p53 and Ceramide as Collaborators in the Stress Response

{kind=link}
{kind=link}
Abstract
:1. Introduction
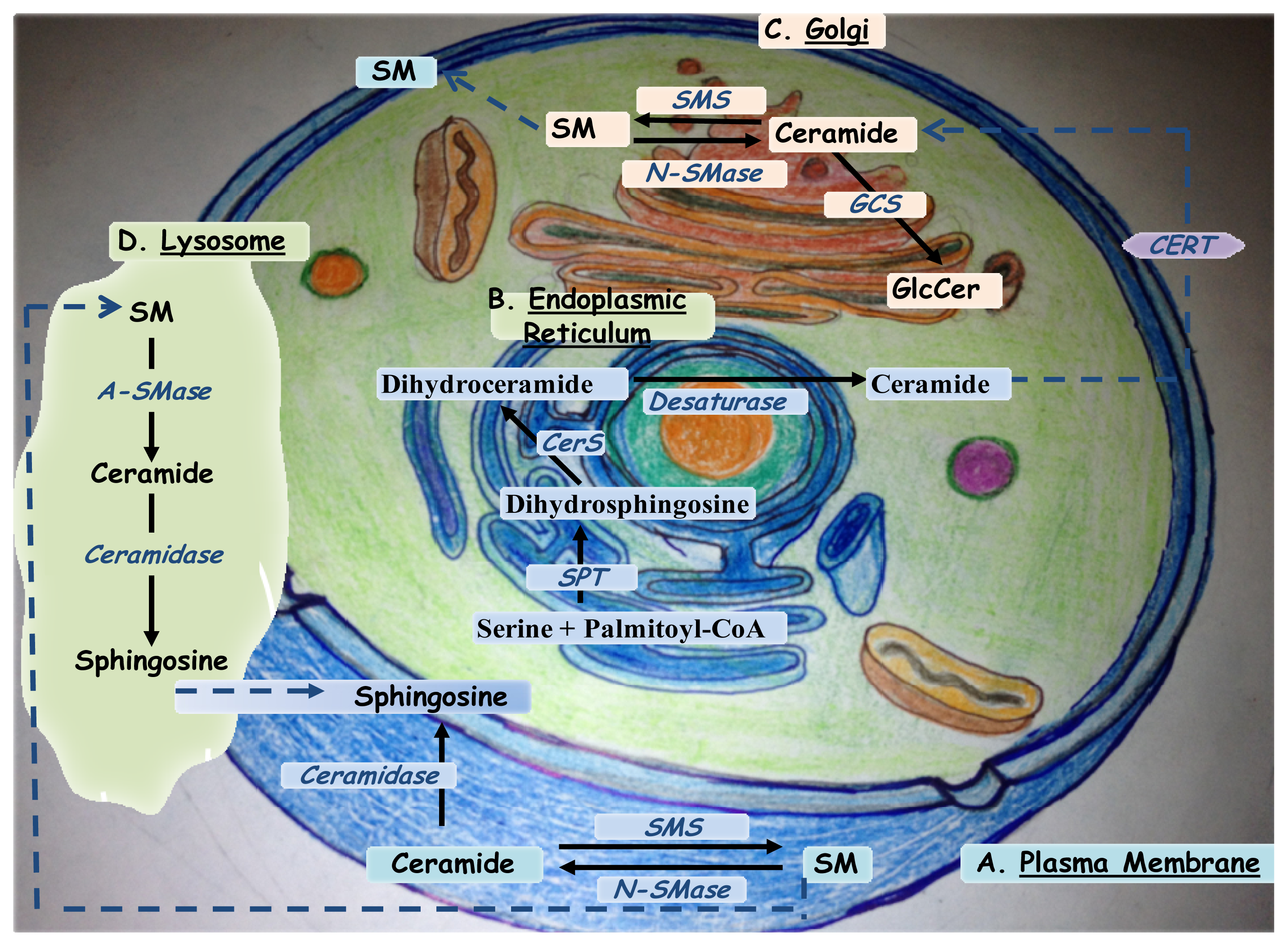
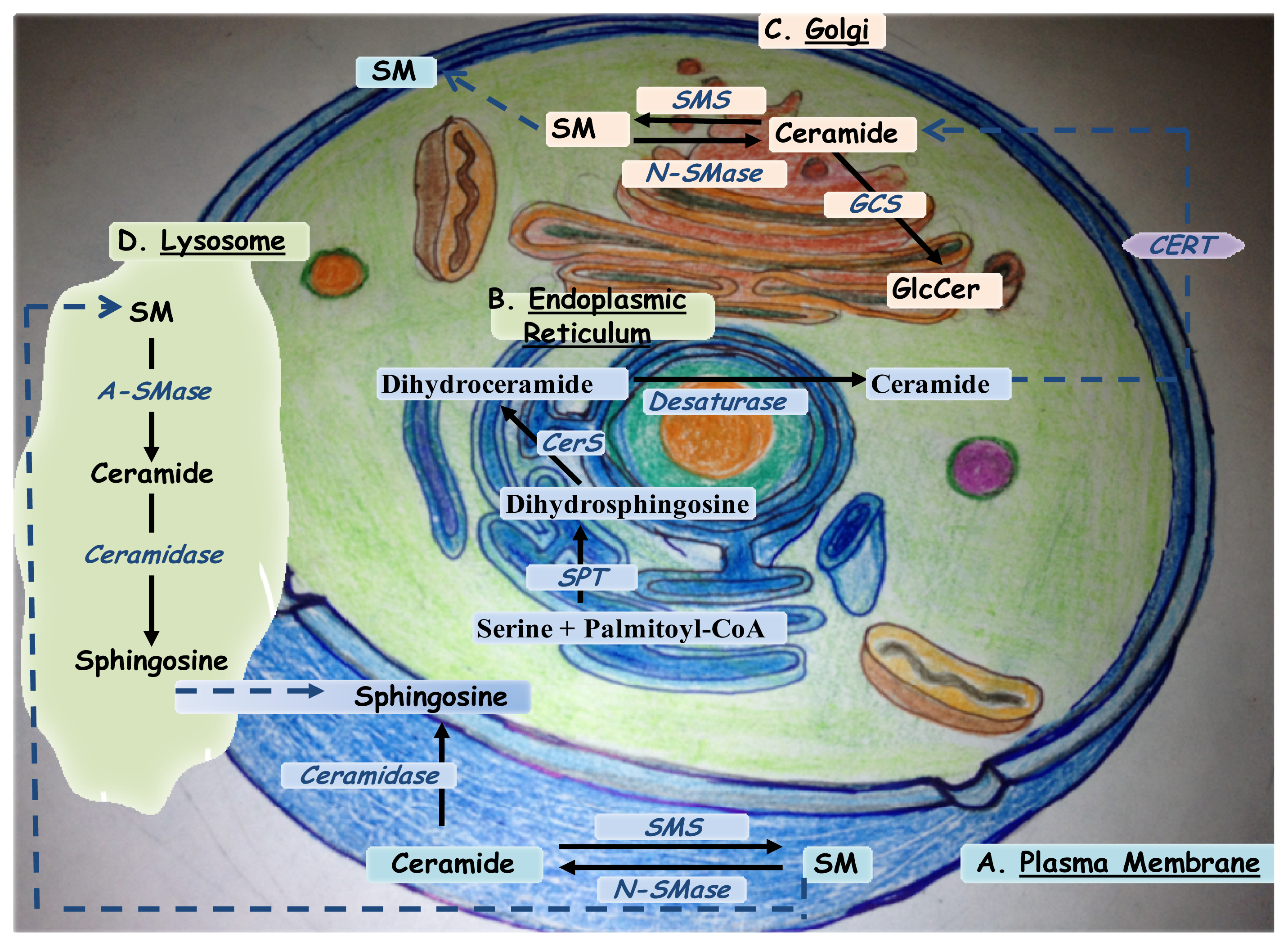
2. Ceramide Biosynthesis
3. Ceramide and Cellular Signaling
3.1. Ceramide and Hypoxia/Hyperoxia
3.2. Ceramide Accumulation and Cell Death
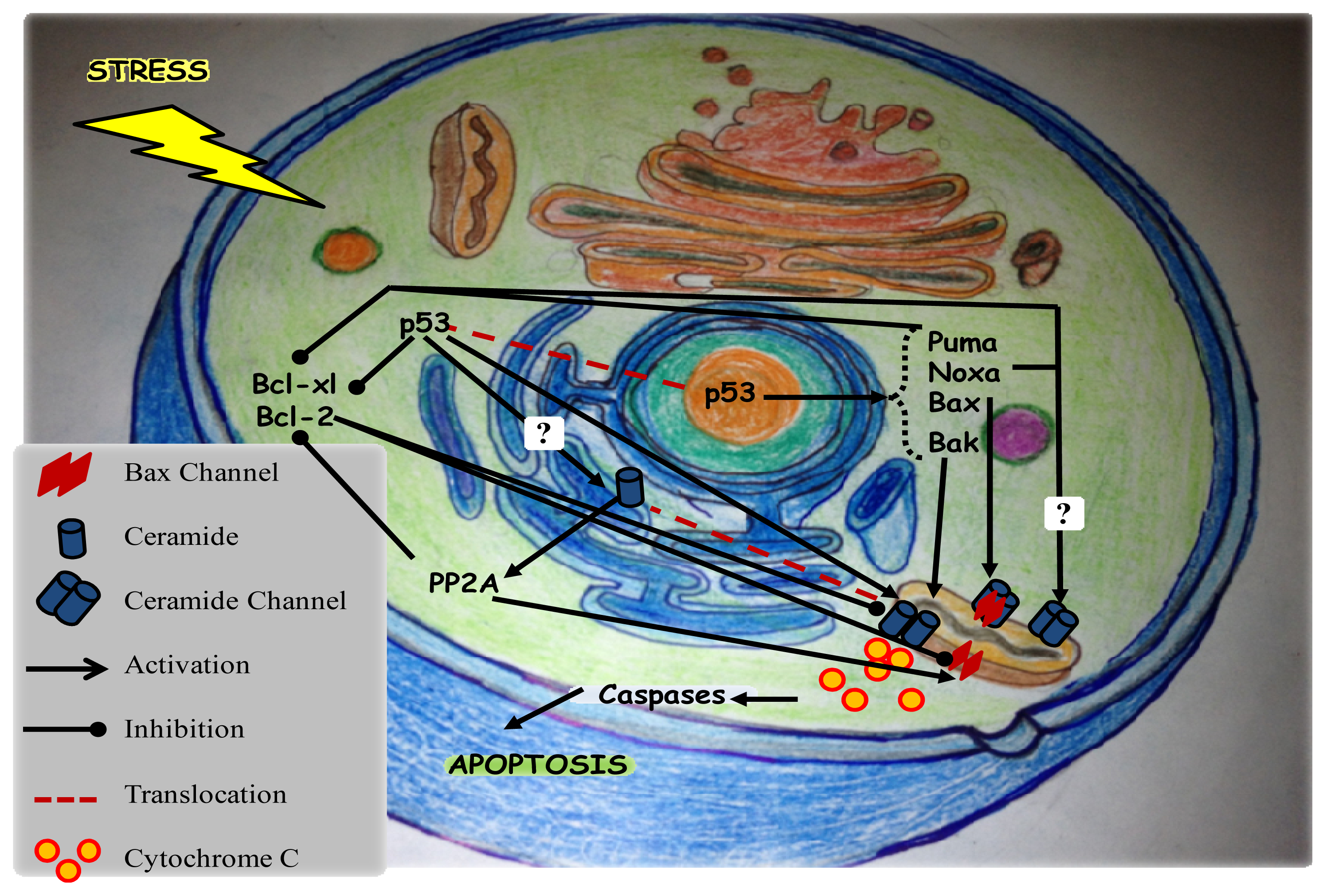
4. Mitochondrial Ceramide Channels
5. Ceramide and Related Molecules
6. Ceramide in Yeast
7. p53 and Apoptosis
7.1. Mechanism of Action
7.2. Regulation of p53
7.3. Apoptosis Independent of p53
8. Ceramide and p53
8.1. Ceramide and p53 in Apoptosis
8.2. Ceramide and p53 in Cell Cycle Arrest
9. Ceramide and Cancer
10. Conclusions
Acknowledgments
Conflict of Interest
References
- Basu, S.; Kolesnick, R. Stress signals for apoptosis: Ceramide and c-Jun kinase. Oncogene 1998, 17, 3277–3285. [Google Scholar]
- Antoine, K.Z.; Hussain, H.; Dongo, E.; Kouam, S.F.; Schulz, B.; Krohn, K. Cameroonemide A: A new ceramide from Helichrysum cameroonense. J. Asian Nat. Products Res 2010, 12, 629–633. [Google Scholar]
- Bankeu, J.J.; Mustafa, S.A.; Gojayev, A.S.; Lenta, B.D.; Tchamo Noungoue, D.; Ngouela, S.A.; Asaad, K.; Choudhary, M.I.; Prigge, S.; Guliyev, A.A.; et al. Ceramide and Cerebroside from the stem bark of Ficus mucuso (Moraceae). Chem. Pharm. Bull 2010, 58, 1661–1665. [Google Scholar]
- Mullen, T.D.; Spassieva, S.; Jenkins, R.W.; Kitatani, K.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Selective knockdown of ceramide synthases reveals complex interregulation of sphingolipid metabolism. J. Lipid Res 2011, 52, 68–77. [Google Scholar]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar]
- Haimovitz-Friedman, A.; Kolesnick, R.N.; Fuks, Z. Ceramide signaling in apoptosis. Br. Med. Bull 1997, 53, 539–553. [Google Scholar]
- Altura, B.M.; Kostellow, A.B.; Zhang, A.; Li, W.; Morrill, G.A.; Gupta, R.K.; Altura, B.T. Expression of the nuclear factor-kappaB and proto-oncogenes c-fos and c-jun are induced by low extracellular Mg2+ in aortic and cerebral vascular smooth muscle cells: Possible links to hypertension, atherogenesis, and stroke. Am. J. Hypertens 2003, 16, 701–707. [Google Scholar]
- Morrill, G.A.; Gupta, R.K.; Kostellow, A.B.; Ma, G.Y.; Zhang, A.; Altura, B.T.; Altura, B.M. Mg2+ modulates membrane lipids in vascular smooth muscle: A link to atherogenesis. FEBS Lett 1997, 408, 191–194. [Google Scholar]
- Morrill, G.A.; Gupta, R.K.; Kostellow, A.B.; Ma, G.Y.; Zhang, A.; Altura, B.T.; Altura, B.M. Mg2+ modulates membrane sphingolipid and lipid second messenger levels in vascular smooth muscle cells. FEBS Lett 1998, 440, 167–171. [Google Scholar]
- Li, J.; Li, W.; Liu, W.; Altura, B.T.; Altura, B.M. Peroxynitrite induces apoptosis and decline in intracellular free Mg with concomitant elevation in [Ca2+]I in rat aortic smooth muscle cells: Possible roles of extracellular and intracellular magnesium ions in peroxynitrite-induced cell death. Drug Metab. Lett 2007, 1, 85–89. [Google Scholar]
- Luberto, C.; Hannun, Y.A. Sphingomyelin synthase, a potential regulator of intracellular levels of ceramide and diacylglycerol during SV40 transformation. Does sphingomyelin synthase account for the putative phosphatidylcholine-specific phospholipase C? J. Biol. Chem 1998, 273, 14550–14559. [Google Scholar]
- Altura, B.M.; Shah, N.C.; Li, Z.; Jiang, X.C.; Zhang, A.; Li, W.; Zheng, T.; Perez-Albela, J.L.; Altura, B.T. Short-term magnesium deficiency upregulates sphingomyelin synthase and p53 in cardiovascular tissues and cells: Relevance to the de novo synthesis of ceramide. Am. J. Physiol 2010, 299, H2046–H2055. [Google Scholar]
- Stancevic, B.; Kolesnick, R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett 2010, 584, 1728–1740. [Google Scholar]
- Birbes, H.; El Bawab, S.; Hannun, Y.A.; Obeid, L.M. Selective hydrolysis of a mitochondrial pool of sphingomyelin induces apoptosis. FASEB J 2001, 15, 2669–2679. [Google Scholar]
- Veldman, R.J.; Klappe, K.; Hoekstra, D.; Kok, J.W. Metabolism and apoptotic properties of elevated ceramide in HT29rev cells. Biochem. J 1998, 331, 563–569. [Google Scholar]
- Zhang, P.; Liu, B.; Jenkins, G.M.; Hannun, Y.A.; Obeid, L.M. Expression of neutral sphingomyelinase identifies a distinct pool of sphingomyelin involved in apoptosis. J. Biol. Chem 1997, 272, 9609–9612. [Google Scholar]
- Bieberich, E. Ceramide in stem cell differentiation and embryo development: Novel functions of a topological cell-signaling lipid and the concept of ceramide compartments. J. Lipids 2011, 2011. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. The Ceramide-centric universe of lipid-mediated cell regulation: Stress encounters of the lipid kind. J. Biol. Chem 2002, 277, 25847–25850. [Google Scholar]
- Pandey, S.; Murphy, R.F.; Agrawal, D.K. Recent advances in the immunobiology of ceramide. Exp. Mol. Pathol 2007, 82, 298–309. [Google Scholar]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev 2008, 9, 162–176. [Google Scholar]
- Webb, L.M.; Arnholt, A.T.; Venable, M.E. Phospholipase D modulation by ceramide in senescence. Mol. Cell. Biochem 2010, 337, 153–158. [Google Scholar]
- Venable, M.E.; Lee, J.Y.; Smyth, M.J.; Bielawska, A.; Obeid, L.M. Role of ceramide in cellular senescence. J. Biol. Chem 1995, 270, 30701–30708. [Google Scholar]
- Choi, J.M.; Chu, S.J.; Ahn, K.H.; Kim, S.K.; Ji, J.E.; Won, J.H.; Kim, H.C.; Back, M.J.; Kim, D.K. C(6)-ceramide enhances phagocytic activity of Kupffer cells through the production of endogenous ceramides. Mol. Cells 2011, 32, 325–331. [Google Scholar]
- Separovic, D.; Kelekar, A.; Nayak, A.K.; Tarca, A.L.; Hanada, K.; Pierce, J.S.; Bielawski, J. Increased ceramide accumulation correlates with downregulation of the autophagy protein ATG-7 in MCF-7 cells sensitized to photodamage. Archives Biochem. Biophys 2010, 494, 101–105. [Google Scholar]
- Turner, L.S.; Cheng, J.C.; Beckham, T.H.; Keane, T.E.; Norris, J.S.; Liu, X. Autophagy is increased in prostate cancer cells overexpressing acid ceramidase and enhances resistance to C6 ceramide. Prostate Cancer Prostatic Dis 2011, 14, 30–37. [Google Scholar]
- Abdel Shakor, A.B.; Atia, M.M.; Kwiatkowska, K.; Sobota, A. Cell surface ceramide controls translocation of transferrin receptor to clathrin-coated pits. Cell. Signal 2012, 24, 677–684. [Google Scholar]
- Castro, B.M.; de Almeida, R.F.; Goormaghtigh, E.; Fedorov, A.; Prieto, M. Organization and dynamics of Fas transmembrane domain in raft membranes and modulation by ceramide. Biophys. J 2011, 101, 1632–1641. [Google Scholar]
- Bitar, F.F.; Bitar, H.; El Sabban, M.; Nasser, M.; Yunis, K.A.; Tawil, A.; Dbaibo, G.S. Modulation of ceramide content and lack of apoptosis in the chronically hypoxic neonatal rat heart. Pediatric Res 2002, 51, 144–149. [Google Scholar]
- Noureddine, L.; Azzam, R.; Nemer, G.; Bielawski, J.; Nasser, M.; Bitar, F.; Dbaibo, G.S. Modulation of total ceramide and constituent ceramide species in the acutely and chronically hypoxic mouse heart at different ages. Prostaglandins Other Lipid Mediat 2008, 86, 49–55. [Google Scholar]
- El Alwani, M.; Usta, J.; Nemer, G.; El Sabban, M.; Nasser, M.; Bitar, H.; Souki, R.; Dbaibo, G.S.; Bitar, F.F. Regulation of the sphingolipid signaling pathways in the growing and hypoxic rat heart. Prostaglandins Other Lipid Mediat 2005, 78, 249–263. [Google Scholar]
- Kang, M.S.; Ahn, K.H.; Kim, S.K.; Jeon, H.J.; Ji, J.E.; Choi, J.M.; Jung, K.M.; Jung, S.Y.; Kim, D.K. Hypoxia-induced neuronal apoptosis is mediated by de novo synthesis of ceramide through activation of serine palmitoyltransferase. Cell. Signal 2010, 22, 610–618. [Google Scholar]
- Husari, A.W.; Dbaibo, G.S.; Bitar, H.; Khayat, A.; Panjarian, S.; Nasser, M.; Bitar, F.F.; El-Sabban, M.; Zaatari, G.; Mroueh, S.M. Apoptosis and the activity of ceramide, Bax and Bcl-2 in the lungs of neonatal rats exposed to limited and prolonged hyperoxia. Resp. Res. 2006, 7. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev 2007, 87, 99–163. [Google Scholar]
- Amaral, J.D.; Xavier, J.M.; Steer, C.J.; Rodrigues, C.M. The role of p53 in apoptosis. Discov. Med 2010, 9, 145–152. [Google Scholar]
- Linardic, C.M.; Hannun, Y.A. Identification of a distinct pool of sphingomyelin involved in the sphingomyelin cycle. J. Biol. Chem 1994, 269, 23530–23537. [Google Scholar]
- Futerman, A.H.; Stieger, B.; Hubbard, A.L.; Pagano, R.E. Sphingomyelin synthesis in rat liver occurs predominantly at the cis and medial cisternae of the Golgi apparatus. J. Biol. Chem 1990, 265, 8650–8657. [Google Scholar]
- Cabral, L.M.; Wengert, M.; Almeida, F.G.; Caruso-Neves, C.; Vieyra, A.; Einicker-Lamas, M. Ceramide-activated protein kinases A and C zeta inhibit kidney proximal tubule cell Na(+)-ATPase. Arch. Biochem. Biophys 2010, 498, 57–61. [Google Scholar]
- Luberto, C.; Yoo, D.S.; Suidan, H.S.; Bartoli, G.M.; Hannun, Y.A. Differential effects of sphingomyelin hydrolysis and resynthesis on the activation of NF-kappa B in normal and SV40-transformed human fibroblasts. J. Biol. Chem 2000, 275, 14760–14766. [Google Scholar]
- Zundel, W.; Swiersz, L.M.; Giaccia, A. Caveolin 1-mediated regulation of receptor tyrosine kinase-associated phosphatidylinositol 3-kinase activity by ceramide. Mol. Cell Biol 2000, 20, 1507–1514. [Google Scholar]
- Mesicek, J.; Lee, H.; Feldman, T.; Jiang, X.; Skobeleva, A.; Berdyshev, E.V.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell. Signal 2010, 22, 1300–1307. [Google Scholar]
- Gao, D.; Pararasa, C.; Dunston, C.R.; Bailey, C.J.; Griffiths, H.R. Palmitate promotes monocyte atherogenicity via de novo ceramide synthesis. Free Radical Biol. Med 2012, 53, 796–806. [Google Scholar]
- Aflaki, E.; Doddapattar, P.; Radovic, B.; Povoden, S.; Kolb, D.; Vujic, N.; Wegscheider, M.; Koefeler, H.; Hornemann, T.; Graier, W.F.; et al. C16 ceramide is crucial for triacylglycerol-induced apoptosis in macrophages. Cell Death Dis 2012, 3, e280. [Google Scholar]
- Senkal, C.E.; Ponnusamy, S.; Bielawski, J.; Hannun, Y.A.; Ogretmen, B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J 2010, 24, 296–308. [Google Scholar]
- Huang, W.C.; Chen, C.L.; Lin, Y.S.; Lin, C.F. Apoptotic sphingolipid ceramide in cancer therapy. J. Lipids 2011, 2011. [Google Scholar] [CrossRef]
- Sridevi, P.; Alexander, H.; Laviad, E.L.; Min, J.; Mesika, A.; Hannink, M.; Futerman, A.H.; Alexander, S. Stress-induced ER to Golgi translocation of ceramide synthase 1 is dependent on proteasomal processing. Exp. Cell Res 2010, 316, 78–91. [Google Scholar]
- Monette, J.S.; Gomez, L.A.; Moreau, R.F.; Dunn, K.C.; Butler, J.A.; Finlay, L.A.; Michels, A.J.; Shay, K.P.; Smith, E.J.; Hagen, T.M. (R)-alpha-Lipoic acid treatment restores ceramide balance in aging rat cardiac mitochondria. Pharmacol. Res. 2011, 63, 23–29. [Google Scholar]
- Sauane, M.; Su, Z.Z.; Dash, R.; Liu, X.; Norris, J.S.; Sarkar, D.; Lee, S.G.; Allegood, J.C.; Dent, P.; Spiegel, S.; et al. Ceramide plays a prominent role in MDA-7/IL-24-induced cancer-specific apoptosis. J. Cell. Physiol 2010, 222, 546–555. [Google Scholar]
- Park, J.Y.; Kim, M.J.; Kim, Y.K.; Woo, J.S. Ceramide induces apoptosis via caspase-dependent and caspase-independent pathways in mesenchymal stem cells derived from human adipose tissue. Arch. Toxicol 2011, 85, 1057–1065. [Google Scholar]
- Colombini, M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2010, 1797, 1239–1244. [Google Scholar]
- Dbaibo, G.S.; Obeid, L.M.; Hannun, Y.A. Tumor necrosis factor-alpha (TNF-alpha) signal transduction through ceramide. Dissociation of growth inhibitory effects of TNF-alpha from activation of nuclear factor-kappa B. J. Biol. Chem 1993, 268, 17762–17766. [Google Scholar]
- Dbaibo, G.S.; El-Assaad, W.; Krikorian, A.; Liu, B.; Diab, K.; Idriss, N.Z.; El-Sabban, M.; Driscoll, T.A.; Perry, D.K.; Hannun, Y.A. Ceramide generation by two distinct pathways in tumor necrosis factor alpha-induced cell death. FEBS Lett 2001, 503, 7–12. [Google Scholar]
- Chandran, S.; Machamer, C.E. Inactivation of ceramide transfer protein during pro-apoptotic stress by Golgi disassembly and caspase cleavage. Biochem. J 2012, 442, 391–401. [Google Scholar]
- Dbaibo, G.S.; Perry, D.K.; Gamard, C.J.; Platt, R.; Poirier, G.G.; Obeid, L.M.; Hannun, Y.A. Cytokine response modifier A (CrmA) inhibits ceramide formation in response to tumor necrosis factor (TNF)-alpha: CrmA and Bcl-2 target distinct components in the apoptotic pathway. J. Exp. Med 1997, 185, 481–490. [Google Scholar]
- El-Assaad, W.; El-Sabban, M.; Awaraji, C.; Abboushi, N.; Dbaibo, G.S. Distinct sites of action of Bcl-2 and Bcl-xL in the ceramide pathway of apoptosis. Biochem. J 1998, 336, 735–741. [Google Scholar]
- Kalo, D.; Roth, Z. Involvement of the sphingolipid ceramide in heat-shock-induced apoptosis of bovine oocytes. Reprod. Fertil. Dev 2011, 23, 876–888. [Google Scholar]
- Kujjo, L.L.; Perez, G.I. Ceramide and mitochondrial function in aging oocytes: Joggling a new hypothesis and old players. Reproduction 2012, 143, 1–10. [Google Scholar]
- Chowdhury, I.; Branch, A.; Olatinwo, M.; Thomas, K.; Matthews, R.; Thompson, W.E. Prohibitin (PHB) acts as a potent survival factor against ceramide induced apoptosis in rat granulosa cells. Life Sci 2011, 89, 295–303. [Google Scholar]
- Rizvi, F.; Heimann, T.; Herrnreiter, A.; O’Brien, W.J. Mitochondrial dysfunction links ceramide activated HRK expression and cell death. PLoS One 2011, 6, e18137. [Google Scholar]
- Stiban, J.; Caputo, L.; Colombini, M. Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J. Lipid Res 2008, 49, 625–634. [Google Scholar]
- Babiychuk, E.B.; Atanassoff, A.P.; Monastyrskaya, K.; Brandenberger, C.; Studer, D.; Allemann, C.; Draeger, A. The targeting of plasmalemmal ceramide to mitochondria during apoptosis. PloS One 2011, 6, e23706. [Google Scholar]
- Novgorodov, S.A.; Chudakova, D.A.; Wheeler, B.W.; Bielawski, J.; Kindy, M.S.; Obeid, L.M.; Gudz, T.I. Developmentally regulated ceramide synthase 6 increases mitochondrial Ca2+ loading capacity and promotes apoptosis. J. Biol. Chem 2011, 286, 4644–4658. [Google Scholar]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J 2012, 441, 789–802. [Google Scholar]
- Bionda, C.; Portoukalian, J.; Schmitt, D.; Rodriguez-Lafrasse, C.; Ardail, D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem. J 2004, 382, 527–533. [Google Scholar]
- Siskind, L.J. Mitochondrial ceramide and the induction of apoptosis. J. Bioenerg. Biomembr 2005, 37, 143–153. [Google Scholar]
- Ardail, D.; Maalouf, M.; Boivin, A.; Chapet, O.; Bodennec, J.; Rousson, R.; Rodriguez-Lafrasse, C. Diversity and complexity of ceramide generation after exposure of jurkat leukemia cells to irradiation. Int. J. Radiat .Oncol. Biol. Phys 2009, 73, 1211–1218. [Google Scholar]
- Ganesan, V.; Colombini, M. Regulation of ceramide channels by Bcl-2 family proteins. FEBS Lett 2010, 584, 2128–2134. [Google Scholar]
- Perera, M.N.; Ganesan, V.; Siskind, L.J.; Szulc, Z.M.; Bielawski, J.; Bielawska, A.; Bittman, R.; Colombini, M. Ceramide channels: Influence of molecular structure on channel formation in membranes. Biochim. Biophys. Acta 2012, 1818, 1291–1301. [Google Scholar]
- Perera, M.N.; Lin, S.H.; Peterson, Y.K.; Bielawska, A.; Szulc, Z.M.; Bittman, R.; Colombini, M. Bax and Bcl-xL exert their regulation on different sites of the ceramide channel. Biochem. J 2012, 445, 81–91. [Google Scholar]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar]
- Siskind, L.J.; Mullen, T.D.; Romero Rosales, K.; Clarke, C.J.; Hernandez-Corbacho, M.J.; Edinger, A.L.; Obeid, L.M. The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. J. Biol. Chem 2010, 285, 11818–11826. [Google Scholar]
- Lee, H.; Rotolo, J.A.; Mesicek, J.; Penate-Medina, T.; Rimner, A.; Liao, W.C.; Yin, X.; Ragupathi, G.; Ehleiter, D.; Gulbins, E.; et al. Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PloS One 2011, 6, e19783. [Google Scholar]
- Ganesan, V.; Perera, M.N.; Colombini, D.; Datskovskiy, D.; Chadha, K.; Colombini, M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 2010, 15, 553–562. [Google Scholar]
- Martinez-Abundis, E.; Correa, F.; Pavon, N.; Zazueta, C. Bax distribution into mitochondrial detergent-resistant microdomains is related to ceramide and cholesterol content in postischemic hearts. FEBS J 2009, 276, 5579–5588. [Google Scholar]
- Lecour, S.; van der Merwe, E.; Opie, L.H.; Sack, M.N. Ceramide attenuates hypoxic cell death via reactive oxygen species signaling. J. Cardiovasc. Pharmacol 2006, 47, 158–163. [Google Scholar]
- Cui, J.; Engelman, R.M.; Maulik, N.; Das, D.K. Role of ceramide in ischemic preconditioning. J. Am. Coll. Surg 2004, 198, 770–777. [Google Scholar]
- Carmona-Gutierrez, D.; Reisenbichler, A.; Heimbucher, P.; Bauer, M.A.; Braun, R.J.; Ruckenstuhl, C.; Buttner, S.; Eisenberg, T.; Rockenfeller, P.; Frohlich, K.U.; et al. Ceramide triggers metacaspase-independent mitochondrial cell death in yeast. Cell Cycle 2011, 10, 3973–3978. [Google Scholar]
- Agudo-Lopez, A.; Miguel, B.G.; Fernandez, I.; Martinez, A.M. Role of protein kinase C and mitochondrial permeability transition pore in the neuroprotective effect of ceramide in ischemia-induced cell death. FEBS Lett 2011, 585, 99–103. [Google Scholar]
- Parihar, A.; Parihar, M.S.; Nazarewicz, R.; Ghafourifar, P. Importance of cytochrome c redox state for ceramide-induced apoptosis of human mammary adenocarcinoma cells. Biochim. Biophys. Acta 2010, 1800, 646–654. [Google Scholar]
- Kizhakkayil, J.; Thayyullathil, F.; Chathoth, S.; Hago, A.; Patel, M.; Galadari, S. Glutathione regulates caspase-dependent ceramide production and curcumin-induced apoptosis in human leukemic cells. Free Radic. Biol. Med 2012, 52, 1854–1864. [Google Scholar]
- Lafont, E.; Dupont, R.; Andrieu-Abadie, N.; Okazaki, T.; Schulze-Osthoff, K.; Levade, T.; Benoist, H.; Segui, B. Ordering of ceramide formation and caspase-9 activation in CD95L-induced Jurkat leukemia T cell apoptosis. Biochim. Biophys. Acta 2012, 1821, 684–693. [Google Scholar]
- Ben-David, O.; Futerman, A.H. The role of the ceramide acyl chain length in neurodegeneration: Involvement of ceramide synthases. Neuromol. Med 2010, 12, 341–350. [Google Scholar]
- Nixon, G.F. Sphingolipids in inflammation: Pathological implications and potential therapeutic targets. Br. J. Pharmacol 2009, 158, 982–993. [Google Scholar]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B. Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol 2010, 6, 1603–1624. [Google Scholar]
- Taha, T.A.; Osta, W.; Kozhaya, L.; Bielawski, J.; Johnson, K.R.; Gillanders, W.E.; Dbaibo, G.S.; Hannun, Y.A.; Obeid, L.M. Down-regulation of sphingosine kinase-1 by DNA damage: Dependence on proteases and p53. J. Biol. Chem 2004, 279, 20546–20554. [Google Scholar]
- Bornancin, F. Ceramide kinase: The first decade. Cell. Signal 2011, 23, 999–1008. [Google Scholar]
- Gomez-Munoz, A.; Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A. Ceramide-1-phosphate in cell survival and inflammatory signaling. Adv. Exp. Med. Biol 2010, 688, 118–130. [Google Scholar]
- Arana, L.; Gangoiti, P.; Ouro, A.; Rivera, I.G.; Ordonez, M.; Trueba, M.; Lankalapalli, R.S.; Bittman, R.; Gomez-Munoz, A. Generation of reactive oxygen species (ROS) is a key factor for stimulation of macrophage proliferation by ceramide 1-phosphate. Exp. Cell Res 2012, 318, 350–360. [Google Scholar]
- Gangoiti, P.; Arana, L.; Ouro, A.; Granado, M.H.; Trueba, M.; Gomez-Munoz, A. Activation of mTOR and RhoA is a major mechanism by which Ceramide 1-phosphate stimulates macrophage proliferation. Cell. Signal 2011, 23, 27–34. [Google Scholar]
- Gangoiti, P.; Bernacchioni, C.; Donati, C.; Cencetti, F.; Ouro, A.; Gomez-Munoz, A.; Bruni, P. Ceramide 1-phosphate stimulates proliferation of C2C12 myoblasts. Biochimie 2012, 94, 597–607. [Google Scholar]
- Bennett, W.F.; Tieleman, D.P. Molecular simulation of rapid translocation of cholesterol, diacylglycerol, and ceramide in model raft and nonraft membranes. J. Lipid Res 2012, 53, 421–429. [Google Scholar]
- Idris, I.; Gray, S.; Donnelly, R. Protein kinase C activation: Isozyme-specific effects on metabolism and cardiovascular complications in diabetes. Diabetologia 2001, 44, 659–673. [Google Scholar]
- Abboushi, N.; El-Hed, A.; El-Assaad, W.; Kozhaya, L.; El-Sabban, M.E.; Bazarbachi, A.; Badreddine, R.; Bielawska, A.; Usta, J.; Dbaibo, G.S. Ceramide inhibits IL-2 production by preventing protein kinase C-dependent NF-kappaB activation: Possible role in protein kinase Ctheta regulation. J. Immunol 2004, 173, 3193–3200. [Google Scholar]
- Perry, D.M.; Kitatani, K.; Roddy, P.; El-Osta, M.; Hannun, Y.A. Identification and characterization of protein phosphatase 2C activation by ceramide. J. Lipid Res 2012, 53, 1513–1521. [Google Scholar]
- Huwiler, A.; Brunner, J.; Hummel, R.; Vervoordeldonk, M.; Stabel, S.; van den Bosch, H.; Pfeilschifter, J. Ceramide-binding and activation defines protein kinase c-Raf as a ceramide-activated protein kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 6959–6963. [Google Scholar]
- Li, T.; Ying, L.; Wang, H.; Li, N.; Fu, W.; Guo, Z.; Xu, L. Microcystin-LR induces ceramide to regulate PP2A and destabilize cytoskeleton in HEK293 cells. Toxicol. Sci 2012, 128, 147–157. [Google Scholar]
- Ruvolo, P.P. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol. Res 2003, 47, 383–392. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol 2008, 9, 139–150. [Google Scholar]
- Cerantola, V.; Guillas, I.; Roubaty, C.; Vionnet, C.; Uldry, D.; Knudsen, J.; Conzelmann, A. Aureobasidin A arrests growth of yeast cells through both ceramide intoxication and deprivation of essential inositolphosphorylceramides. Mol. Microbiol 2009, 71, 1523–1537. [Google Scholar] [Green Version]
- Vionnet, C.; Roubaty, C.; Ejsing, C.S.; Knudsen, J.; Conzelmann, A. Yeast cells lacking all known ceramide synthases continue to make complex sphingolipids and to incorporate ceramides into glycosylphosphatidylinositol (GPI) anchors. J. Biol. Chem 2011, 286, 6769–6779. [Google Scholar]
- Funato, K.; Riezman, H. Vesicular and nonvesicular transport of ceramide from ER to the Golgi apparatus in yeast. J. Cell Biol 2001, 155, 949–959. [Google Scholar]
- Epstein, S.; Kirkpatrick, C.L.; Castillon, G.A.; Muniz, M.; Riezman, I.; David, F.P.; Wollheim, C.B.; Riezman, H. Activation of the unfolded protein response pathway causes ceramide accumulation in yeast and INS-1E insulinoma cells. J. Lipid Res 2012, 53, 412–420. [Google Scholar]
- Shimanuki, M.; Kinoshita, N.; Ohkura, H.; Yoshida, T.; Toda, T.; Yanagida, M. Isolation and characterization of the fission yeast protein phosphatase gene ppe1+ involved in cell shape control and mitosis. Mol. Biol. Cell 1993, 4, 303–313. [Google Scholar]
- Nickels, J.T.; Broach, J.R. A ceramide-activated protein phosphatase mediates ceramide-induced G1 arrest of Saccharomyces cerevisiae. Genes Dev 1996, 10, 382–394. [Google Scholar]
- Guillas, I.; Jiang, J.C.; Vionnet, C.; Roubaty, C.; Uldry, D.; Chuard, R.; Wang, J.; Jazwinski, S.M.; Conzelmann, A. Human homologues of LAG1 reconstitute Acyl-CoA-dependent ceramide synthesis in yeast. J. Biol. Chem 2003, 278, 37083–37091. [Google Scholar]
- Horvath, A.; Sutterlin, C.; Manning-Krieg, U.; Movva, N.R.; Riezman, H. Ceramide synthesis enhances transport of GPI-anchored proteins to the Golgi apparatus in yeast. EMBO J 1994, 13, 3687–3695. [Google Scholar]
- Zink, S.; Mehlgarten, C.; Kitamoto, H.K.; Nagase, J.; Jablonowski, D.; Dickson, R.C.; Stark, M.J.; Schaffrath, R. Mannosyl-diinositolphospho-ceramide, the major yeast plasma membrane sphingolipid, governs toxicity of Kluyveromyces lactis zymocin. Eukaryot Cell 2005, 4, 879–889. [Google Scholar]
- Lee, M.C.; Hamamoto, S.; Schekman, R. Ceramide biosynthesis is required for the formation of the oligomeric H+-ATPase Pma1p in the yeast endoplasmic reticulum. J. Biol. Chem 2002, 277, 22395–22401. [Google Scholar]
- Futerman, A.H.; Hannun, Y.A. The complex life of simple sphingolipids. EMBO Rep 2004, 5, 777–782. [Google Scholar]
- Mullen, T.D.; Jenkins, R.W.; Clarke, C.J.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Ceramide synthase-dependent ceramide generation and programmed cell death: Involvement of salvage pathway in regulating postmitochondrial events. J. Biol. Chem 2011, 286, 15929–15942. [Google Scholar]
- Carmona-Gutierrez, D.; Eisenberg, T.; Buttner, S.; Meisinger, C.; Kroemer, G.; Madeo, F. Apoptosis in yeast: Triggers, pathways, subroutines. Cell Death Differ 2010, 17, 763–773. [Google Scholar]
- Kuerbitz, S.J.; Plunkett, B.S.; Walsh, W.V.; Kastan, M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc. Natl. Acad. Sci. USA 1992, 89, 7491–7495. [Google Scholar]
- Debbas, M.; White, E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev 1993, 7, 546–554. [Google Scholar]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993, 74, 957–967. [Google Scholar]
- Zhan, Q.; Bae, I.; Kastan, M.B.; Fornace, A.J., Jr. The p53-dependent gamma-ray response of GADD45. Cancer Res. 1994, 54, 2755–2760. [Google Scholar]
- Smith, M.L.; Chen, I.T.; Zhan, Q.; Bae, I.; Chen, C.Y.; Gilmer, T.M.; Kastan, M.B.; O’Connor, P.M.; Fornace, A.J., Jr. Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 1994, 266, 1376–1380. [Google Scholar]
- Mummenbrauer, T.; Janus, F.; Muller, B.; Wiesmuller, L.; Deppert, W.; Grosse, F. p53 protein exhibits 3′-to-5′ exonuclease activity. Cell 1996, 85, 1089–1099. [Google Scholar]
- Amaral, J.D.; Xavier, J.M.; Steer, C.J.; Rodrigues, C.M. Targeting the p53 pathway of apoptosis. Curr. Pharm. Design 2010, 16, 2493–2503. [Google Scholar]
- Liu, Y.Y. Resuscitating wild-type p53 expression by disrupting ceramide glycosylation: A novel approach to target mutant p53 tumors. Cancer Res 2011, 71, 6295–6299. [Google Scholar]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar]
- Martin, M.E.; Berk, A.J. Adenovirus E1B 55K represses p53 activation in vitro. J. Virol 1998, 72, 3146–3154. [Google Scholar]
- Kashuba, E.; Yurchenko, M.; Yenamandra, S.P.; Snopok, B.; Szekely, L.; Bercovich, B.; Ciechanover, A.; Klein, G. Epstein-Barr virus-encoded EBNA-5 forms trimolecular protein complexes with MDM2 and p53 and inhibits the transactivating function of p53. Int. J. Cancer 2011, 128, 817–825. [Google Scholar]
- Muench, P.; Probst, S.; Schuetz, J.; Leiprecht, N.; Busch, M.; Wesselborg, S.; Stubenrauch, F.; Iftner, T. Cutaneous papillomavirus E6 proteins must interact with p300 and block p53-mediated apoptosis for cellular immortalization and tumorigenesis. Cancer Res 2010, 70, 6913–6924. [Google Scholar]
- Pietsch, E.C.; Sykes, S.M.; McMahon, S.B.; Murphy, M.E. The p53 family and programmed cell death. Oncogene 2008, 27, 6507–6521. [Google Scholar]
- Chen, D.; Zheng, X.; Kang, D.; Yan, B.; Liu, X.; Gao, Y.; Zhang, K. Apoptosis and expression of the Bcl-2 family of proteins and P53 in human pancreatic ductal adenocarcinoma. Med. Princ. Pract 2012, 21, 68–73. [Google Scholar]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar]
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N.; et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003, 4, 321–328. [Google Scholar]
- Altin, S.E.; Schulze, P.C. p53-upregulated modulator of apoptosis (PUMA): A novel proapoptotic molecule in the failing heart. Circulation 2011, 124, 7–8. [Google Scholar]
- Miyashita, T.; Harigai, M.; Hanada, M.; Reed, J.C. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res 1994, 54, 3131–3135. [Google Scholar]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem 2002, 277, 3247–3257. [Google Scholar]
- Bouvard, V.; Zaitchouk, T.; Vacher, M.; Duthu, A.; Canivet, M.; Choisy-Rossi, C.; Nieruchalski, M.; May, E. Tissue and cell-specific expression of the p53-target genes: Bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene 2000, 19, 649–660. [Google Scholar]
- Bennett, M.; Macdonald, K.; Chan, S.W.; Luzio, J.P.; Simari, R.; Weissberg, P. Cell surface trafficking of Fas: A rapid mechanism of p53-mediated apoptosis. Science 1998, 282, 290–293. [Google Scholar]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genetics 1997, 17, 141–143. [Google Scholar]
- Maldonado, M.E.; Bousserouel, S.; Gosse, F.; Lobstein, A.; Raul, F. Implication of NF-kappaB and p53 in the expression of TRAIL-death receptors and apoptosis by apple procyanidins in human metastatic SW620 cells. Biomedica 2010, 30, 577–586. [Google Scholar]
- Song, G.; Wang, W.; Hu, T. p53 facilitates BH3-only BID nuclear export to induce apoptosis in the irrepairable DNA damage response. Med. Hypotheses 2011, 77, 850–852. [Google Scholar]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol 2008, 10, 676–687. [Google Scholar]
- Trinh, D.L.; Elwi, A.N.; Kim, S.W. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget 2010, 1, 396–404. [Google Scholar]
- Speidel, D. Transcription-independent p53 apoptosis: An alternative route to death. Trends Cell Biol 2010, 20, 14–24. [Google Scholar]
- Schuler, M.; Bossy-Wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J. Biol. Chem 2000, 275, 7337–7342. [Google Scholar]
- Geng, Y.; Walls, K.C.; Ghosh, A.P.; Akhtar, R.S.; Klocke, B.J.; Roth, K.A. Cytoplasmic p53 and activated Bax regulate p53-dependent, transcription-independent neural precursor cell apoptosis. J. Histochem. Cytochem 2010, 58, 265–275. [Google Scholar]
- Lee, S.K.; Kim, Y.C.; Song, S.B.; Kim, Y.S. Stabilization and translocation of p53 to mitochondria is linked to Bax translocation to mitochondria in simvastatin-induced apoptosis. Biochem. Biophys. Res. Commun 2010, 391, 1592–1597. [Google Scholar]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar]
- Mossalam, M.; Matissek, K.J.; Okal, A.; Constance, J.E.; Lim, C.S. Direct induction of apoptosis using an optimal mitochondrially targeted p53. Mol. Pharm 2012, 9, 1449–1458. [Google Scholar]
- Chipuk, J.E.; Bouchier-Hayes, L.; Kuwana, T.; Newmeyer, D.D.; Green, D.R. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 2005, 309, 1732–1735. [Google Scholar]
- Muer, A.; Overkamp, T.; Gillissen, B.; Richter, A.; Pretzsch, T.; Milojkovic, A.; Dorken, B.; Daniel, P.T.; Hemmati, P. p14(ARF)-induced apoptosis in p53 protein-deficient cells is mediated by BH3-only protein-independent derepression of Bak protein through down-regulation of Mcl-1 and Bcl-xL proteins. J. Biol. Chem 2012, 287, 17343–17352. [Google Scholar]
- Chen, Q.; Takeyama, N.; Brady, G.; Watson, A.J.; Dive, C. Blood cells with reduced mitochondrial membrane potential and cytosolic cytochrome C can survive and maintain clonogenicity given appropriate signals to suppress apoptosis. Blood 1998, 92, 4545–4553. [Google Scholar]
- Zhang, J.; Krishnamurthy, P.K.; Johnson, G.V. Cdk5 phosphorylates p53 and regulates its activity. J. Neurochem 2002, 81, 307–313. [Google Scholar]
- Ajay, A.K.; Upadhyay, A.K.; Singh, S.; Vijayakumar, M.V.; Kumari, R.; Pandey, V.; Boppana, R.; Bhat, M.K. Cdk5 phosphorylates non-genotoxically overexpressed p53 following inhibition of PP2A to induce cell cycle arrest/apoptosis and inhibits tumor progression. Mol. Cancer 2010, 9, 204. [Google Scholar]
- Puca, R.; Nardinocchi, L.; Starace, G.; Rechavi, G.; Sacchi, A.; Givol, D.; D’Orazi, G. Nox1 is involved in p53 deacetylation and suppression of its transcriptional activity and apoptosis. Free Radic. Biol. Med 2010, 48, 1338–1346. [Google Scholar]
- Goloudina, A.R.; Mazur, S.J.; Appella, E.; Garrido, C.; Demidov, O.N. Wip1 sensitizes p53-negative tumors to apoptosis by regulating the Bax/Bcl-xL ratio. Cell Cycle 2012, 11, 1883–1887. [Google Scholar]
- How, P.C.; Shields, D. Tethering function of the caspase cleavage fragment of Golgi protein p115 promotes apoptosis via a p53-dependent pathway. J. Biol. Chem 2011, 286, 8565–8576. [Google Scholar]
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B.; et al. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol. Cell 2011, 42, 584–596. [Google Scholar]
- Li, M.; Luo, J.; Brooks, C.L.; Gu, W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem 2002, 277, 50607–50611. [Google Scholar]
- Brooks, C.L.; Li, M.; Gu, W. Mechanistic studies of MDM2-mediated ubiquitination in p53 regulation. J. Biol. Chem 2007, 282, 22804–22815. [Google Scholar]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev 2008, 9, 402–412. [Google Scholar]
- Zou, B.; Chim, C.S.; Pang, R.; Zeng, H.; Dai, Y.; Zhang, R.; Lam, C.S.; Tan, V.P.; Hung, I.F.; Lan, H.Y.; Wong, B.C. XIAP-associated factor 1 (XAF1), a novel target of p53, enhances p53-mediated apoptosis via post-translational modification. Mol. Carcinogen 2012, 51, 422–432. [Google Scholar]
- McNamee, L.M.; Brodsky, M.H. p53-independent apoptosis limits DNA damage-induced aneuploidy. Genetics 2009, 182, 423–435. [Google Scholar]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar]
- Sidi, S.; Sanda, T.; Kennedy, R.D.; Hagen, A.T.; Jette, C.A.; Hoffmans, R.; Pascual, J.; Imamura, S.; Kishi, S.; Amatruda, J.F.; et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008, 133, 864–877. [Google Scholar]
- Dirisina, R.; Katzman, R.B.; Goretsky, T.; Managlia, E.; Mittal, N.; Williams, D.B.; Qiu, W.; Yu, J.; Chandel, N.S.; Zhang, L.; et al. p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology 2011, 141, 1036–1045. [Google Scholar]
- Gurzov, E.N.; Germano, C.M.; Cunha, D.A.; Ortis, F.; Vanderwinden, J.M.; Marchetti, P.; Zhang, L.; Eizirik, D.L. p53 up-regulated modulator of apoptosis (PUMA) activation contributes to pancreatic beta-cell apoptosis induced by proinflammatory cytokines and endoplasmic reticulum stress. J. Biol. Chem. y 2010, 285, 19910–19920. [Google Scholar]
- Donauer, J.; Schreck, I.; Liebel, U.; Weiss, C. Role and interaction of p53, BAX and the stress-activated protein kinases p38 and JNK in benzo(a)pyrene-diolepoxide induced apoptosis in human colon carcinoma cells. Arch. Toxicol 2012, 86, 329–337. [Google Scholar]
- Tong, Q.S.; Zheng, L.D.; Wang, L.; Liu, J.; Qian, W. BAK overexpression mediates p53-independent apoptosis inducing effects on human gastric cancer cells. BMC Cancer 2004, 4. [Google Scholar] [CrossRef] [Green Version]
- Lanni, J.S.; Lowe, S.W.; Licitra, E.J.; Liu, J.O.; Jacks, T. p53-independent apoptosis induced by paclitaxel through an indirect mechanism. Proc. Natl. Acad. Sci. USA 1997, 94, 9679–9683. [Google Scholar]
- Koike, M.; Fujita, F.; Komori, K.; Katoh, F.; Sugimoto, T.; Sakamoto, Y.; Matsuda, M.; Fujita, M. Dependence of chemotherapy response on p53 mutation status in a panel of human cancer lines maintained in nude mice. Cancer Sci 2004, 95, 541–546. [Google Scholar]
- Dbaibo, G.S.; Pushkareva, M.Y.; Rachid, R.A.; Alter, N.; Smyth, M.J.; Obeid, L.M.; Hannun, Y.A. p53-dependent ceramide response to genotoxic stress. J. Clin. Invest 1998, 102, 329–339. [Google Scholar]
- Panjarian, S.; Kozhaya, L.; Arayssi, S.; Yehia, M.; Bielawski, J.; Bielawska, A.; Usta, J.; Hannun, Y.A.; Obeid, L.M.; Dbaibo, G.S. De novo N-palmitoylsphingosine synthesis is the major biochemical mechanism of ceramide accumulation following p53 up-regulation. Prostaglandins Other Lipid Mediat 2008, 86, 41–48. [Google Scholar]
- Bose, R.; Verheij, M.; Haimovitz-Friedman, A.; Scotto, K.; Fuks, Z.; Kolesnick, R. Ceramide synthase mediates daunorubicin-induced apoptosis: An alternative mechanism for generating death signals. Cell 1995, 82, 405–414. [Google Scholar]
- Jaffrezou, J.P.; Levade, T.; Bettaieb, A.; Andrieu, N.; Bezombes, C.; Maestre, N.; Vermeersch, S.; Rousse, A.; Laurent, G. Daunorubicin-induced apoptosis: Triggering of ceramide generation through sphingomyelin hydrolysis. EMBO J 1996, 15, 2417–2424. [Google Scholar]
- Deng, X.; Yin, X.; Allan, R.; Lu, D.D.; Maurer, C.W.; Haimovitz-Friedman, A.; Fuks, Z.; Shaham, S.; Kolesnick, R. Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science 2008, 322, 110–115. [Google Scholar]
- Rotolo, J.A.; Mesicek, J.; Maj, J.; Truman, J.P.; Haimovitz-Friedman, A.; Kolesnick, R.; Fuks, Z. Regulation of ceramide synthase-mediated crypt epithelium apoptosis by DNA damage repair enzymes. Cancer Res 2010, 70, 957–967. [Google Scholar]
- Kim, S.S.; Chae, H.S.; Bach, J.H.; Lee, M.W.; Kim, K.Y.; Lee, W.B.; Jung, Y.M.; Bonventre, J.V.; Suh, Y.H. P53 mediates ceramide-induced apoptosis in SKN-SH cells. Oncogene 2002, 21, 2020–2028. [Google Scholar]
- El-Assaad, W.; Kozhaya, L.; Araysi, S.; Panjarian, S.; Bitar, F.F.; Baz, E.; El-Sabban, M.E.; Dbaibo, G.S. Ceramide and glutathione define two independently regulated pathways of cell death initiated by p53 in Molt-4 leukaemia cells. Biochem. J 2003, 376, 725–732. [Google Scholar]
- Liu, Y.Y.; Patwardhan, G.A.; Bhinge, K.; Gupta, V.; Gu, X.; Jazwinski, S.M. Suppression of glucosylceramide synthase restores p53-dependent apoptosis in mutant p53 cancer cells. Cancer Res 2011, 71, 2276–2285. [Google Scholar]
- Shultz, J.C.; Goehe, R.W.; Wijesinghe, D.S.; Murudkar, C.; Hawkins, A.J.; Shay, J.W.; Minna, J.D.; Chalfant, C.E. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res 2010, 70, 9185–9196. [Google Scholar]
- Massiello, A.; Roesser, J.R.; Chalfant, C.E. SAP155 Binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5′ splice site selection of Bcl-x pre-mRNA. FASEB J 2006, 20, 1680–1682. [Google Scholar]
- Castro, M.E.; Ferrer, I.; Cascon, A.; Guijarro, M.V.; Lleonart, M.; Ramon y Cajal, S.; Leal, J.F.; Robledo, M.; Carnero, A. PPP1CA contributes to the senescence program induced by oncogenic Ras. Carcinogenesis 2008, 29, 491–499. [Google Scholar]
- Heffernan-Stroud, L.A.; Obeid, L.M. p53 and regulation of bioactive sphingolipids. Adv. Enzyme Regul 2011, 51, 219–228. [Google Scholar]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar]
- El-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 1994, 54, 1169–1174. [Google Scholar]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar]
- Dbaibo, G.S.; Pushkareva, M.Y.; Jayadev, S.; Schwarz, J.K.; Horowitz, J.M.; Obeid, L.M.; Hannun, Y.A. Retinoblastoma gene product as a downstream target for a ceramide-dependent pathway of growth arrest. Proc. Natl. Acad. Sci. USA 1995, 92, 1347–1351. [Google Scholar]
- Kishikawa, K.; Chalfant, C.E.; Perry, D.K.; Bielawska, A.; Hannun, Y.A. Phosphatidic acid is a potent and selective inhibitor of protein phosphatase 1 and an inhibitor of ceramide-mediated responses. J. Biol. Chem 1999, 274, 21335–21341. [Google Scholar]
- Struckhoff, A.P.; Patel, B.; Beckman, B.S. Inhibition of p53 sensitizes MCF-7 cells to ceramide treatment. Int. J. Oncol 2010, 37, 21–30. [Google Scholar]
- Villani, M.G.; Appierto, V.; Cavadini, E.; Bettiga, A.; Prinetti, A.; Clagett-Dame, M.; Curley, R.W.; Formelli, F. 4-oxo-fenretinide, a recently identified fenretinide metabolite, induces marked G2-M cell cycle arrest and apoptosis in fenretinide-sensitive and fenretinide-resistant cell lines. Cancer Res 2006, 66, 3238–3247. [Google Scholar]
- Nasr, R.; El-Sabban, M.E.; Karam, J.A.; Dbaibo, G.; Kfoury, Y.; Arnulf, B.; Lepelletier, Y.; Bex, F.; de The, H.; Hermine, O.; et al. Efficacy and mechanism of action of the proteasome inhibitor PS-341 in T-cell lymphomas and HTLV-I associated adult T-cell leukemia/lymphoma. Oncogene 2005, 24, 419–430. [Google Scholar]
- Yang, J.; Duerksen-Hughes, P.J. Activation of a p53-independent, sphingolipid-mediated cytolytic pathway in p53-negative mouse fibroblast cells treated with N-methyl-N-nitro-N-nitrosoguanidine. J. Biol. Chem 2001, 276, 27129–27135. [Google Scholar]
- Deng, X.; Gao, F.; May, W.S. Protein phosphatase 2A inactivates Bcl2’s antiapoptotic function by dephosphorylation and up-regulation of Bcl2-p53 binding. Blood 2009, 113, 422–428. [Google Scholar]
- Haynes, T.A.; Filippov, V.; Filippova, M.; Yang, J.; Zhang, K.; Duerksen-Hughes, P.J. DNA damage induces down-regulation of UDP-glucose ceramide glucosyltransferase, increases ceramide levels and triggers apoptosis in p53-deficient cancer cells. Biochim. Biophys. Acta 2012, 1821, 943–953. [Google Scholar]
- Henry, B.; Moller, C.; Dimanche-Boitrel, M.T.; Gulbins, E.; Becker, K.A. Targeting the ceramide system in cancer. Cancer Lett. 2011, 2011. [Google Scholar] [CrossRef]
- Ahn, E.H.; Schroeder, J.J. Induction of apoptosis by sphingosine, sphinganine, and C(2)-ceramide in human colon cancer cells, but not by C(2)-dihydroceramide. Anticancer Res 2010, 30, 2881–2884. [Google Scholar]
- Aureli, M.; Bassi, R.; Prinetti, A.; Chiricozzi, E.; Pappalardi, B.; Chigorno, V.; Di Muzio, N.; Loberto, N.; Sonnino, S. Ionizing radiations increase the activity of the cell surface glycohydrolases and the plasma membrane ceramide content. Glycoconjugate J 2012, 29, 585–597. [Google Scholar]
- Camgoz, A.; Gencer, E.B.; Ural, A.U.; Avcu, F.; Baran, Y. Roles of ceramide synthase and ceramide clearence genes in nilotinib-induced cell death in chronic myeloid leukemia cells. Leukemia Lymphoma 2011, 52, 1574–1584. [Google Scholar]
- Chapman, J.V.; Gouaze-Andersson, V.; Messner, M.C.; Flowers, M.; Karimi, R.; Kester, M.; Barth, B.M.; Liu, X.; Liu, Y.Y.; Giuliano, A.E.; et al. Metabolism of short-chain ceramide by human cancer cells—Implications for therapeutic approaches. Biochem. Pharmacol 2010, 80, 308–315. [Google Scholar]
- Bektas, M.; Jolly, P.S.; Muller, C.; Eberle, J.; Spiegel, S.; Geilen, C.C. Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells: Role of Bcl-2 expression. Oncogene 2005, 24, 178–187. [Google Scholar]
- Flowers, M.; Fabrias, G.; Delgado, A.; Casas, J.; Abad, J.L.; Cabot, M.C. C6-ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res. Treat 2012, 133, 447–458. [Google Scholar]
- Zhu, Q.Y.; Wang, Z.; Ji, C.; Cheng, L.; Yang, Y.L.; Ren, J.; Jin, Y.H.; Wang, Q.J.; Gu, X.J.; Bi, Z.G.; et al. C6-ceramide synergistically potentiates the anti-tumor effects of histone deacetylase inhibitors via AKT dephosphorylation and alpha-tubulin hyperacetylation both in vitro and in vivo. Cell Death Dis 2011, 2, e117. [Google Scholar]
- Jiang, L.; Pan, X.; Chen, Y.; Wang, K.; Du, Y.; Zhang, J. Preferential involvement of both ROS and ceramide in fenretinide-induced apoptosis of HL60 rather than NB4 and U937 cells. Biochem. Biophys. Res. Commun 2011, 405, 314–318. [Google Scholar]
- Chatzakos, V.; Rundlof, A.K.; Ahmed, D.; de Verdier, P.J.; Flygare, J. Inhibition of sphingosine kinase 1 enhances cytotoxicity, ceramide levels and ROS formation in liver cancer cells treated with selenite. Biochem. Pharmacol 2012, 84, 712–721. [Google Scholar]
- Kartal, M.; Saydam, G.; Sahin, F.; Baran, Y. Resveratrol triggers apoptosis through regulating ceramide metabolizing genes in human K562 chronic myeloid leukemia cells. Nutr. Cancer 2011, 63, 637–644. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hage-Sleiman, R.; Esmerian, M.O.; Kobeissy, H.; Dbaibo, G. p53 and Ceramide as Collaborators in the Stress Response. Int. J. Mol. Sci. 2013, 14, 4982-5012. https://doi.org/10.3390/ijms14034982
Hage-Sleiman R, Esmerian MO, Kobeissy H, Dbaibo G. p53 and Ceramide as Collaborators in the Stress Response. International Journal of Molecular Sciences. 2013; 14(3):4982-5012. https://doi.org/10.3390/ijms14034982
Chicago/Turabian StyleHage-Sleiman, Rouba, Maria O. Esmerian, Hadile Kobeissy, and Ghassan Dbaibo. 2013. "p53 and Ceramide as Collaborators in the Stress Response" International Journal of Molecular Sciences 14, no. 3: 4982-5012. https://doi.org/10.3390/ijms14034982
APA StyleHage-Sleiman, R., Esmerian, M. O., Kobeissy, H., & Dbaibo, G. (2013). p53 and Ceramide as Collaborators in the Stress Response. International Journal of Molecular Sciences, 14(3), 4982-5012. https://doi.org/10.3390/ijms14034982