Early Exercise Protects against Cerebral Ischemic Injury through Inhibiting Neuron Apoptosis in Cortex in Rats

Abstract
:1. Introduction
2. Results and Discussion
2.1. Physiological Variables
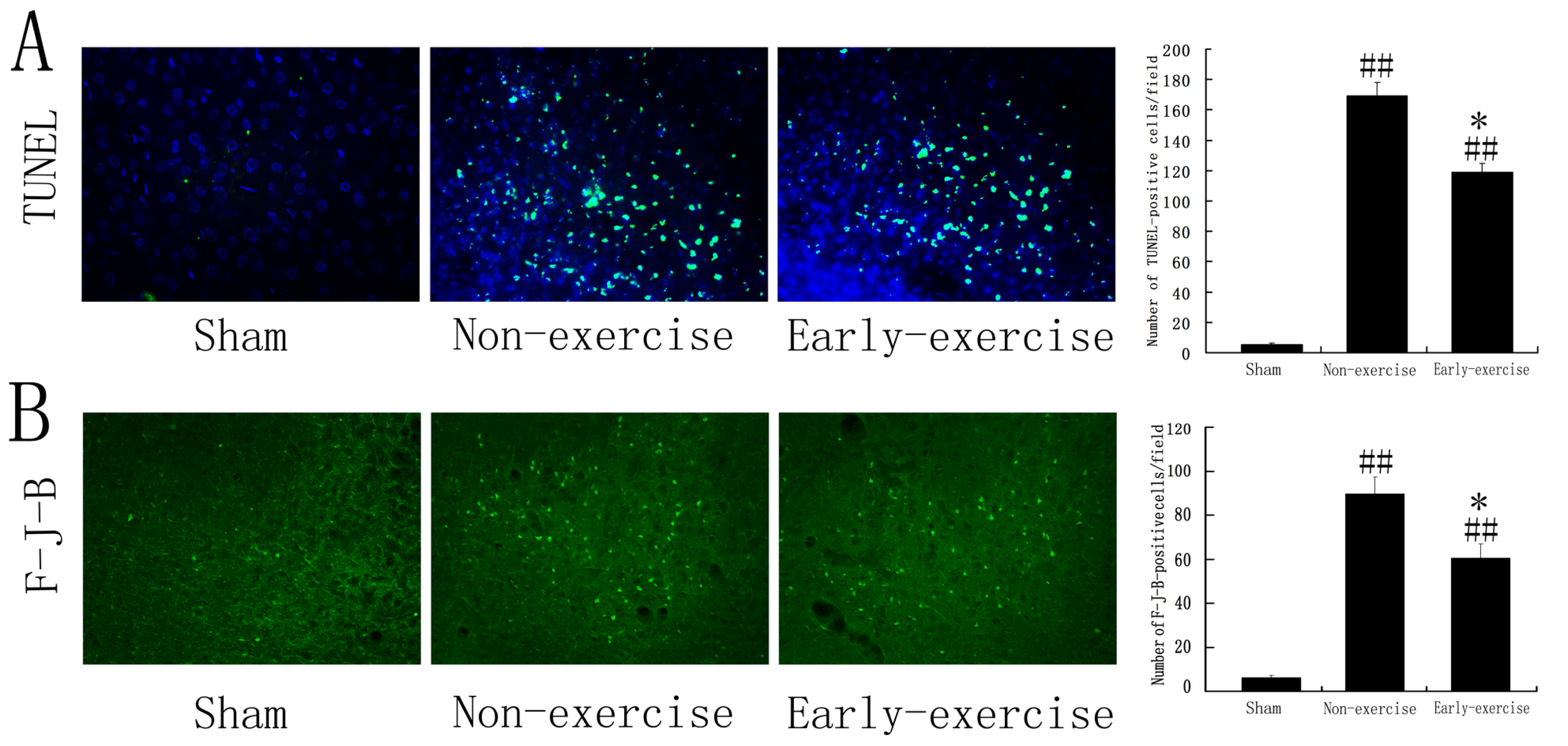
2.2. Early Exercise Reduced Apoptotic Cells after MCAO
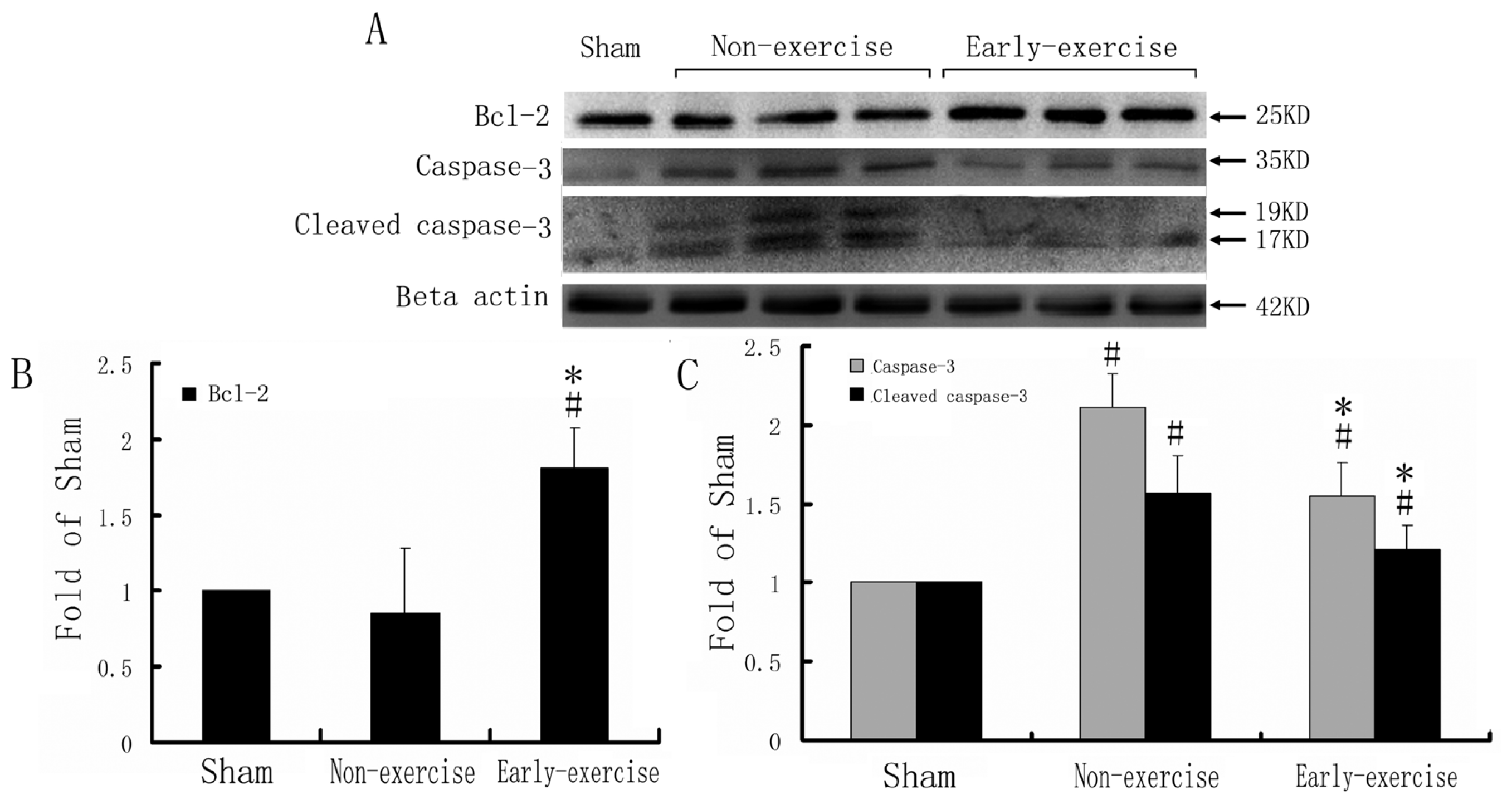
2.3. Expression of Caspase-3, Cleaved Caspase-3 and Bcl-2
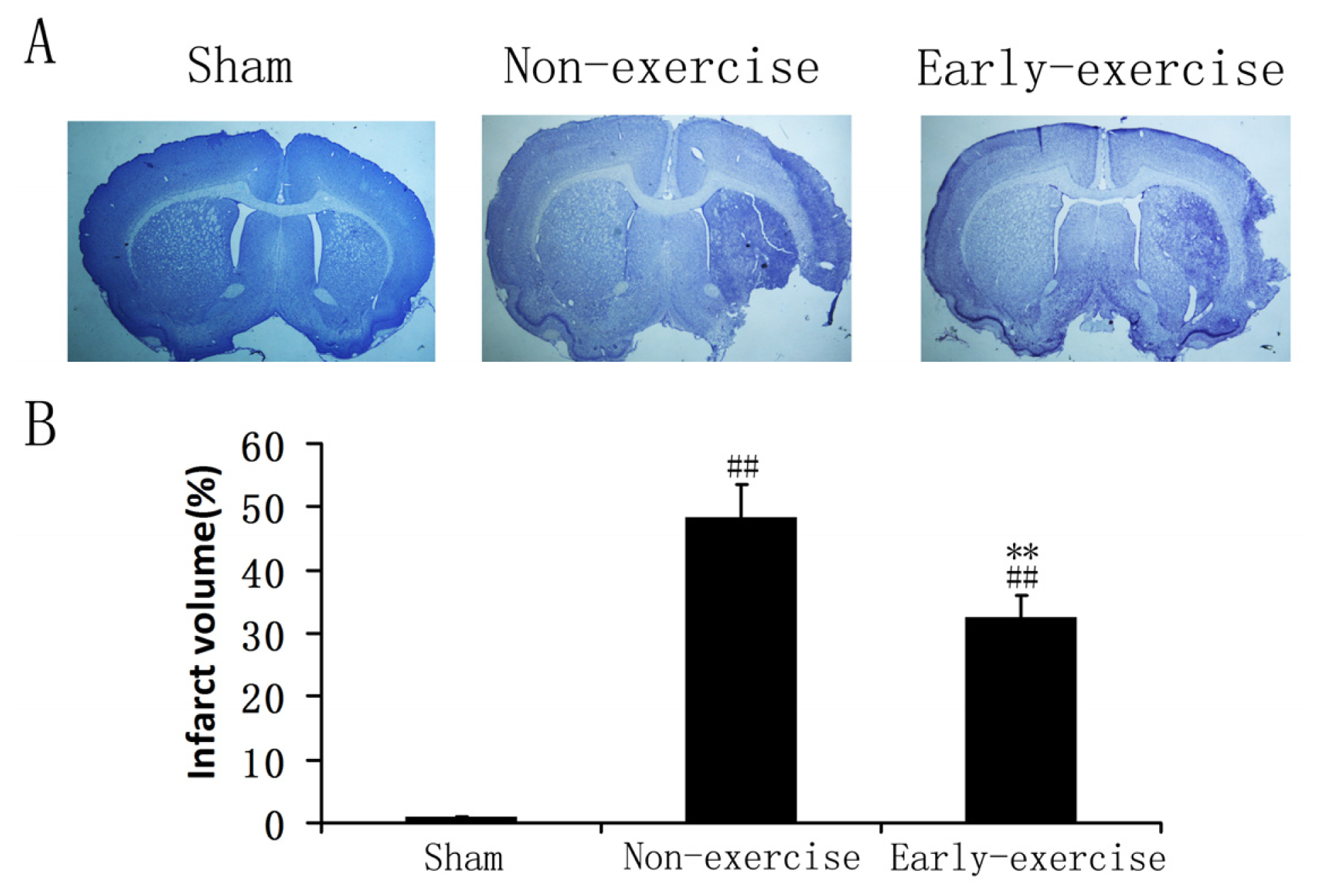
2.4. Early Exercise Reduced the Infarct Volume after MCAO
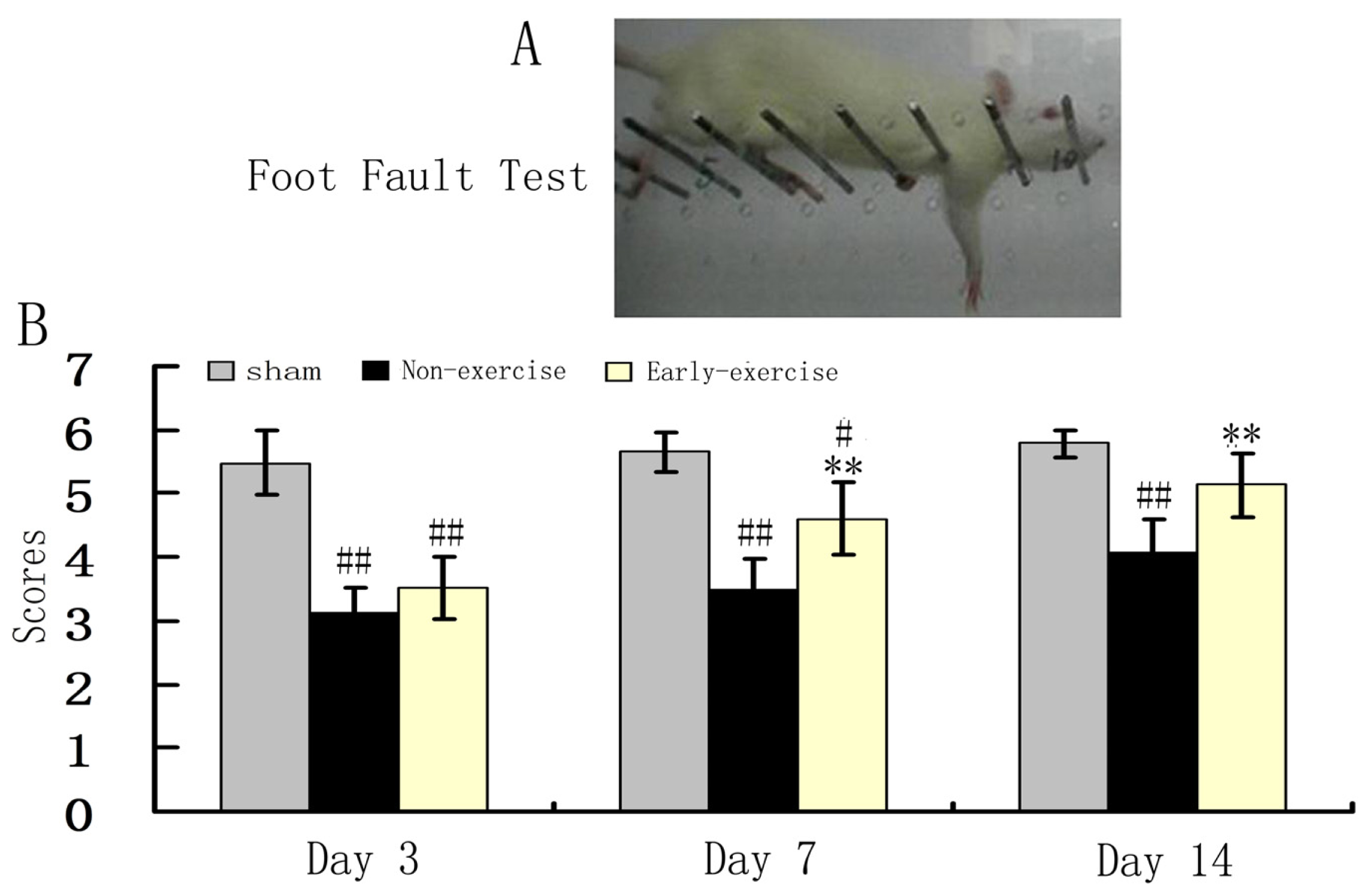
2.5. Early Exercise Improved the Recovery of Coordinated Locomotor Function after MCAO
2.6. Discussion
3. Experimental Section
3.1. Rat Middle Cerebral Artery Occlusion (MCAO) Model
3.2. Group and Treadmill Training Protocol
3.3. Tissue Section Preparation
3.4. TUNEL and F-J-B Staining
3.5. Protein Isolation and Western Blotting
3.6. Determination of Brain Infarct Volume
3.7. Foot Fault Test
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Knecht, S.; Hesse, S.; Oster, P. Rehabilitation after stroke. Dtsch. Arztebl. Int 2011, 108, 600–606. [Google Scholar]
- The atlas of heart disease and stroke. Available online: http://www.who.int/cardiovascular_diseases/resources/atlas/en/ accessed on 13 March 2013.
- De la Perez, O.N.; Davalos, A. Neuroprotection in cerebral infarction: The opportunity of new studies. Cerebrovasc. Dis 2007, 24, 153–156. [Google Scholar]
- Shimodozono, M.; Noma, T.; Nomoto, Y.; Hisamatsu, N.; Kamada, K.; Miyata, R.; Matsumoto, S.; Ogata, A.; Etoh, S.; Basford, J.R.; et al. Benefits of a repetitive facilitative exercise program for the upper paretic extremity after subacute stroke: A randomized controlled trial. Neurorehabil. Neural Repair 2012. [Google Scholar] [CrossRef]
- Dragert, K.; Zehr, E.P. High-intensity unilateral dorsiflexor resistance training results in bilateral neuromuscular plasticity after stroke. Exp. Brain Res 2013, 225, 93–104. [Google Scholar]
- Bernhardt, J. Very early mobilization following acute stroke: Controversies, the unknowns, and a way forward. Ann. Indian Acad. Neurol 2008, 11, 88–98. [Google Scholar]
- Bland, S.T.; Schallert, T.; Strong, R.; Aronowski, J.; Grotta, J.C.; Feeney, D.M. Early exclusive use of the affected forelimb after moderate transient focal ischemia in rats: Functional and anatomic outcome. Stroke 2000, 31, 1144–1152. [Google Scholar]
- Scopel, D.; Fochesatto, C.; Cimarosti, H.; Rabbo, M.; Bello-Klein, A.; Salbego, C.; Netto, C.A.; Siqueira, I.R. Exercise intensity influences cell injury in rat hippocampal slices exposed to oxygen and glucose deprivation. Brain Res. Bull 2006, 71, 155–159. [Google Scholar]
- Seo, H.G.; Kim, D.Y.; Park, H.W.; Lee, S.U.; Park, S.H. Early motor balance and coordination training increased synaptophysin in subcortical regions of the ischemic rat brain. J. Korean Med. Sci 2010, 25, 1638–1645. [Google Scholar]
- Lee, S.U.; Kim, D.Y.; Park, S.H.; Choi, D.H.; Park, H.W.; Han, T.R. Mild to moderate early exercise promotes recovery from cerebral ischemia in rats. Can. J. Neurol. Sci 2009, 36, 443–449. [Google Scholar]
- Purvis, T.; Cadilhac, D.; Donnan, G.; Bernhardt, J. Systematic review of process indicators: Including early rehabilitation interventions used to measure quality of acute stroke care. Int. J. Stroke 2009, 4, 72–80. [Google Scholar]
- Bernhardt, J.; Dewey, H.; Thrift, A.; Collier, J.; Donnan, G. A very early rehabilitation trial for stroke (avert): Phase II safety and feasibility. Stroke 2008, 39, 390–396. [Google Scholar]
- Cumming, T.B.; Thrift, A.G.; Collier, J.M.; Churilov, L.; Dewey, H.M.; Donnan, G.A.; Bernhardt, J. Very early mobilization after stroke fast-tracks return to walking: Further results from the phase II avert randomized controlled trial. Stroke 2011, 42, 153–158. [Google Scholar]
- Clinical guidelines for acute stroke management. Available online: http://www.strokefoundation.com.au/news/welcome/clinicalguidelines-for-acute-stroke-management accessed on 13 March 2013.
- Yuan, J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis 2009, 14, 469–477. [Google Scholar]
- Ribe, E.M.; Serrano-Saiz, E.; Akpan, N.; Troy, C.M. Mechanisms of neuronal death in disease: Defining the models and the players. Biochem. J 2008, 415, 165–182. [Google Scholar]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell. Biol 2008, 9, 231–241. [Google Scholar]
- Legos, J.J.; Barone, F.C. Update on pharmacological strategies for stroke: Prevention, acute intervention and regeneration. Curr. Opin. Investig. Drugs 2003, 4, 847–858. [Google Scholar]
- Mitsios, N.; Gaffney, J.; Krupinski, J.; Mathias, R.; Wang, Q.; Hayward, S.; Rubio, F.; Kumar, P.; Kumar, S.; Slevin, M. Expression of signaling molecules associated with apoptosis in human ischemic stroke tissue. Cell Biochem. Biophys 2007, 47, 73–86. [Google Scholar]
- Singh, M.H.; Brooke, S.M.; Zemlyak, I.; Sapolsky, R.M. Evidence for caspase effects on release of cytochrome c and aif in a model of ischemia in cortical neurons. Neurosci. Lett 2010, 469, 179–183. [Google Scholar]
- Martinou, J.C.; Dubois-Dauphin, M.; Staple, J.K.; Rodriguez, I.; Frankowski, H.; Missotten, M.; Albertini, P.; Talabot, D.; Catsicas, S.; Pietra, C. Overexpression of bcl-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 1994, 13, 1017–1030. [Google Scholar]
- Zhao, H.; Yenari, M.A.; Cheng, D.; Barreto-Chang, O.L.; Sapolsky, R.M.; Steinberg, G.K. Bcl-2 transfection via herpes simplex virus blocks apoptosis-inducing factor translocation after focal ischemia in the rat. J. Cereb. Blood Flow Metab 2004, 24, 681–692. [Google Scholar]
- De Waard, M.C.; Duncker, D.J. Prior exercise improves survival, infarct healing, and left ventricular function after myocardial infarction. J. Appl. Physiol 2009, 107, 928–936. [Google Scholar]
- Haack, D.; Luu, H.; Cho, J.; Chen, M.J.; Russo-Neustadt, A. Exercise reverses chronic stress-induced bax oligomer formation in the cerebral cortex. Neurosci. Lett 2008, 438, 290–294. [Google Scholar]
- Kavazis, A.N.; Smuder, A.J.; Min, K.; Tumer, N.; Powers, S.K. Short-term exercise training protects against doxorubicin-induced cardiac mitochondrial damage independent of hsp72. Am. J. Physiol. Heart Circ. Physiol 2010, 299, H1515–H1524. [Google Scholar]
- Ascensao, A.; Magalhaes, J.; Soares, J.M.; Ferreira, R.; Neuparth, M.J.; Marques, F.; Oliveira, P.J.; Duarte, J.A. Moderate endurance training prevents doxorubicin-induced in vivo mitochondriopathy and reduces the development of cardiac apoptosis. Am. J. Physiol. Heart Circ. Physiol 2005, 289, H722–H731. [Google Scholar]
- Kavazis, A.N.; McClung, J.M.; Hood, D.A.; Powers, S.K. Exercise induces a cardiac mitochondrial phenotype that resists apoptotic stimuli. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H928–H935. [Google Scholar]
- Quindry, J.; French, J.; Hamilton, K.; Lee, Y.; Mehta, J.L.; Powers, S. Exercise training provides cardioprotection against ischemia-reperfusion induced apoptosis in young and old animals. Exp. Gerontol 2005, 40, 416–425. [Google Scholar]
- Quindry, J.C.; Hamilton, K.L.; French, J.P.; Lee, Y.; Murlasits, Z.; Tumer, N.; Powers, S.K. Exercise-induced hsp-72 elevation and cardioprotection against infarct and apoptosis. J. Appl. Physiol 2007, 103, 1056–1062. [Google Scholar]
- Ghosh, S.; Khazaei, M.; Moien-Afshari, F.; Ang, L.S.; Granville, D.J.; Verchere, C.B.; Dunn, S.R.; McCue, P.; Mizisin, A.; Sharma, K.; et al. Moderate exercise attenuates caspase-3 activity, oxidative stress, and inhibits progression of diabetic renal disease in db/db mice. Am. J. Physiol. Renal. Physiol 2009, 296, F700–F708. [Google Scholar]
- French, J.P.; Hamilton, K.L.; Quindry, J.C.; Lee, Y.; Upchurch, P.A.; Powers, S.K. Exercise-induced protection against myocardial apoptosis and necrosis: Mnsod, calcium-handling proteins, and calpain. FASEB J 2008, 22, 2862–2871. [Google Scholar]
- Kwak, H.B.; Song, W.; Lawler, J.M. Exercise training attenuates age-induced elevation in bax/bcl-2 ratio, apoptosis, and remodeling in the rat heart. FASEB J 2006, 20, 791–793. [Google Scholar]
- Peterson, J.M.; Bryner, R.W.; Sindler, A.; Frisbee, J.C.; Alway, S.E. Mitochondrial apoptotic signaling is elevated in cardiac but not skeletal muscle in the obese zucker rat and is reduced with aerobic exercise. J. Appl. Physiol 2008, 105, 1934–1943. [Google Scholar]
- Ascensao, A.; Lumini-Oliveira, J.; Machado, N.G.; Ferreira, R.M.; Goncalves, I.O.; Moreira, A.C.; Marques, F.; Sardao, V.A.; Oliveira, P.J.; Magalhaes, J. Acute exercise protects against calcium-induced cardiac mitochondrial permeability transition pore opening in doxorubicin-treated rats. Clin. Sci. (Lond. ) 2011, 120, 37–49. [Google Scholar]
- Steensberg, A.; Dalsgaard, M.K.; Secher, N.H.; Pedersen, B.K. Cerebrospinal fluid il-6, hsp72, and tnf-alpha in exercising humans. Brain Behav. Immun 2006, 20, 585–589. [Google Scholar]
- Packer, N.; Pervaiz, N.; Hoffman-Goetz, L. Does exercise protect from cognitive decline by altering brain cytokine and apoptotic protein levels? A systematic review of the literature. Exerc. Immunol. Rev 2010, 16, 138–162. [Google Scholar]
- Um, H.S.; Kang, E.B.; Leem, Y.H.; Cho, I.H.; Yang, C.H.; Chae, K.R.; Hwang, D.Y.; Cho, J.Y. Exercise training acts as a therapeutic strategy for reduction of the pathogenic phenotypes for Alzheimer’s disease in an nse/appsw-transgenic model. Int. J. Mol. Med 2008, 22, 529–539. [Google Scholar]
- Zhang, P.; Zhang, Q.; Pu, H.; Wu, Y.; Bai, Y.; Vosler, P.S.; Chen, J.; Shi, H.; Gao, Y.; Hu, Y. Very early-initiated physical rehabilitation protects against ischemic brain injury. Front. Biosci (Elite Ed.) 2012, 4, 2476–2489. [Google Scholar]
- Zhang, Q.; Wu, Y.; Sha, H.; Zhang, P.; Jia, J.; Hu, Y.; Zhu, J. Early exercise affects mitochondrial transcription factors expression after cerebral ischemia in rats. Int. J. Mol. Sci 2012, 13, 1670–1679. [Google Scholar]
- Schmued, L.C.; Hopkins, K.J. Fluoro-jade b: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 2000, 874, 123–130. [Google Scholar]
- Broughton, B.R.S.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar]
- Namura, S.; Zhu, J.; Fink, K.; Endres, M.; Srinivasan, A.; Tomaselli, K.J.; Yuan, J.; Moskowitz, M.A. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci 1998, 18, 3659–3668. [Google Scholar]
- Rami, A.; Sims, J.; Botez, G.; Winckler, J. Spatial resolution of phospholipid scramblase 1 (plscr1), caspase-3 activation and dna-fragmentation in the human hippocampus after cerebral ischemia. Neurochem. Int 2003, 43, 79–87. [Google Scholar]
- Le, D.A.; Wu, Y.; Huang, Z.; Matsushita, K.; Plesnila, N.; Augustinack, J.C.; Hyman, B.T.; Yuan, J.; Kuida, K.; Flavell, R.A.; et al. Caspase activation and neuroprotection in caspase-3- deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15188–15193. [Google Scholar]
- Fink, K.; Zhu, J.; Namura, S.; Shimizu-Sasamata, M.; Endres, M.; Ma, J.; Dalkara, T.; Yuan, J.; Moskowitz, M.A. Prolonged therapeutic window for ischemic brain damage caused by delayed caspase activation. J. Cereb. Blood Flow Metab 1998, 18, 1071–1076. [Google Scholar]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar]
- Adams, J.M.; Cory, S. Life-or-death decisions by the bcl-2 protein family. Trends Biochem. Sci 2001, 26, 61–66. [Google Scholar]
- Chen, J.; Graham, S.H.; Chan, P.H.; Lan, J.; Zhou, R.L.; Simon, R.P. Bcl-2 is expressed in neurons that survive focal ischemia in the rat. Neuroreport 1995, 6, 394–398. [Google Scholar]
- Shinoura, N.; Satou, R.; Yoshida, Y.; Asai, A.; Kirino, T.; Hamada, H. Adenovirus-mediated transfer of bcl-x(l) protects neuronal cells from bax-induced apoptosis. Exp. Cell Res 2000, 254, 221–231. [Google Scholar]
- Zhao, H.; Yenari, M.A.; Cheng, D.; Sapolsky, R.M.; Steinberg, G.K. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J. Neurochem 2003, 85, 1026–1036. [Google Scholar]
- Okazaki, T.; Magaki, T.; Takeda, M.; Kajiwara, Y.; Hanaya, R.; Sugiyama, K.; Arita, K.; Nishimura, M.; Kato, Y.; Kurisu, K. Intravenous administration of bone marrow stromal cells increases survivin and bcl-2 protein expression and improves sensorimotor function following ischemia in rats. Neurosci. Lett 2008, 430, 109–114. [Google Scholar]
- Xing, B.; Chen, H.; Zhang, M.; Zhao, D.; Jiang, R.; Liu, X.; Zhang, S. Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke 2008, 39, 2362–2369. [Google Scholar]
- Graham, S.H.; Chen, J. Programmed cell death in cerebral ischemia. J. Cereb. Blood Flow Metab 2001, 21, 99–109. [Google Scholar]
- Chen, S.D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, pgc-1alpha and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci 2011, 12, 7199–7215. [Google Scholar]
- Hood, D.A.; Chabi, B.; Menzies, K.; O’Leary, M.; Walkinshaw, D. Exercise-Induced Mitochondrial Biogenesis in Skeletal Muscle. In Role of Physical Exercise in Preventing Disease and Improving the Quality of Life; Stocchi, V., de Feo, P., Hood, D.A., Eds.; Springer Milan: New York, NY, USA, 2007; pp. 37–60. [Google Scholar]
- Wright, D.C.; Han, D.H.; Garcia-Roves, P.M.; Geiger, P.C.; Jones, T.E.; Holloszy, J.O. Exercise-induced mitochondrial biogenesis begins before the increase in muscle pgc-1alpha expression. J. Biol. Chem 2007, 282, 194–199. [Google Scholar]
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise training increases mitochondrial biogenesis in the brain. J. Appl. Physiol 2011, 111, 1066–1071. [Google Scholar]
- Bayod, S.; Del, V.J.; Canudas, A.M.; Lalanza, J.F.; Sanchez-Roige, S.; Camins, A.; Escorihuela, R.M.; Pallas, M. Long-term treadmill exercise induces neuroprotective molecular changes in rat brain. J. Appl. Physiol 2011, 111, 1380–1390. [Google Scholar]
- Minami, M.; Katayama, T.; Satoh, M. Brain cytokines and chemokines: Roles in ischemic injury and pain. J. Pharmacol. Sci 2006, 100, 461–470. [Google Scholar]
- Offner, H.; Subramanian, S.; Parker, S.M.; Afentoulis, M.E.; Vandenbark, A.A.; Hurn, P.D. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb. Blood Flow Metab 2006, 26, 654–665. [Google Scholar]
- Srivastava, K.D.; Rom, W.N.; Jagirdar, J.; Yie, T.A.; Gordon, T.; Tchou-Wong, K.M. Crucial role of interleukin-1beta and nitric oxide synthase in silica-induced inflammation and apoptosis in mice. Am. J. Respir. Crit. Care. Med 2002, 165, 527–533. [Google Scholar]
- Marcinkiewicz, J.L.; Balchak, S.K.; Morrison, L.J. The involvement of tumor necrosis factor-alpha (tnf) as an intraovarian regulator of oocyte apoptosis in the neonatal rat. Front. Biosci 2002, 7, d1997–d2005. [Google Scholar]
- Agard, N.J.; Maltby, D.; Wells, J.A. Inflammatory stimuli regulate caspase substrate profiles. Mol. Cell Proteomics 2010, 9, 880–893. [Google Scholar]
- Bennett, M.; Yu, H.; Clarke, M. Signalling from dead cells drives inflammation and vessel remodelling. Vascul. Pharmacol 2012, 56, 187–192. [Google Scholar]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar]
- Tureyen, K.; Vemuganti, R.; Sailor, K.A.; Dempsey, R.J. Infarct volume quantification in mouse focal cerebral ischemia: A comparison of triphenyltetrazolium chloride and cresyl violet staining techniques. J. Neurosci. Methods 2004, 139, 203–207. [Google Scholar]
- Ding, Y.; Zhou, Y.; Lai, Q.; Li, J.; Park, H.; Diaz, F.G. Impaired motor activity and motor learning function in rat with middle cerebral artery occlusion. Behav. Brain Res 2002, 132, 29–36. [Google Scholar]
- Liebelt, B.; Papapetrou, P.; Ali, A.; Guo, M.; Ji, X.; Peng, C.; Rogers, R.; Curry, A.; Jimenez, D.; Ding, Y. Exercise preconditioning reduces neuronal apoptosis in stroke by up-regulating heat shock protein-70 (heat shock protein-72) and extracellular-signal-regulated-kinase 1/2. Neuroscience 2010, 166, 1091–1100. [Google Scholar]
- NIH Image software, version. Available online: http://rsb.info.nih.gov/nih-image/ accessed on 13 March 2013.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | pH | PaCO2 (mm Hg) | PaO2 (mm Hg) | MABP (mm Hg) | Rectal temperature (°C) |
|---|---|---|---|---|---|
| sham (n = 6) | 7.36 ± 0.02 | 44.3 ± 1.8 | 95.8 ± 2.8 | 96.1 ± 6.7 | 37.3 ± 0.1 |
| exercise (n = 10) | |||||
| pre-ischemia | 7.30 ± 0.07 | 40.5 ± 6.7 | 89.9 ± 2.8 | 95.0 ± 4.5 | 37.6 ± 0.3 |
| during-ischemia | 7.32 ± 0.05 | 42.7 ± 8.1 | 84.3 ± 5.3 | 84.4 ± 9.4 | 37.5 ± 0.2 |
| post-ischemia | 7.30 ± 0.09 | 38.2 ± 8.4 | 89.5 ± 6.7 | 89.2 ± 10.0 | 37.6 ± 0.3 |
| non-exercise (n = 10) | |||||
| pre-ischemia | 7.39 ± 0.01 | 37.95 ± 2.48 | 87.65 ± 2.97 | 95.87 ± 3.60 | 37.5 ± 0.4 |
| during-ischemia | 7.36 ± 0.01 | 47.63 ± 8.24 | 82.86 ± 2.89 | 88.5 ± 2.78 | 37.3 ± 0.3 |
| post-ischemia | 7.4 ± 0.006 | 34.43 ± 1.8 | 88.4 ± 3.98 | 90.4 ± 3.5 | 37.6 ± 0.2 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, P.; Zhang, Y.; Zhang, J.; Wu, Y.; Jia, J.; Wu, J.; Hu, Y. Early Exercise Protects against Cerebral Ischemic Injury through Inhibiting Neuron Apoptosis in Cortex in Rats. Int. J. Mol. Sci. 2013, 14, 6074-6089. https://doi.org/10.3390/ijms14036074
Zhang P, Zhang Y, Zhang J, Wu Y, Jia J, Wu J, Hu Y. Early Exercise Protects against Cerebral Ischemic Injury through Inhibiting Neuron Apoptosis in Cortex in Rats. International Journal of Molecular Sciences. 2013; 14(3):6074-6089. https://doi.org/10.3390/ijms14036074
Chicago/Turabian StyleZhang, Pengyue, Yuling Zhang, Jie Zhang, Yi Wu, Jie Jia, Junfa Wu, and Yongshan Hu. 2013. "Early Exercise Protects against Cerebral Ischemic Injury through Inhibiting Neuron Apoptosis in Cortex in Rats" International Journal of Molecular Sciences 14, no. 3: 6074-6089. https://doi.org/10.3390/ijms14036074