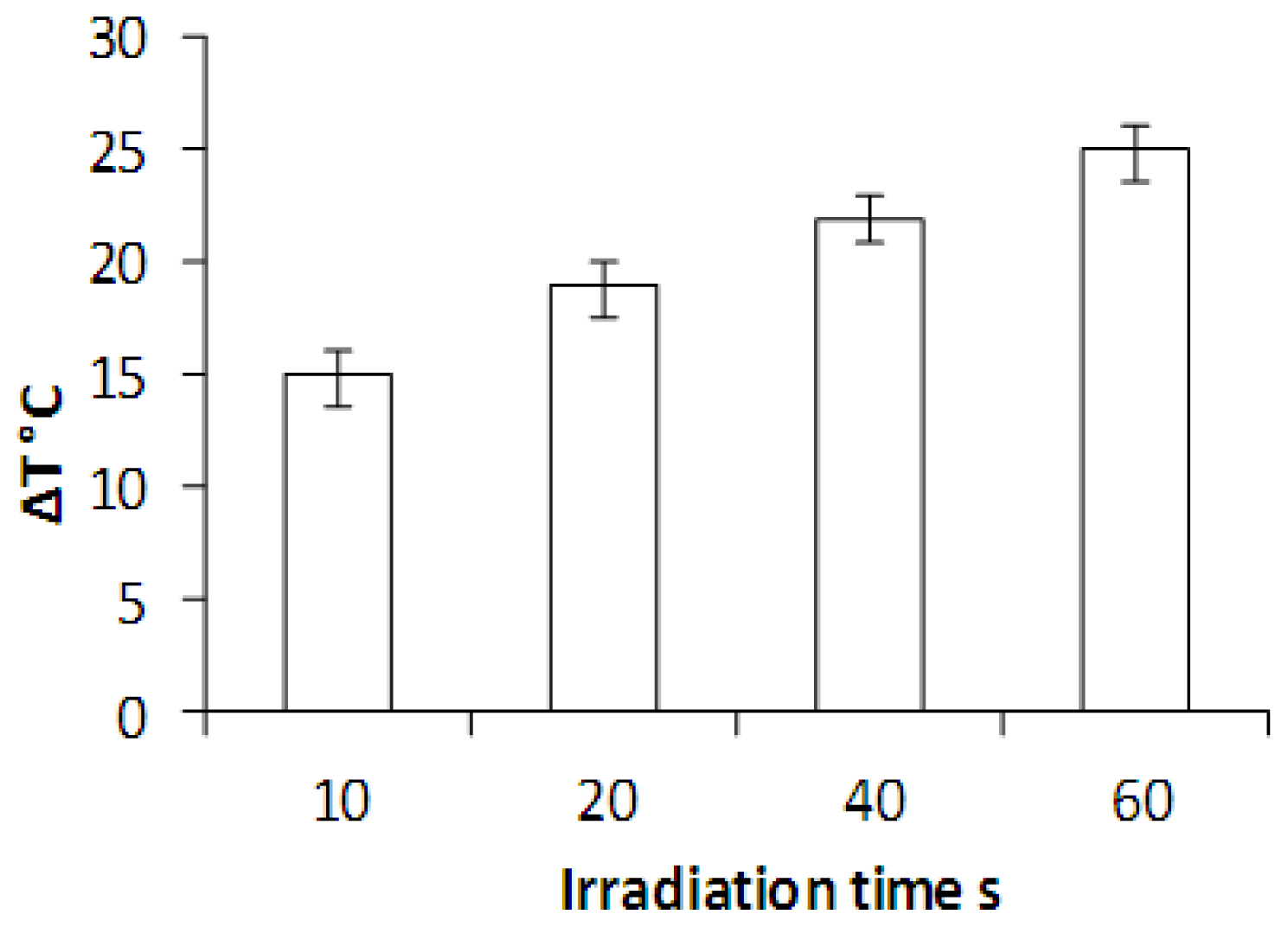
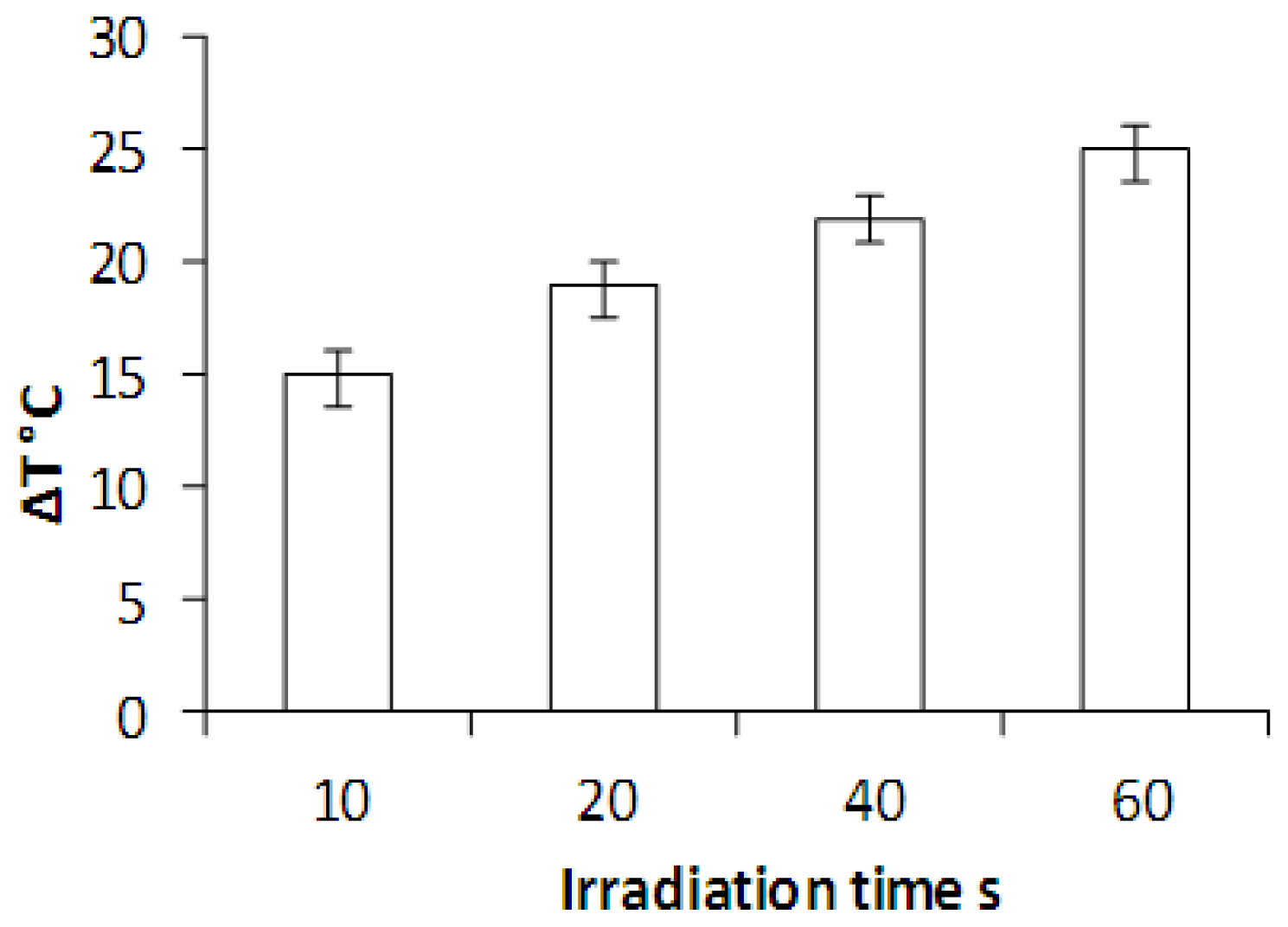
2.4. Laser Irradiation of HNP-PEG Phantoms
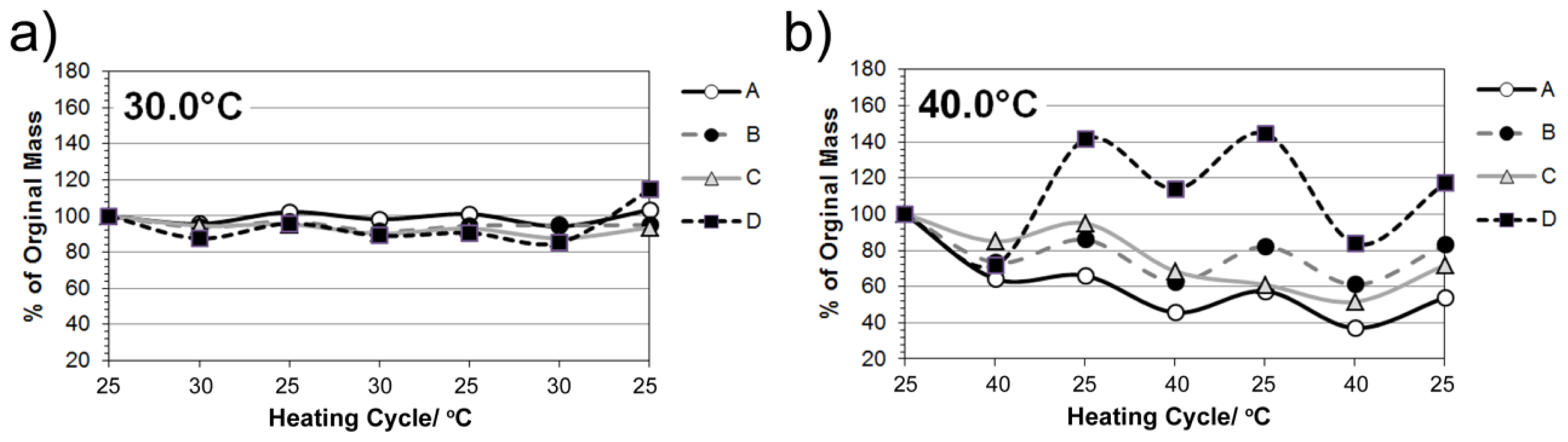
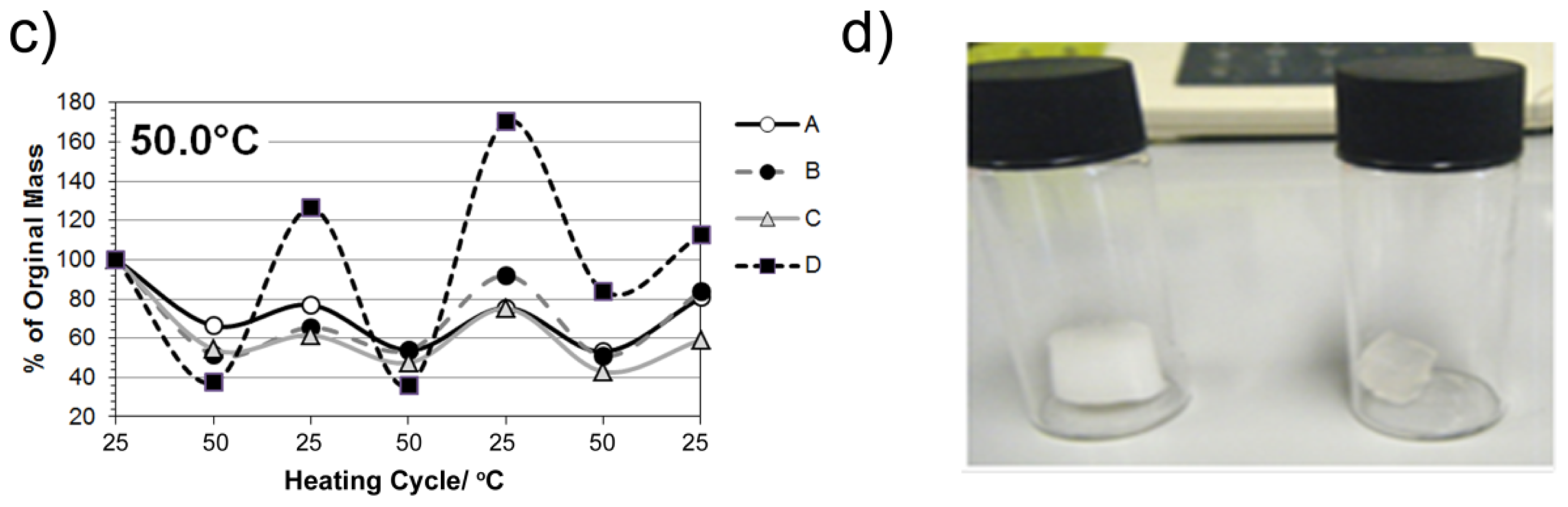
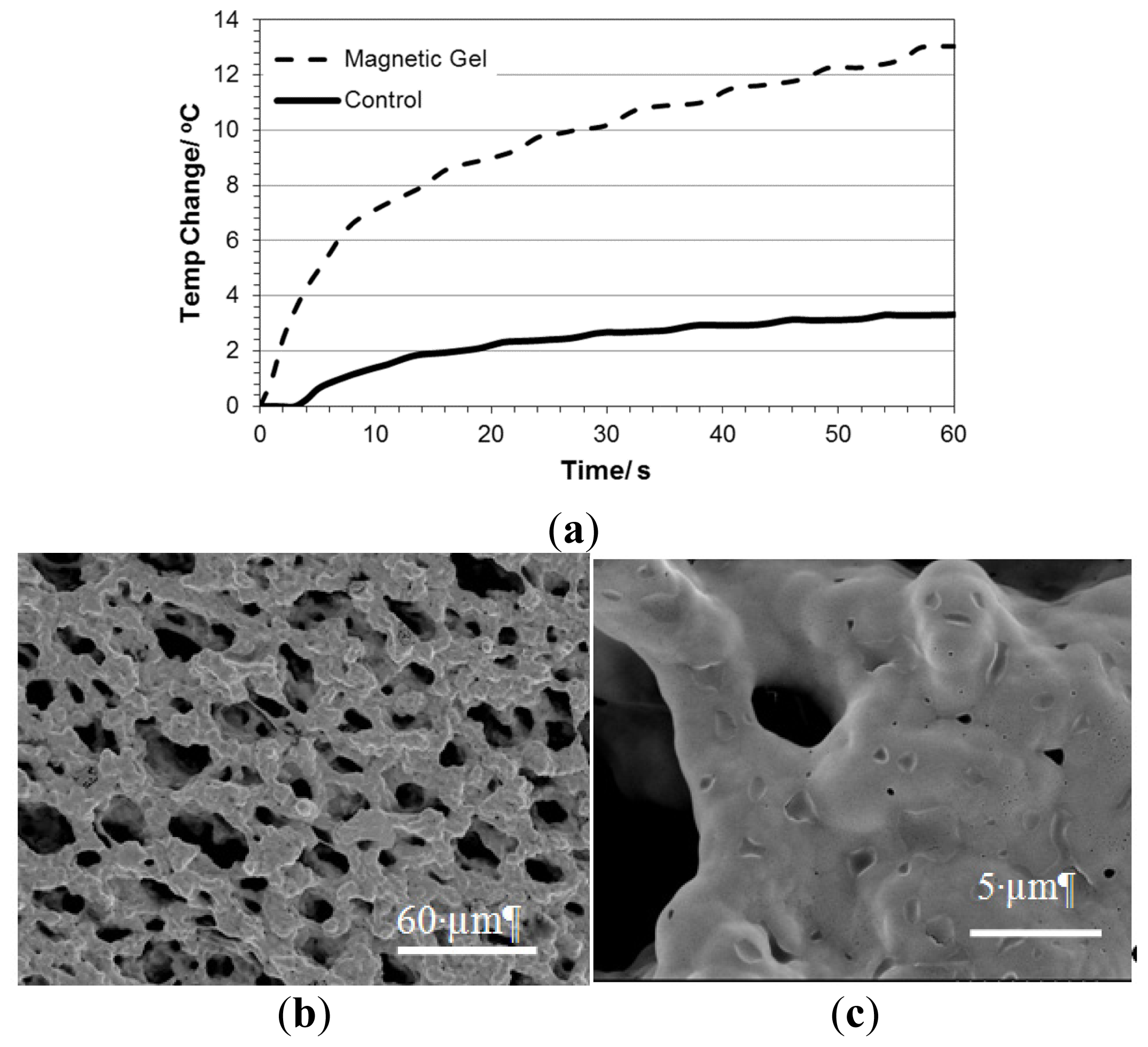
2.5. Studies on pNiPAM-HNP Composites
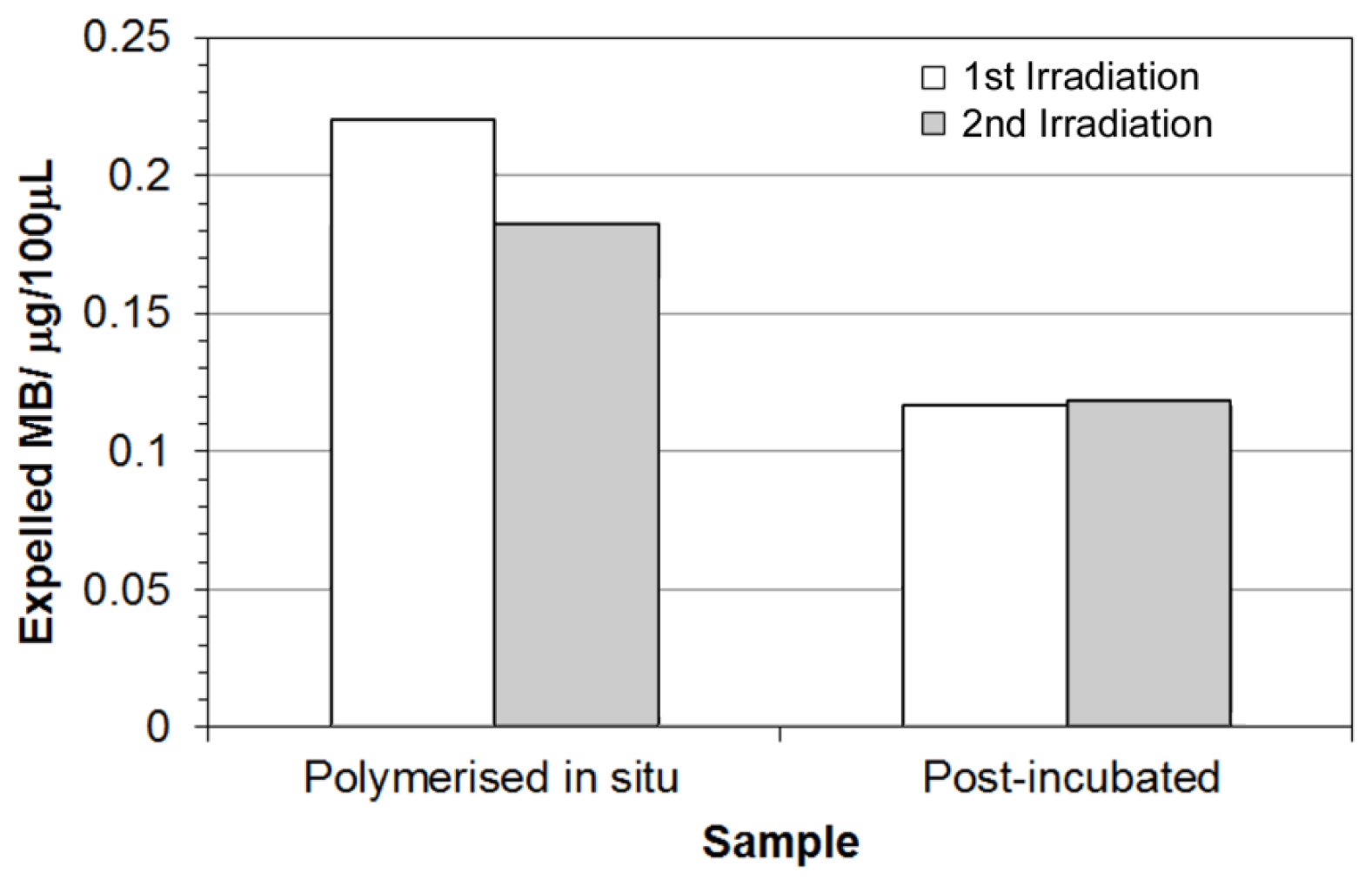
2.6 Stimulated Release of Model Compound
3. Experimental Section
3.1. Scaffold Formation
3.2. Synthesis of HNPs
3.3. Characterization of HNPs
3.4. Biological Characterization of HNPs
3.5. Laser Irradiation of HNPs in Agar Phantom
3.6. Incorporation of HNPs into pNiPAM Scaffolds
3.7 Laser Irradiation of pNiPAM-HNP Scaffolds
3.8. Model Pharmaceutical Release
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Chrzanowski, W.; Khademhosseini, A. Biologically inspired “smart” materials. Adv. Drug Deliv. Rev. 2013. [Google Scholar] [CrossRef]
- Atala, A. Regenerative medicine strategies. J. Paediat. Surg 2012, 47, 17–28. [Google Scholar]
- Panagiotou, N. Forces driving change in medical diagnostics. Clin. Chim. Acta 2013, 415, 31–34. [Google Scholar]
- Yang, Y.; Basu, S.; Tomasko, D.L.; Lee, L.J.; Yang, S.T. Fabrication of well-defined PLGA scaffolds using novel microembossing and carbon dioxide bonding. Biomaterials 2005, 26, 2585–2594. [Google Scholar]
- Tai, H.; Mather, M.L.; Howard, D.; Wang, W.; White, L.J.; Crowe, J.A.; Morgan, S.P.; Chandra, A.; Williams, D.J.; Howdle, S.M.; et al. Control of pore size and structure of tissue engineering scaffolds produced by supercritical fluid processing. Eur. Cells Mater 2007, 14, 64–77. [Google Scholar]
- Mohan, N.; Nair, P.D. Novel porous, polysaccharide scaffolds for tissue engineering applications. Trends Biomater. Artif. Org 2005, 18, 219–224. [Google Scholar]
- Salgado, A.J.; Countinho, O.P.; Reis, R.L. Bone Tissue Engineering: State of the Art and Future Trends. Macromol. Biosci 2004, 4, 743–765. [Google Scholar] [Green Version]
- Upadhyay, D.; Scalia, S.; Vogel, R.; Salama, R.O.; Young, P.M.; Traini, D.; Chrzanowski, W. Magnetised thermo responsive lipid vehicles for targeted and controlled lung drug delivery. Pharm. Res 2012, 29, 2456–2467. [Google Scholar]
- Hoskins, C.; Min, Y.; Gueorguieva, M.; McDougall, C.; Volovick, A.; Prentice, P.; Wang, Z.; Melzer, A.; Cuschieri, A.; Wang, L. Hybrid gold-iron oxide nanoparticles as a multifunctional platform for biomedical application. J. Nanobiotechnol 2012, 10, 27. [Google Scholar]
- Barnett, C.M.; Gueorguieva, M.; Lees, M.R.; McGarvey, D.J.; Darton, R.J.; Hoskins, C. Effect of the hybrid composition on the physicochemical properties and morphology of iron oxide–gold nanoparticles. J. Nanopart. Res 2012, 14, 1170. [Google Scholar]
- Wagstaff, A.J.; Brown, S.D.; Holden, M.R.; Craig, G.E.; Plumb, J.A.; Brown, R.E.; Schreiter, N.; Chrzanowski, W.; Wheate, N.J. Cisplatin drug delivery using gold-coated iron oxide nanoparticles for enhanced tumour targeting with external magnetic fields. Inorg. Chim. Acta 2012, 393, 328–333. [Google Scholar]
- Riggio, C.; Calatayud, M.P.; Hoskins, C.; Pinkernelle, J.; Sanz, B.; Torres, T.E.; Ibarra, M.R.; Wang, L.; Keilhoff, G.; Goya, G.F.; et al. Poly-l-lysine coated magnetic nanoparticles as intracellular actuators for neural guidance. Int. J. Nanomed 2012, 7, 1–12. [Google Scholar]
- Laurent, S.; Boutry, S.; Mahieu, I.; Vander Elst, L.; Muller, R.N. Iron oxide based MR contrast agents: From chemistry to cell labelling. Curr. Med. Chem 2009, 16, 4712–4727. [Google Scholar]
- Pissuwan, D.; Valenzueka, S.M.; Cortie, M.B. Therapeutic possibilities of plasmonically heated gold nanoparticles. Trends Biotechnol 2006, 24, 62–67. [Google Scholar]
- Jain, P.K.; El-Sayed, M.A. Au nanoparticles target cancer. Nanotoday 2007, 2, 18–29. [Google Scholar]
- Chan, A.; Orme, R.P.; Fricker, R.A.; Roach, P. Remote and local control of stimuli responsive materials for therapeutic delivery. Adv. Drug Deliv. Rev. 2012, in press. [Google Scholar]
- Tian, H.; Tang, Z.; Zhuang, X.; Chen, X.; Jing, X. Biodegradable synthetic polymers: Preparation, functionalization and biomedical application. Prog. Polym. Sci 2012, 37, 237–280. [Google Scholar]
- Plunkett, K.N.; Zhu, X.; Moore, J.S.; Leckband, D.E. PNIPAM chain collapse depends on the molecular weight and grafting density. Langmuir 2006, 22, 4259–4266. [Google Scholar]
- Dimitrov, I.; Trzebika, B.; Müller, A.H.E.; Dworak, A.; Tsvetanov, C.B. Thermosensitive water-soluble copolymers with doubly responsive reversibly interacting entities. Prog. Polym. Sci 2007, 32, 1275–1343. [Google Scholar]
- Abulateefah, S.R.; Spain, S.G.; Aylott, J.W.; Chan, W.; Garnett, M.C.; Alexander, C. Thermoresponsive colloids in cancer therapy. Macromol. Biosci 2011, 11, 1722–1734. [Google Scholar]
- Purushotham, S.; Chang, P.E.; Rumpel, H.; Kee, I.H.; Ng, R.T.; Chow, P.K.; Tan, C.K.; Ramanujan, R.V. Thermoresponsive core-shell magnetic nanoparticles for combined modalities of cancer therapy. Nanotechnology 2009, 20, 305101. [Google Scholar]
- Kim, J.-H.; Lee, T.R. Thermo-responsive hydrogel-coated gold nanoshells for in vivo drug delivery. J. Pharm. Biomed. Eng 2008, 2, 29–35. [Google Scholar]
- McGillicuddy, F.C.; Lynch, I.; Rochev, Y.A.; Burke, M.; Dawson, K.A.; Gallagher, W.M.; Keenan, A.K. Novel ‘plum pudding’ gels as potential drug-eluting stent coatings: Controlled release of fluvastatin. J. Biomed. Mater. Res 2006, 79A, 923–933. [Google Scholar]
- You, J.-O.; Almeda, D.; Ye, G.J.C.; Auguste, D.T. Bioresponsive matrices in drug delivery. J. Biol. Eng 2010, 4, 15. [Google Scholar]
- Campbell, S.; Hoare, T. Injectable, Biodegradable PNiPAM-Magnetite Nanoparticle Hydrogels. Proceedings of 12th Annual American Institute of Chemical Engineers Meeting: Nanoscale Science and Engineering Forum, Pittsburg, PA, USA, 30 October 2012.
- Ramanan, V.V.; Hribar, K.C.; Katz, J.S.; Burdick, J.A. Nanofibre-nanorod composites exhibiting light-induced reversible LCST transitions. Nanotechnology 2011, 22, 494009. [Google Scholar]
- Sugimoto, T.; Matijevíc, E. Formation of uniform spherical magnetite particles by crystallization from ferrous hydroxide gels. J. Colloid Interface Sci 1980, 74, 227–243. [Google Scholar]
- Andrés Vergés, M.; Costo, R.; Roca, A.G.; Marco, J.P.; Goya, G.F.; Serna, C.J.; Morales, M.P. Uniform and water stable magnetite nanoparticles with diameters around the monodomain-multidomain limit. J. Phys. D 2008, 41, 134003. [Google Scholar]
- Goon, I.Y.; Lai, L.M.H.; Lim, M.; Munroe, P.; Gooding, J.J.; Amal, R. Fabrication of gold-shell-protected magnetite nanoparticles: Systematic control using polyethyleneimine. Chem. Mater 2009, 21, 673–681. [Google Scholar]
- Józejczak, A.; Skumiel, A. Ultrasonic investigation of magnetic nanoparticles suspension with PEG biocompatible coating. J. Magn. Magn. Mater 2011, 11, 1509–1516. [Google Scholar]
- Manson, J.; Kumar, D.; Meenan, B.J.; Dixon, D. Polyethylene glycol functionalized gold nanoparticles: The influence of capping density on stability in various media. Gold Bull 2011, 4, 99–105. [Google Scholar]
- Peng, J.; Zou, F.; Liu, L.; Tang, L.; Yu, L.; Chen, W.; Liu, H.; Tang, J.-B.; Wu, L.-X. Preparation and characterization of PEG-PEI/Fe3o4 nano-magnetic fluid by co-precipitation method. Trans. Nonferrous Met. Soc. China 2008, 18, 393–398. [Google Scholar]
- Mishra, S.; Webster, P.; Davis, M.E. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur. J. Cell Biol 2004, 83, 97–111. [Google Scholar]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem 2010, 49, 6288–6308. [Google Scholar]
- Hamming, L.M.; Messersmith, P.B. Fouling resistant biomimetic poly(ethylene glycol) based grafted polymer coatings. Mater. Matters 2008, 3, 52. [Google Scholar]
- Barnett, C.; Gueorguieva, M.; Lees, M.; McGarvey, D.; Hoskins, C. Physical stability, biocompatibility and potential use of hybrid iron oxide-gold nanoparticles as drug carriers. J. Nanopart. Res 2013.
- JagadeeshBabu, P.E.; Kumar, R.S.; Maheswari, B. Synthesis and characterization of temperature sensitive P-NIPAM macro/micro hydrogels. Colloid Surface A 2011, 384, 466–472. [Google Scholar]
- Vila, A.; Gill, H.; McCallion, O.; Alonso, M.J. Transport of PLA-PEG particles across the nasal mucosa: Effect of particle size and PEG coating density. J. Control Release 2004, 98, 231–244. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Constiutent component | Required amount for 1 mL solvent | |||
|---|---|---|---|---|
| 0.7 M (A) | 0.5M (B) | 0.3M (C) | 0.1M (D) | |
| Water | 1 mL | 1 mL | 1 mL | 1 mL |
| NiPAM | 0.0795 g | 0.0568 g | 0.03408 g | 0.01136 g |
| MBA | 1.32 mg | 0.943 mg | 0.5658 mg | 0.1896 mg |
| APS | 0.01 mg | 0.01 mg | 0.01 mg | 0.01 mg |
| TMEDA | 4 μL | 4 μL | 4 μL | 4 μL |
| Particle | Metal content analysis ug mL−1 | Size nm (±SD) | PDI (±SD) | Zeta potential mV (±SD) | |
|---|---|---|---|---|---|
| Fe | Au | ||||
| Fe3O4 | 7000 | - | 2250 (125) | 0.540 (0.125) | −16.5 (1) |
| Fe3O4-PEI | 1920 | - | 270 (11) | 0.125 (0.004) | +47.4 (3) |
| Fe3O4-PEI-AuSEED | 1025 | 45 | 190 (9) | 0.254 (0.001) | +27.5 (1) |
| Fe3O4-PEI-AuCOAT (HNP) | 990 | 370 | 115(5) | 0.258 (0.011) | +10.5 (0) |
| HNP-Cysteamine | 868 | 323 | 413 (13) | 0.679 (0.082) | +30.4 (1) |
| HNP-allyl methyl sulfide | 885 | 385 | 392 (59) | 0.738 (0.147) | +39.1 (4) |
| HNP-PEG | 898 | 377 | 141.9 (4) | 0.321 (0.054) | +20.5 (1) |
| HNP-mercaptodecane | 924 | 370 | 442 (21) | 0.921 (0.137) | +23.6 (2) |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roach, P.; McGarvey, D.J.; Lees, M.R.; Hoskins, C. Remotely Triggered Scaffolds for Controlled Release of Pharmaceuticals. Int. J. Mol. Sci. 2013, 14, 8585-8602. https://doi.org/10.3390/ijms14048585
Roach P, McGarvey DJ, Lees MR, Hoskins C. Remotely Triggered Scaffolds for Controlled Release of Pharmaceuticals. International Journal of Molecular Sciences. 2013; 14(4):8585-8602. https://doi.org/10.3390/ijms14048585
Chicago/Turabian StyleRoach, Paul, David J. McGarvey, Martin R. Lees, and Clare Hoskins. 2013. "Remotely Triggered Scaffolds for Controlled Release of Pharmaceuticals" International Journal of Molecular Sciences 14, no. 4: 8585-8602. https://doi.org/10.3390/ijms14048585