Synthesis and Characterization of Naphthalenediimide-Functionalized Flavin Derivatives




Abstract
:
1. Introduction
2. Results and Discussion
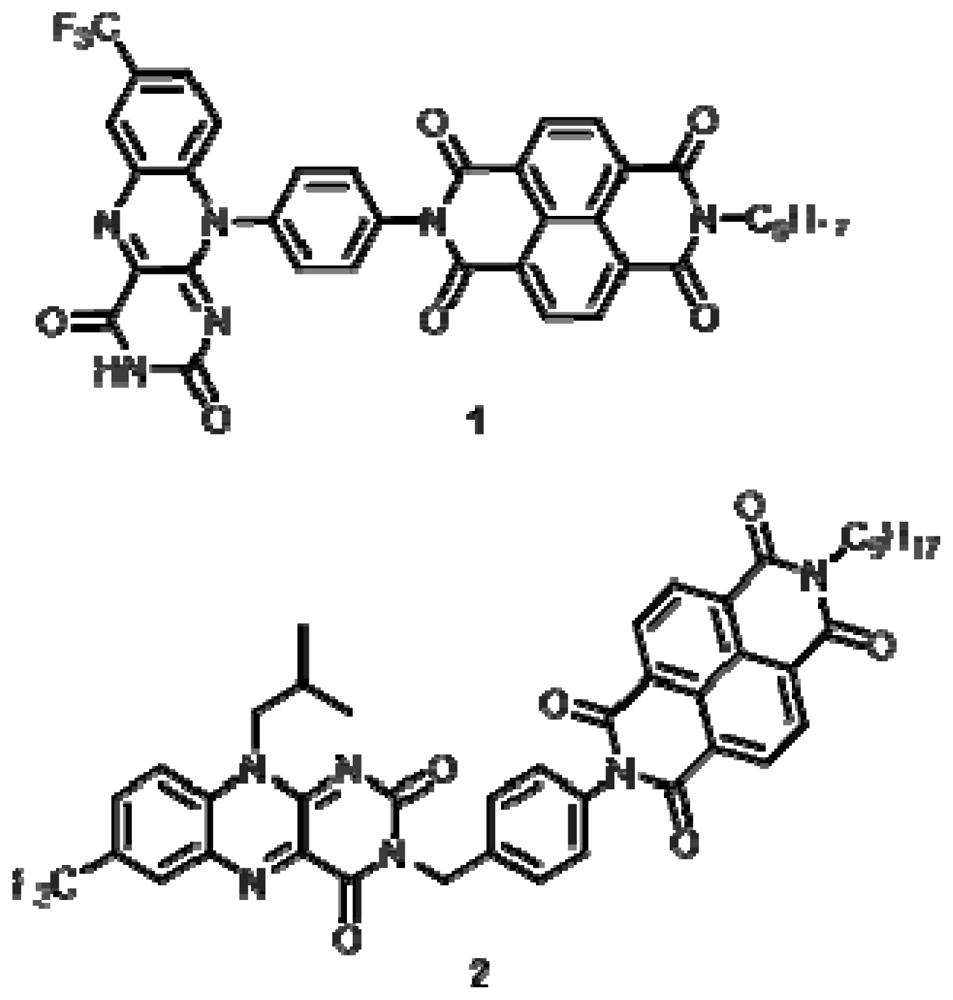
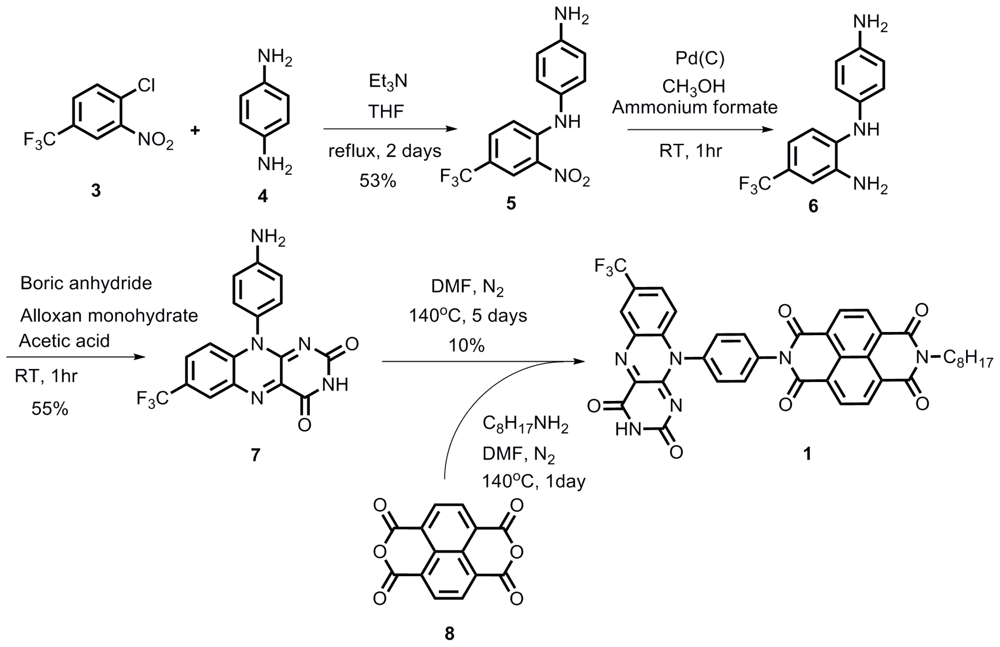
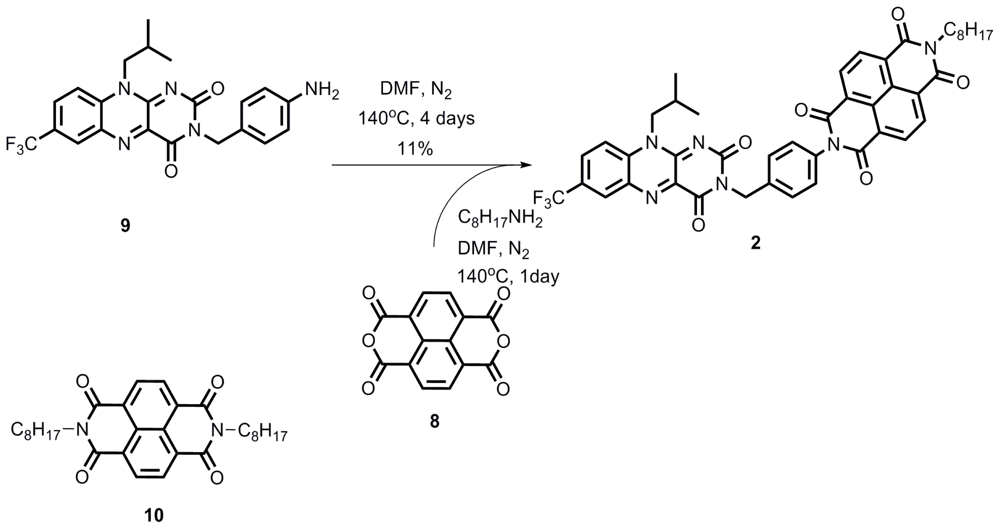
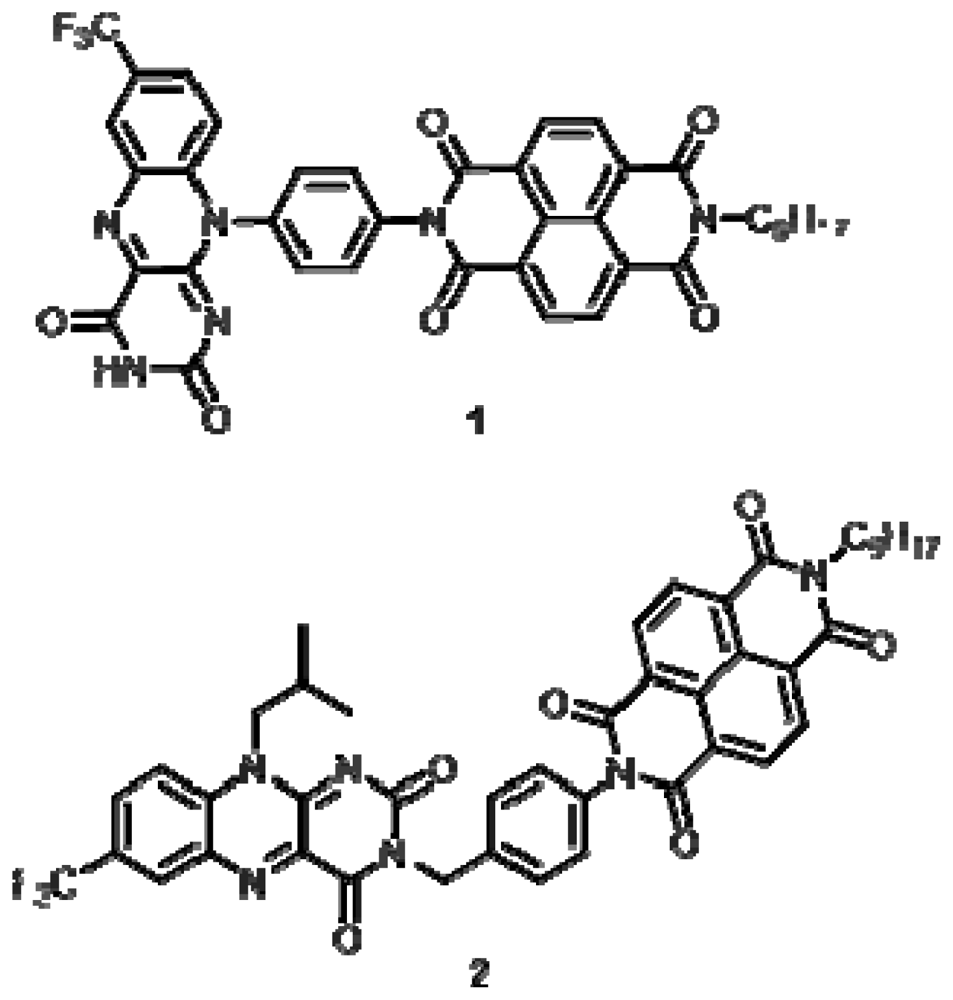
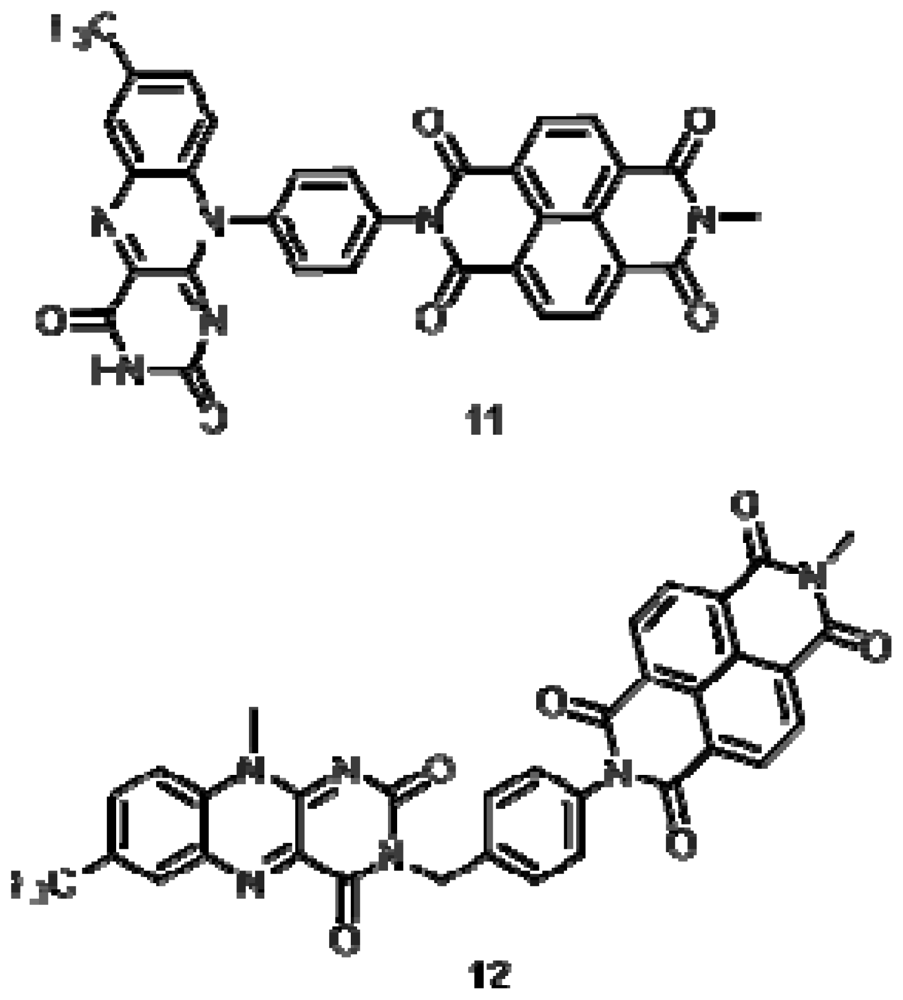
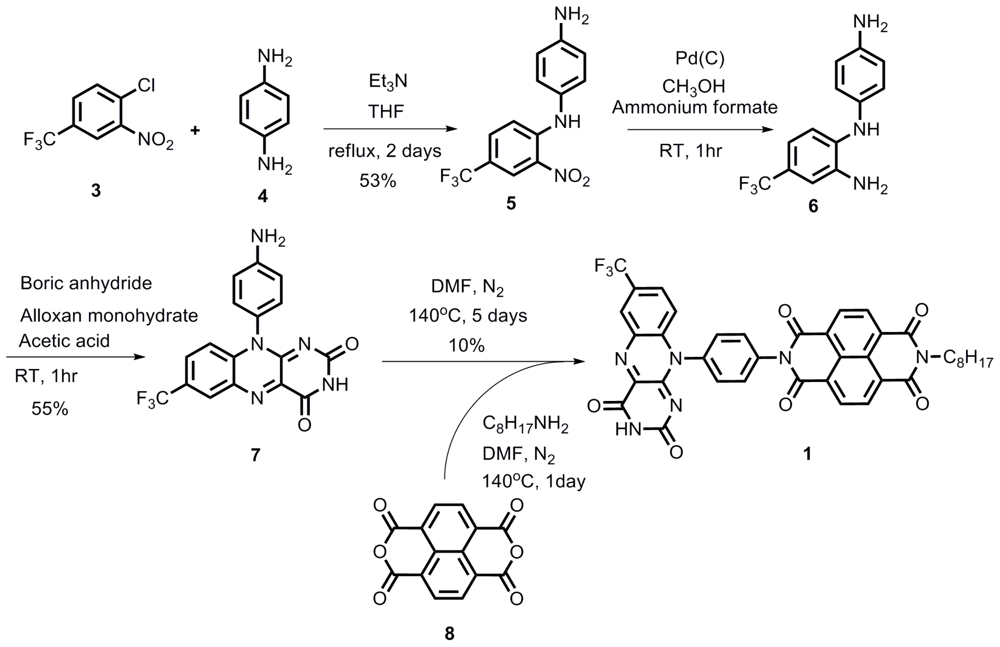
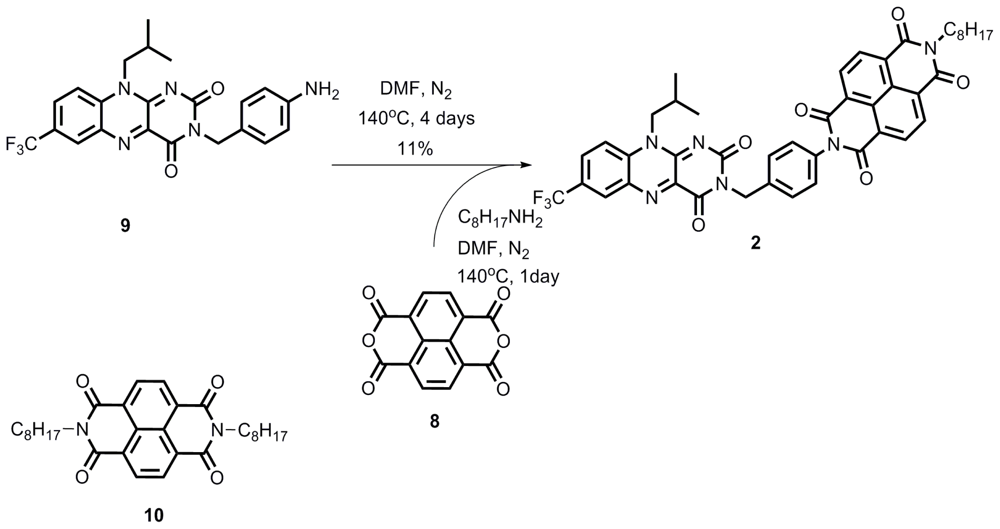
2.1. Synthesis
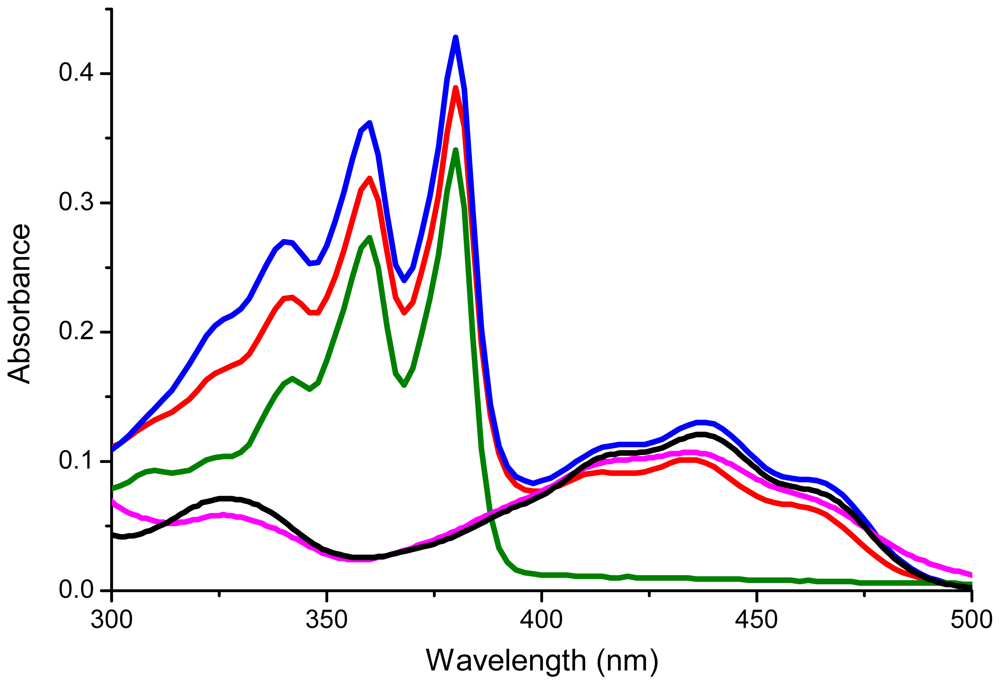
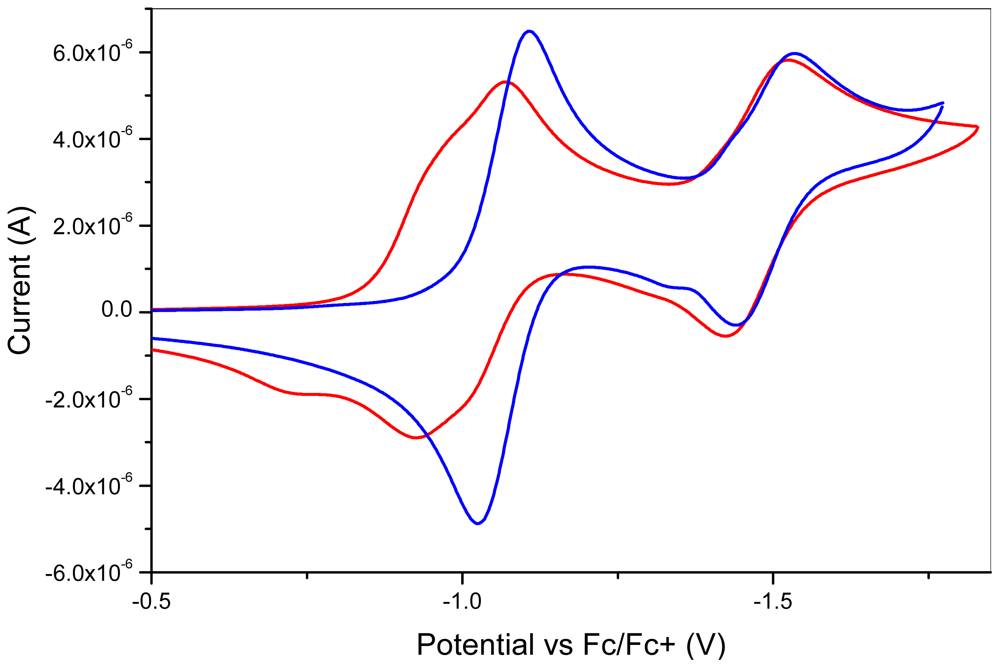
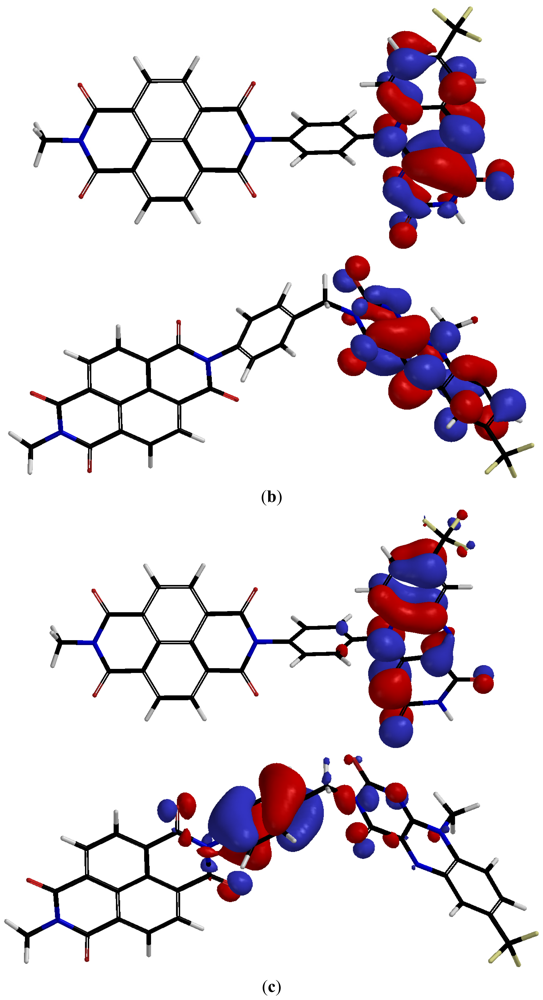
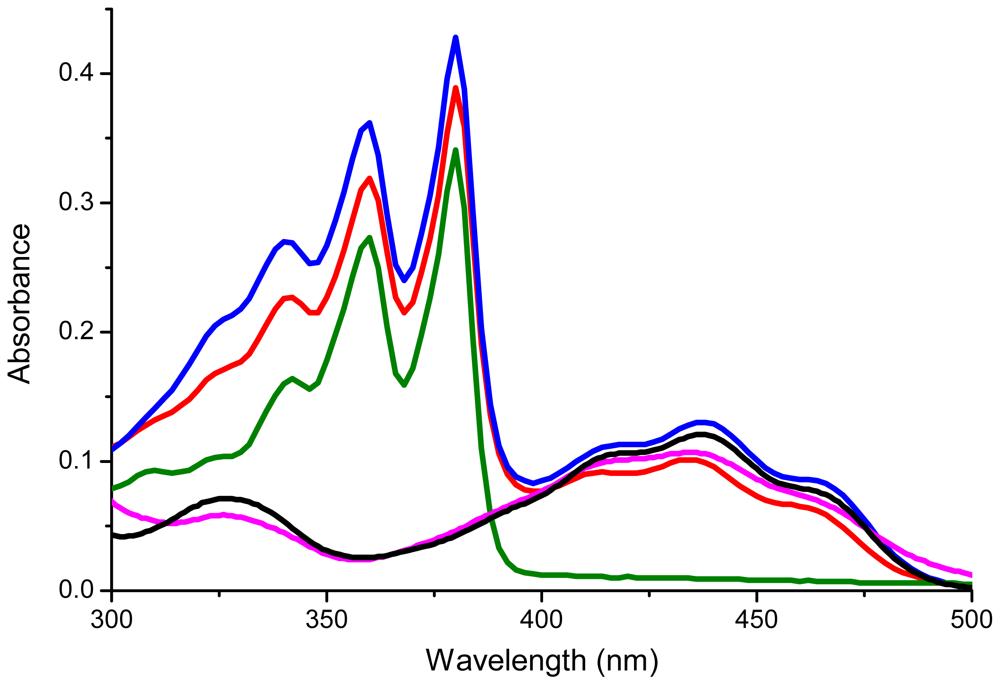
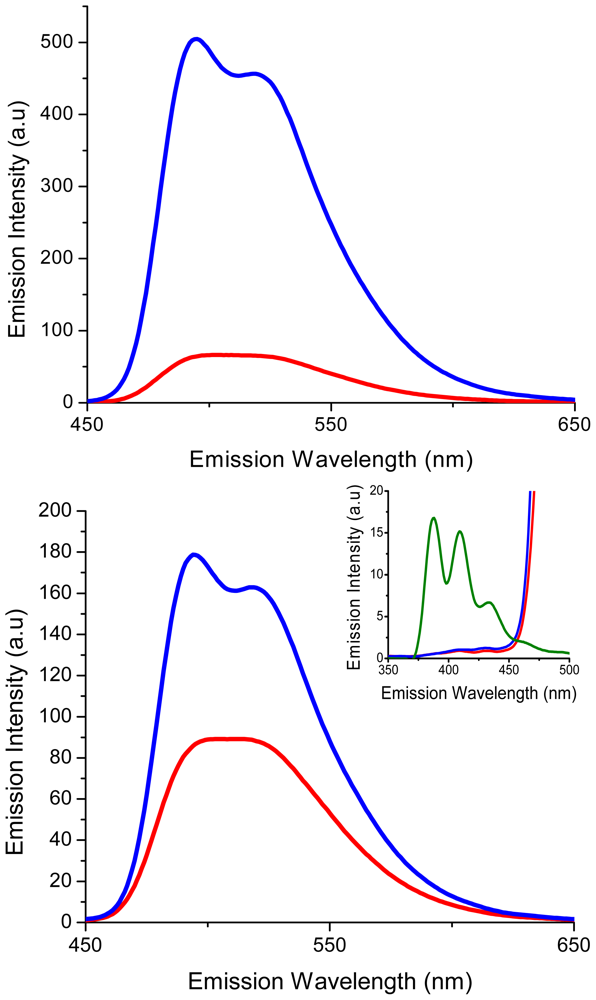
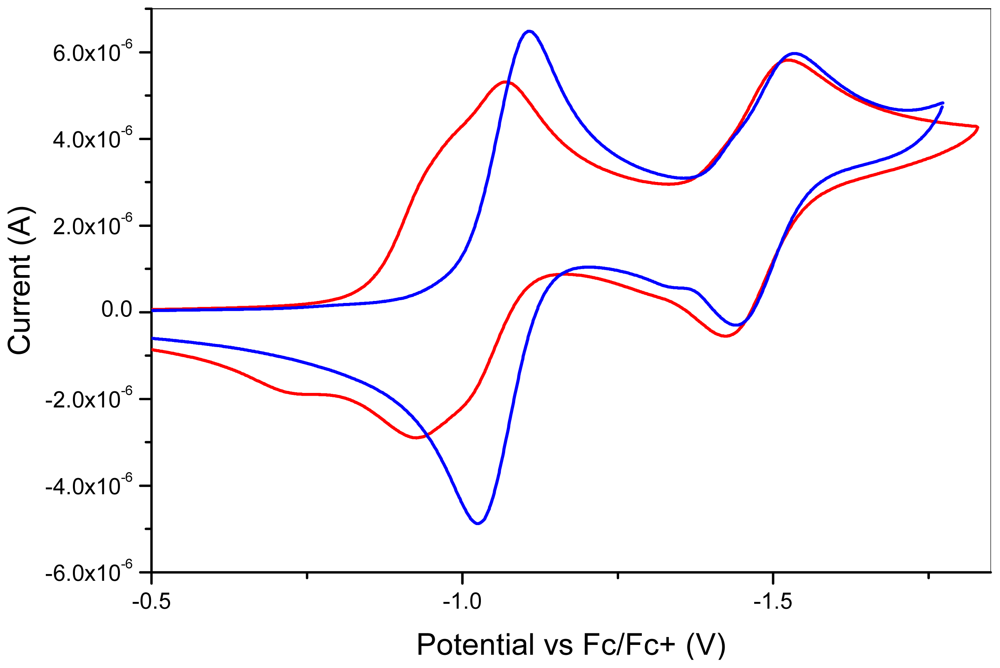
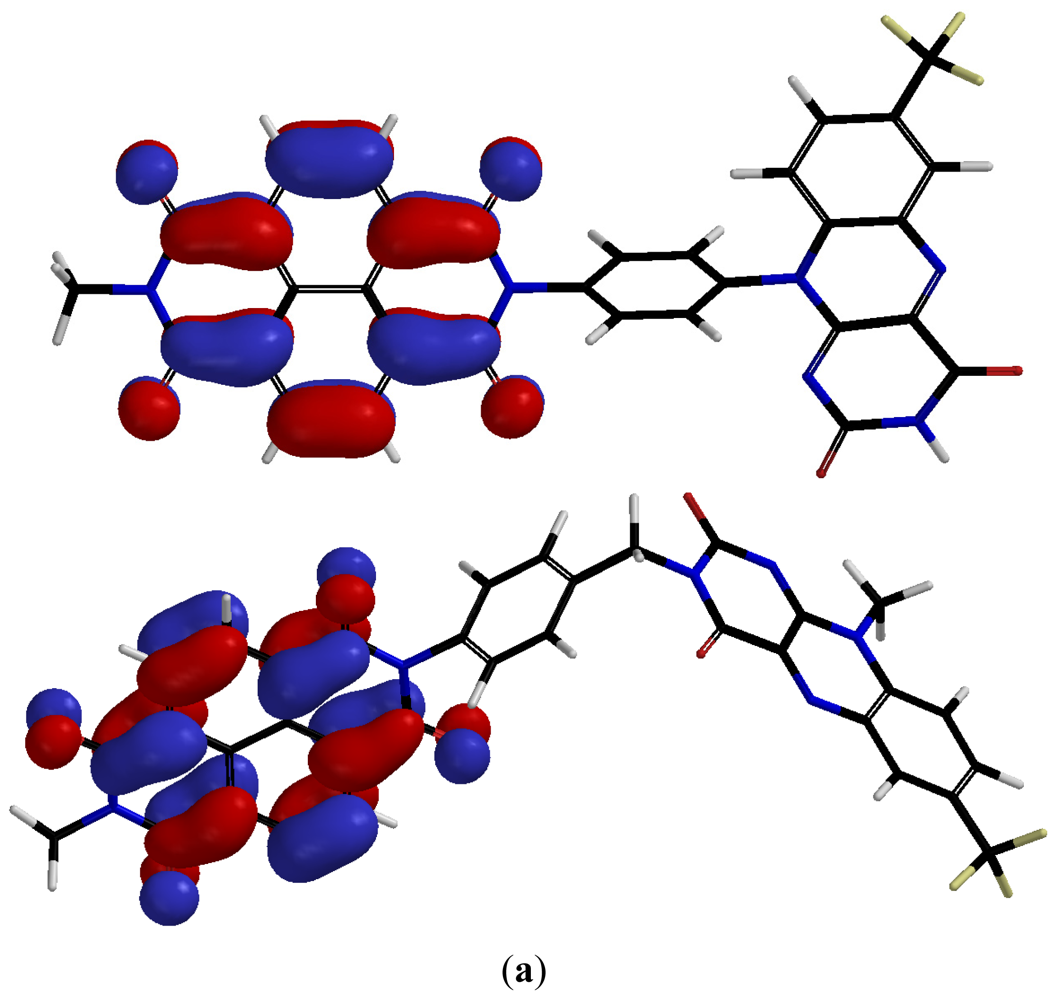
2.2. Characterization
3. Experimental Section
Compound 5
Compound 6
Compound 7
Compound 1
Compound 2
4. Conclusions
Acknowledgments
References
- Stevenson, K.; Massey, V.; Williams, C. Flavins and Flavoproteins; University of Calgary: Calgary, AB, Canada, 1997. [Google Scholar]
- Van Berkel, W.J.H. Chemistry of Flavoenzymes. In Encyclopedia of Chemical Biology; Begley, T.P., Ed.; John Wiley & Sons Inc: Hoboken, NJ, USA, 2008; pp. 17–27. [Google Scholar]
- Stankovich, M.T. Redox Properties of Flavins and Flavoproteins. In Chemisty Biochemistry of Flavoenzymes; Müller, F., Ed.; CRC Press Inc.: Boca Raton, FL, USA, 1991; Volume 1, pp. 401–425. [Google Scholar]
- Mataga, N.; Chosrowian, H.; Shibata, Y.; Tanaka, F.; Nishina, Y.; Shiga, K. Dynamics and mechanisms of ultrafast fluorescence quenching reactions of flavin chromophores in protein nanospace. J. Phys. Chem. B 2000, 104, 10667–10677. [Google Scholar]
- Niemz, A.; Rotello, V.M. From enzyme to molecular device. Exploring the interdependence of redox and molecular recognition. Acc. Chem. Res 1998, 32, 44–52. [Google Scholar]
- Greaves, M.D.; Rotello, V.M. Model systems for flavoenzyme activity. Specific hydrogen bond recognition of flavin in a silicate sol-gel. J. Am. Chem. Soc 1997, 119, 10569–10572. [Google Scholar]
- Sang-Yong, J.; Papadimitrakopoulos, F. Synthesis and redox behavior of flavin mononucleotide-functionalized single-walled carbon nanotubes. J. Am. Chem. Soc 2008, 130, 655–664. [Google Scholar]
- Carroll, J.B.; Cooke, G.; Garety, J.F.; Jordan, B.J.; Mabruk, S.; Rotello, V.M. The electrochemically-tuneable interactions between flavin-functionalised C60 derivatives and 2,6-diethylamidopyridine. Chem. Commun. 2005, 3838–3840. [Google Scholar]
- Bhosale, S.V.; Jani, C.H.; Langford, S.J. Chemistry of naphthalene diimides. Chem. Soc. Rev 2008, 37, 331–342. [Google Scholar]
- Zhan, X.; Facchetti, A.; Barlow, S.; Marks, T.J.; Ratner, M.A.; Wasielewski, M.R.; Marder, S. Rylene and related diimides for organic electronics. Adv. Mater 2011, 23, 268–284. [Google Scholar]
- Rahe, N.; Rinn, C.; Carell, T. Development of donor-acceptor modified DNA hairpins for the investigation of charge hopping kinetics in DNA. Chem. Commun. 2003, 2120–2121. [Google Scholar]
- Sandanaraj, B.S.; Demont, R.; Thayumanavan, S. Generating patterns for sensing using a single receptor scaffold. J. Am. Chem. Soc 2007, 129, 3506–3507. [Google Scholar]
- Cooke, G.; Garety, J.F.; Jordan, B.; Kryvokhyzha, N.; Parkin, A.; Rabani, G.; Rotello, V.M. Flavin-based [2]rotaxanes. Org. Lett 2006, 8, 2297–2300. [Google Scholar]
- Andric, G.; Boas, J.F.; Bond, A.M.; Fallon, G.D.; Ghiggino, K.P.; Hogan, C.F.; Hutchison, J.A.; Lee, M.A.-P.; Langford, S.J.; Pilbrow, J.R.; et al. Spectroscopy of naphthalene diimides and their anion radicals. Aust. J. Chem 2004, 57, 1011–1019. [Google Scholar]
- Harbury, H.A.; LaNoue, K.F.; Loach, P.A.; Amick, R.M. Molecular interaction of isoalloxazine derivatives. Proc. Natl. Acad. Sci. USA 1959, 45, 1708–1717. [Google Scholar]
- Heelis, P.F. The photophysical and photochemical properties of flavins. Chem. Soc. Rev 1982, 11, 15–39. [Google Scholar]
- Koziol, J. Studies on flavins in organic solvents. 3. Spectral behaviour of lumiflavin. Photochem. Photobiol 1969, 9, 45–53. [Google Scholar]
- Kotaki, A.; Yagi, K. Fluorescence properties of flavins in various solvents. J. Biochem 1970, 68, 509–516. [Google Scholar]
- Niemz, A.; Imbriglio, J.; Rotello, V.M. Model systems for flavoenzyme activity: One- and two-electron reduction of flavins in aprotic hydrophobic environments. J. Am. Chem. Soc 1997, 119, 892–897. [Google Scholar]
- Guha, S.; Goodson, F.S.; Roy, S.; Corson, L.J.; Gravenmier, C.A.; Saha, S. Electronically regulated thermally and light-gated electron transfer from anions to naphthalenediimides. J. Am. Chem. Soc 2011, 133, 15256–15259. [Google Scholar]
- Yu, X.; Eymur, S.; Singh, V.; Yang, B.; Tonga, M.; Bheemaraju, A.; Cooke, G.; Subramani, C.; Venkataraman, D.; Stanley, R.J.; Rotello, V.M. Flavin as a photo-active acceptor for efficient energy and charge transfer in a model donor-acceptor system. Phys. Chem. Chem. Phys. 2012, 14, 6749–6754. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λ1 (nm) | λ2(nm) | λ3(nm) | λ4(nm) | λ5(nm) |
|---|---|---|---|---|---|
| 10 | 360 | 380 | |||
| 7 | 418 | 434 | 459 | ||
| 1 | 359 | 379 | 414 | 434 | 455 |
| 9 | 419 | 437 | 462 | ||
| 2 | 359 | 380 | 420 | 437 | 462 |
| Compounds | E1½ (V) | E2½ (V) | E3½ (V) |
|---|---|---|---|
| 10 | −1.07 | −1.52 | |
| 7 | −1.02 | ||
| 1 | −0.95 | −0.99 | −1.47 |
| 9 | −1.12 | ||
| 2 | −1.07 | −1.53 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zainalabdeen, N.; Fitzpatrick, B.; Kareem, M.M.; Nandwana, V.; Cooke, G.; Rotello, V.M. Synthesis and Characterization of Naphthalenediimide-Functionalized Flavin Derivatives. Int. J. Mol. Sci. 2013, 14, 7468-7479. https://doi.org/10.3390/ijms14047468
Zainalabdeen N, Fitzpatrick B, Kareem MM, Nandwana V, Cooke G, Rotello VM. Synthesis and Characterization of Naphthalenediimide-Functionalized Flavin Derivatives. International Journal of Molecular Sciences. 2013; 14(4):7468-7479. https://doi.org/10.3390/ijms14047468
Chicago/Turabian StyleZainalabdeen, Nada, Brian Fitzpatrick, Mohanad Mousa Kareem, Vikas Nandwana, Graeme Cooke, and Vincent M. Rotello. 2013. "Synthesis and Characterization of Naphthalenediimide-Functionalized Flavin Derivatives" International Journal of Molecular Sciences 14, no. 4: 7468-7479. https://doi.org/10.3390/ijms14047468