Non-Coding RNAs: Functional Aspects and Diagnostic Utility in Oncology

Abstract
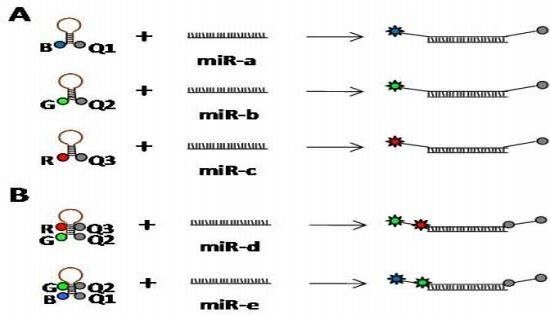
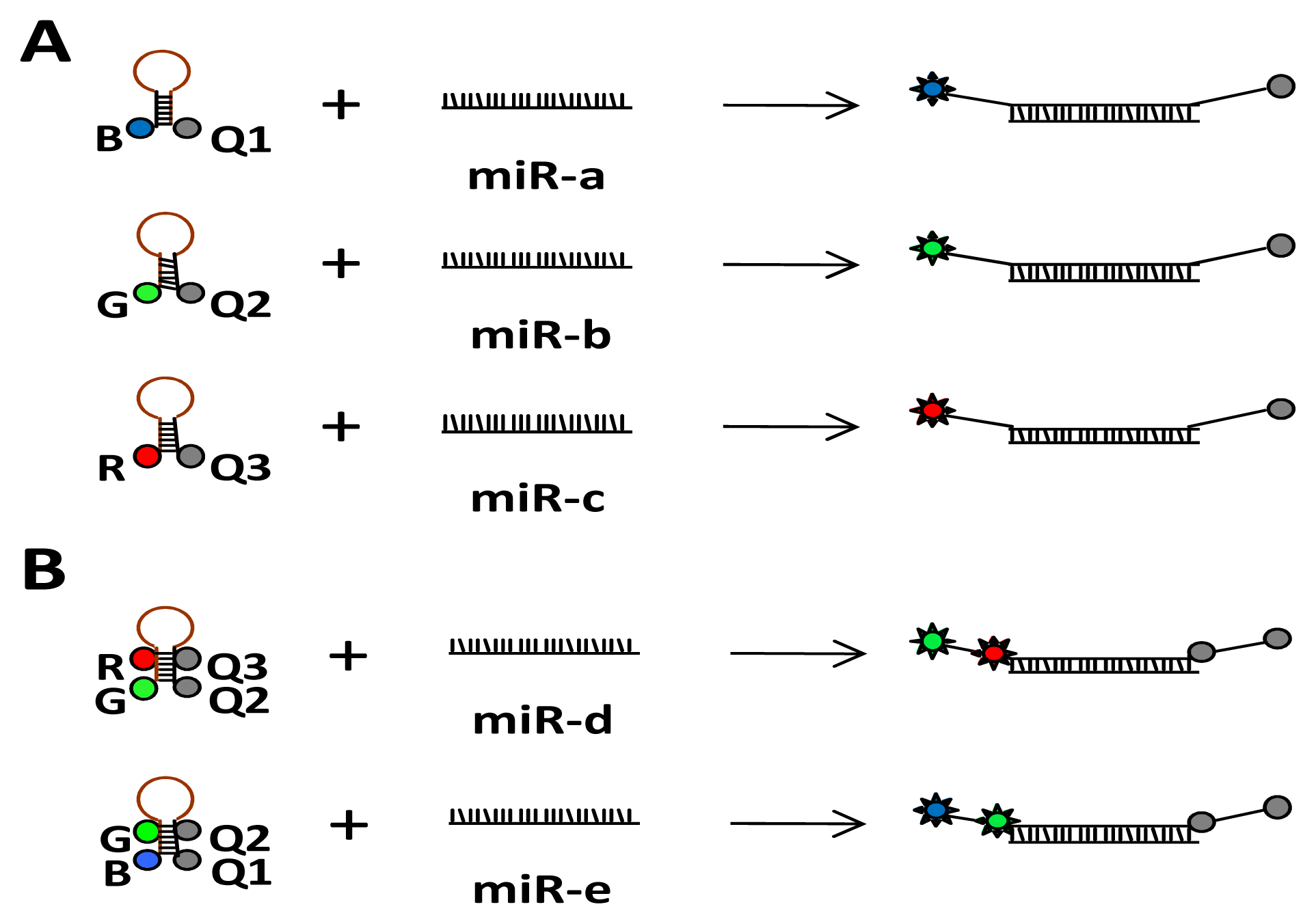
:
1. Introduction
2. Non-Coding RNAs in Oncology
3. Diagnostic Utility of MicroRNAs as Biomarkers for Cancer Detection and Prognosis
3.1. Prostate Cancer
3.2. Breast Cancer
3.3. Lung Cancer
3.4. Colorectal Cancer
4. MicroRNA Detection Technologies: State-of-the-Art and Future Perspectives
4.1. Microarray-Based Methods
4.2. qRT-PCR-Based Methods
4.3. Deep Sequencing-Based Methods
4.4. Single Molecule Detection Methods
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Costa, F.F. Non-coding RNAs: Meet thy masters. Bioessays 2010, 32, 599–608. [Google Scholar]
- Skipper, M.; Dhand, R.; Campbell, P. Presenting ENCODE. Nature 2012, 489, 45. [Google Scholar]
- Washietl, S.; Hofacker, I.L.; Lukasser, M.; Huttenhofer, A.; Stadler, P.F. Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome. Nat. Biotechnol 2005, 23, 1383–1390. [Google Scholar]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet 2006, 15, R17–R29. [Google Scholar]
- Khachane, A.N.; Harrison, P.M. Mining mammalian transcript data for functional long non-coding RNAs. PLoS One 2010, 5, e10316. [Google Scholar]
- Kapranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet 2007, 8, 413–423. [Google Scholar]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar]
- Frith, M.C.; Pheasant, M.; Mattick, J.S. The amazing complexity of the human transcriptome. Eur. J. Hum. Genet 2005, 13, 894–897. [Google Scholar]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol 2012, 30, 253–260. [Google Scholar]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar]
- Pei, B.; Sisu, C.; Frankish, A.; Howald, C.; Habegger, L.; Mu, X.J.; Harte, R.; Balasubramanian, S.; Tanzer, A.; Diekhans, M.; et al. The GENCODE pseudogene resource. Genome Biol 2012, 13, R51. [Google Scholar]
- Howald, C.; Tanzer, A.; Chrast, J.; Kokocinski, F.; Derrien, T.; Walters, N.; Gonzalez, J.M.; Frankish, A.; Aken, B.L.; Hourlier, T.; et al. Combining RT-PCR-seq and RNA-seq to catalog all genic elements encoded in the human genome. Genome Res 2012, 22, 1698–1710. [Google Scholar]
- Djebali, S.; Lagarde, J.; Kapranov, P.; Lacroix, V.; Borel, C.; Mudge, J.M.; Howald, C.; Foissac, S.; Ucla, C.; Chrast, J.; et al. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One 2012, 7, e28213. [Google Scholar]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar]
- Bernstein, B.E.; Birney, E.; Dunham, I.; Green, E.D.; Gunter, C.; Snyder, M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 2012, 22, 1775–1789. [Google Scholar]
- The National Human Genome Research Institute. GENCODE: The reference human genome annotation for The ENCODE Project. Available online: http://www.gencodegenes.org/stats.html accessed on 24 October 2012.
- Ebisuya, M.; Yamamoto, T.; Nakajima, M.; Nishida, E. Ripples from neighbouring transcription. Nat. Cell. Biol 2008, 10, 1106–1113. [Google Scholar]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev 2009, 23, 1494–1504. [Google Scholar]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet 2009, 10, 155–159. [Google Scholar]
- Van Bakel, H.; Nislow, C.; Blencowe, B.J.; Hughes, T.R. Most “dark matter” transcripts are associated with known genes. PLoS Biol 2010, 8, e1000371. [Google Scholar]
- Mattick, J.S. Long noncoding RNAs in cell and developmental biology. Semin. Cell. Dev. Biol 2011, 22, 327. [Google Scholar]
- Morey, C.; Avner, P. Employment opportunities for non-coding RNAs. FEBS Lett 2004, 567, 27–34. [Google Scholar]
- Taft, R.J.; Kaplan, C.D.; Simons, C.; Mattick, J.S. Evolution, biogenesis and function of promoter-associated RNAs. Cell. Cycle 2009, 8, 2332–2338. [Google Scholar]
- Chekulaeva, M.; Filipowicz, W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell. Biol 2009, 21, 452–460. [Google Scholar]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet 2010, 11, 597–610. [Google Scholar]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem 2010, 79, 351–379. [Google Scholar]
- Pers, Y.M.; Fabre, S.; Djouad, F.; Duroux-Richard, I.; Apparailly, F.; Noel, D.; Jorgensen, C. The micrornas as biomarkers specific of knee osteoarthritis. Osteoarthritis Cartilage 2010, 18, S37–S38. [Google Scholar]
- Murata, K.; Yoshitomi, H.; Tanida, S.; Ishikawa, M.; Nishitani, K.; Ito, H.; Nakamura, T. Plasma and synovial fluid microRNAs as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Res. Ther 2010, 12, R86. [Google Scholar]
- Nakasa, T.; Nagata, Y.; Yamasaki, K.; Ochi, M. A mini-review: Microrna in arthritis. Physiol. Genomics 2011, 43, 566–570. [Google Scholar]
- Yu, C.; Chen, W.P.; Wang, X.H. MicroRNA in osteoarthritis. J. Int. Med. Res 2011, 39, 1–9. [Google Scholar]
- Filkova, M.; Jungel, A.; Gay, R.E.; Gay, S. MicroRNAs in rheumatoid arthritis: Potential role in diagnosis and therapy. BioDrugs 2012, 26, 131–141. [Google Scholar]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers. Dis 2008, 14, 27–41. [Google Scholar]
- Geekiyanage, H.; Jicha, G.A.; Nelson, P.T.; Chan, C. Blood serum miRNA: Non-invasive biomarkers for Alzheimer’s disease. Exp. Neurol 2011, 235, 491–496. [Google Scholar]
- Culpan, D.; Kehoe, P.G.; Love, S. Tumour necrosis factor-alpha (TNF-alpha) and miRNA expression in frontal and temporal neocortex in Alzheimer’s disease and the effect of TNF-alpha on miRNA expression in vitro. Int. J. Mol. Epidemiol. Genet 2011, 2, 156–162. [Google Scholar]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci 2008, 28, 1213–1223. [Google Scholar]
- Jin, P.; Levey, A.I.; Szulwach, K. Micro RNA Markers and Methods Related Thereto. U.S. Patent 20120108650, 3 May 2012. [Google Scholar]
- Salta, E.; De Strooper, B. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol 2012, 11, 189–200. [Google Scholar]
- Wang, G.K.; Zhu, J.Q.; Zhang, J.T.; Li, Q.; Li, Y.; He, J.; Qin, Y.W.; Jing, Q. Circulating microRNA: A novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J 2010, 31, 659–666. [Google Scholar]
- D’Alessandra, Y.; Devanna, P.; Limana, F.; Straino, S.; Di Carlo, A.; Brambilla, P.G.; Rubino, M.; Carena, M.C.; Spazzafumo, L.; De Simone, M.; et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J 2010, 31, 2765–2773. [Google Scholar]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet 2010, 3, 499–506. [Google Scholar]
- Cheng, Y.; Tan, N.; Yang, J.; Liu, X.; Cao, X.; He, P.; Dong, X.; Qin, S.; Zhang, C. A translational study of circulating cell-free microRNA-1 in acute myocardial infarction. Clin. Sci. (Lond. ) 2010, 119, 87–95. [Google Scholar]
- Ai, J.; Zhang, R.; Li, Y.; Pu, J.; Lu, Y.; Jiao, J.; Li, K.; Yu, B.; Li, Z.; Wang, R.; et al. Circulating microRNA-1 as a potential novel biomarker for acute myocardial infarction. Biochem. Biophys. Res. Commun 2010, 391, 73–77. [Google Scholar]
- Divakaran, V.; Mann, D.L. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ. Res 2008, 103, 1072–1083. [Google Scholar]
- Adachi, T.; Nakanishi, M.; Otsuka, Y.; Nishimura, K.; Hirokawa, G.; Goto, Y.; Nonogi, H.; Iwai, N. Plasma microRNA 499 as a biomarker of acute myocardial infarction. Clin. Chem 2010, 56, 1183–1185. [Google Scholar]
- Van Empel, V.P.; de Windt, L.J.; Martins, P.A. Circulating miRNAs: Reflecting or affecting cardiovascular disease? Curr. Hypertens. Rep 2012, 14, 498–509. [Google Scholar]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar]
- Ruan, K.; Fang, X.; Ouyang, G. MicroRNAs: Novel regulators in the hallmarks of human cancer. Cancer Lett 2009, 285, 116–126. [Google Scholar]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol 2010, 220, 126–139. [Google Scholar]
- Setoyama, T.; Ling, H.; Natsugoe, S.; Calin, G.A. Non-coding RNAs for medical practice in oncology. Keio J. Med 2011, 60, 106–113. [Google Scholar]
- Wang, D.; Garcia-Bassets, I.; Benner, C.; Li, W.; Su, X.; Zhou, Y.; Qiu, J.; Liu, W.; Kaikkonen, M.U.; Ohgi, K.A.; et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474, 390–394. [Google Scholar]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar]
- Bussemakers, M.J.; van Bokhoven, A.; Verhaegh, G.W.; Smit, F.P.; Karthaus, H.F.; Schalken, J.A.; Debruyne, F.M.; Ru, N.; Isaacs, W.B. DD3: A new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res 1999, 59, 5975–5979. [Google Scholar]
- Groskopf, J.; Aubin, S.M.; Deras, I.L.; Blase, A.; Bodrug, S.; Clark, C.; Brentano, S.; Mathis, J.; Pham, J.; Meyer, T.; et al. APTIMA PCA3 molecular urine test: Development of a method to aid in the diagnosis of prostate cancer. Clin. Chem 2006, 52, 1089–1095. [Google Scholar]
- Shappell, S.B. Clinical utility of prostate carcinoma molecular diagnostic tests. Rev. Urol 2008, 10, 44–69. [Google Scholar]
- Marks, L.S.; Bostwick, D.G. Prostate cancer specificity of PCA3 gene testing: Examples from clinical practice. Rev. Urol 2008, 10, 175–181. [Google Scholar]
- Lee, G.L.; Dobi, A.; Srivastava, S. Prostate cancer: Diagnostic performance of the PCA3 urine test. Nat. Rev. Urol 2011, 8, 123–124. [Google Scholar]
- Gabory, A.; Jammes, H.; Dandolo, L. The H19 locus: Role of an imprinted non-coding RNA in growth and development. Bioessays 2010, 32, 473–480. [Google Scholar]
- Berteaux, N.; Lottin, S.; Monte, D.; Pinte, S.; Quatannens, B.; Coll, J.; Hondermarck, H.; Curgy, J.J.; Dugimont, T.; Adriaenssens, E. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J. Biol. Chem 2005, 280, 29625–29636. [Google Scholar]
- Matouk, I.J.; DeGroot, N.; Mezan, S.; Ayesh, S.; Abu-lail, R.; Hochberg, A.; Galun, E. The H19 non-coding RNA is essential for human tumor growth. PLoS One 2007, 2, e845. [Google Scholar]
- Fellig, Y.; Ariel, I.; Ohana, P.; Schachter, P.; Sinelnikov, I.; Birman, T.; Ayesh, S.; Schneider, T.; de Groot, N.; Czerniak, A.; et al. H19 expression in hepatic metastases from a range of human carcinomas. J. Clin. Pathol 2005, 58, 1064–1068. [Google Scholar]
- Arima, T.; Matsuda, T.; Takagi, N.; Wake, N. Association of IGF2 and H19 imprinting with choriocarcinoma development. Cancer Genet. Cytogenet 1997, 93, 39–47. [Google Scholar]
- Hibi, K.; Nakamura, H.; Hirai, A.; Fujikake, Y.; Kasai, Y.; Akiyama, S.; Ito, K.; Takagi, H. Loss of H19 imprinting in esophageal cancer. Cancer Res 1996, 56, 480–482. [Google Scholar]
- Cunnington, M.S.; Santibanez Koref, M.; Mayosi, B.M.; Burn, J.; Keavney, B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet 2010, 6, e1000899. [Google Scholar]
- Yap, K.L.; Li, S.; Munoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2010, 30, 1956–1962. [Google Scholar]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar]
- Diehl, J.A. Cycling to cancer with cyclin D1. Cancer Biol. Ther 2002, 1, 226–231. [Google Scholar]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar]
- Coccia, E.M.; Cicala, C.; Charlesworth, A.; Ciccarelli, C.; Rossi, G.B.; Philipson, L.; Sorrentino, V. Regulation and expression of a growth arrest-specific gene (gas5) during growth, differentiation, and development. Mol. Cell. Biol 1992, 12, 3514–3521. [Google Scholar]
- Mourtada-Maarabouni, M.; Pickard, M.R.; Hedge, V.L.; Farzaneh, F.; Williams, G.T. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 2009, 28, 195–208. [Google Scholar]
- Calin, G.A.; Liu, C.G.; Ferracin, M.; Hyslop, T.; Spizzo, R.; Sevignani, C.; Fabbri, M.; Cimmino, A.; Lee, E.J.; Wojcik, S.E.; et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 2007, 12, 215–229. [Google Scholar]
- Allen, T.A.; Von Kaenel, S.; Goodrich, J.A.; Kugel, J.F. The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat. Struct. Mol. Biol 2004, 11, 816–821. [Google Scholar]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved elements in the human genome. Science 2004, 304, 1321–1325. [Google Scholar]
- Elgar, G.; Vavouri, T. Tuning in to the signals: Noncoding sequence conservation in vertebrate genomes. Trends Genet 2008, 24, 344–352. [Google Scholar]
- Garzon, R.; Fabbri, M.; Cimmino, A.; Calin, G.A.; Croce, C.M. MicroRNA expression and function in cancer. Trends Mol. Med 2006, 12, 580–587. [Google Scholar]
- Martens-Uzunova, E.S.; Jalava, S.E.; Dits, N.F.; van Leenders, G.J.; Moller, S.; Trapman, J.; Bangma, C.H.; Litman, T.; Visakorpi, T.; Jenster, G. Diagnostic and prognostic signatures from the small non-coding RNA transcriptome in prostate cancer. Oncogene 2011, 31, 978–991. [Google Scholar]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res 2005, 65, 7065–7070. [Google Scholar]
- Cummins, J.M.; Velculescu, V.E. Implications of micro-RNA profiling for cancer diagnosis. Oncogene 2006, 25, 6220–6227. [Google Scholar]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar]
- He, H.; Jazdzewski, K.; Li, W.; Liyanarachchi, S.; Nagy, R.; Volinia, S.; Calin, G.A.; Liu, C.G.; Franssila, K.; Suster, S.; et al. The role of microRNA genes in papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 19075–19080. [Google Scholar]
- Ciafre, S.A.; Galardi, S.; Mangiola, A.; Ferracin, M.; Liu, C.G.; Sabatino, G.; Negrini, M.; Maira, G.; Croce, C.M.; et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem. Biophys. Res. Commun 2005, 334, 1351–1358. [Google Scholar]
- Murakami, Y.; Yasuda, T.; Saigo, K.; Urashima, T.; Toyoda, H.; Okanoue, T.; Shimotohno, K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006, 25, 2537–2545. [Google Scholar]
- Roldo, C.; Missiaglia, E.; Hagan, J.P.; Falconi, M.; Capelli, P.; Bersani, S.; Calin, G.A.; Volinia, S.; Liu, C.G.; Scarpa, A.; et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J. Clin. Oncol 2006, 24, 4677–4684. [Google Scholar]
- Bottoni, A.; Zatelli, M.C.; Ferracin, M.; Tagliati, F.; Piccin, D.; Vignali, C.; Calin, G.A.; Negrini, M.; Croce, C.M.; Degli Uberti, E.C. Identification of differentially expressed microRNAs by microarray: A possible role for microRNA genes in pituitary adenomas. J. Cell Physiol 2007, 210, 370–377. [Google Scholar]
- Nicoloso, M.S.; Spizzo, R.; Shimizu, M.; Rossi, S.; Calin, G.A. MicroRNAs—The micro steering wheel of tumour metastases. Nat. Rev. Cancer 2009, 9, 293–302. [Google Scholar]
- Nagel, S.; Venturini, L.; Przybylski, G.K.; Grabarczyk, P.; Schmidt, C.A.; Meyer, C.; Drexler, H.G.; Macleod, R.A.; Scherr, M. Activation of miR-17–92 by NK-like homeodomain proteins suppresses apoptosis via reduction of E2F1 in T-cell acute lymphoblastic leukemia. Leuk. Lymphoma 2009, 50, 101–108. [Google Scholar]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar]
- Kim, H.H.; Kuwano, Y.; Srikantan, S.; Lee, E.K.; Martindale, J.L.; Gorospe, M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 2009, 23, 1743–1748. [Google Scholar]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar]
- Wittmann, J.; Jack, H.M. Serum microRNAs as powerful cancer biomarkers. Biochim. Biophys. Acta 2010, 1806, 200–207. [Google Scholar]
- Brase, J.C.; Wuttig, D.; Kuner, R.; Sultmann, H. Serum microRNAs as non-invasive biomarkers for cancer. Mol. Cancer 2010, 9, 306. [Google Scholar]
- Blenkiron, C.; Miska, E.A. miRNAs in cancer: Approaches, aetiology, diagnostics and therapy. Hum. Mol. Genet 2007, 16, R106–R113. [Google Scholar]
- Cortez, M.A.; Calin, G.A. MicroRNA identification in plasma and serum: A new tool to diagnose and monitor diseases. Expert Opin. Biol. Ther 2009, 9, 703–711. [Google Scholar]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med 2005, 353, 1793–1801. [Google Scholar]
- Schetter, A.J.; Leung, S.Y.; Sohn, J.J.; Zanetti, K.A.; Bowman, E.D.; Yanaihara, N.; Yuen, S.T.; Chan, T.L.; Kwong, D.L.; Au, G.K.; et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 2008, 299, 425–436. [Google Scholar]
- Yu, S.L.; Chen, H.Y.; Chang, G.C.; Chen, C.Y.; Chen, H.W.; Singh, S.; Cheng, C.L.; Yu, C.J.; Lee, Y.C.; Chen, H.S.; et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 2008, 13, 48–57. [Google Scholar]
- Sana, J.; Faltejskova, P.; Svoboda, M.; Slaby, O. Novel classes of non-coding RNAs and cancer. J. Transl. Med 2012, 10, 103. [Google Scholar]
- Caby, M.P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol 2005, 17, 879–887. [Google Scholar]
- Orozco, A.F.; Lewis, D.E. Flow cytometric analysis of circulating microparticles in plasma. Cytometry A 2010, 77, 502–514. [Google Scholar]
- Record, M.; Subra, C.; Silvente-Poirot, S.; Poirot, M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem. Pharmacol 2011, 81, 1171–1182. [Google Scholar]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell. Res 2008, 18, 997–1006. [Google Scholar]
- Teplyuk, N.M.; Mollenhauer, B.; Gabriely, G.; Giese, A.; Kim, E.; Smolsky, M.; Kim, R.Y.; Saria, M.G.; Pastorino, S.; Kesari, S.; et al. MicroRNAs in cerebrospinal fluid identify glioblastoma and metastatic brain cancers and reflect disease activity. Neuro. Oncol 2012, 14, 689–700. [Google Scholar]
- Baraniskin, A.; Kuhnhenn, J.; Schlegel, U.; Maghnouj, A.; Zollner, H.; Schmiegel, W.; Hahn, S.; Schroers, R. Identification of microRNAs in the cerebrospinal fluid as biomarker for the diagnosis of glioma. Neuro. Oncol 2011, 14, 29–33. [Google Scholar]
- Baraniskin, A.; Kuhnhenn, J.; Schlegel, U.; Schmiegel, W.; Hahn, S.; Schroers, R. MicroRNAs in cerebrospinal fluid as biomarker for disease course monitoring in primary central nervous system lymphoma. J. Neurooncol 2012, 109, 239–244. [Google Scholar]
- Qu, H.; Xu, W.; Huang, Y.; Yang, S. Circulating miRNAs: Promising biomarkers of human cancer. Asian Pac. J. Cancer Prev 2011, 12, 1117–1125. [Google Scholar]
- Mostert, B.; Sieuwerts, A.M.; Martens, J.W.; Sleijfer, S. Diagnostic applications of cell-free and circulating tumor cell-associated miRNAs in cancer patients. Expert Rev. Mol. Diagn 2011, 11, 259–275. [Google Scholar]
- Fabbri, M.; Croce, C.M.; Calin, G.A. MicroRNAs in the ontogeny of leukemias and lymphomas. Leuk. Lymphoma 2009, 50, 160–170. [Google Scholar]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar]
- Kroh, E.M.; Parkin, R.K.; Mitchell, P.S.; Tewari, M. Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR). Methods 2010, 50, 298–301. [Google Scholar]
- Gilad, S.; Meiri, E.; Yogev, Y.; Benjamin, S.; Lebanony, D.; Yerushalmi, N.; Benjamin, H.; Kushnir, M.; Cholakh, H.; Melamed, N.; et al. Serum microRNAs are promising novel biomarkers. PLoS One 2008, 3, e3148. [Google Scholar]
- Bhattacharjya, S.; Nath, S.; Ghose, J.; Maiti, G.P.; Biswas, N.; Bandyopadhyay, S.; Panda, C.K.; Bhattacharyya, N.P.; Roychoudhury, S. miR-125b promotes cell death by targeting spindle assembly checkpoint gene MAD1 and modulating mitotic progression. Cell. Death Differ 2013, 20, 430–442. [Google Scholar]
- Wang, H.; Tan, G.; Dong, L.; Cheng, L.; Li, K.; Wang, Z.; Luo, H. Circulating MiR-125b as a marker predicting chemoresistance in breast cancer. PLoS One 2012, 7, e34210. [Google Scholar]
- Bracken, C.P.; Gregory, P.A.; Khew-Goodall, Y.; Goodall, G.J. The role of microRNAs in metastasis and epithelial-mesenchymal transition. Cell. Mol. Life Sci 2009, 66, 1682–1699. [Google Scholar]
- Mateescu, B.; Batista, L.; Cardon, M.; Gruosso, T.; de Feraudy, Y.; Mariani, O.; Nicolas, A.; Meyniel, J.P.; Cottu, P.; Sastre-Garau, X.; et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med 2011, 17, 1627–1635. [Google Scholar]
- Cheng, H.; Zhang, L.; Cogdell, D.E.; Zheng, H.; Schetter, A.J.; Nykter, M.; Harris, C.C.; Chen, K.; Hamilton, S.R.; Zhang, W. Circulating plasma MiR-141 is a novel biomarker for metastatic colon cancer and predicts poor prognosis. PLoS One 2011, 6, e17745. [Google Scholar]
- Brase, J.C.; Johannes, M.; Schlomm, T.; Falth, M.; Haese, A.; Steuber, T.; Beissbarth, T.; Kuner, R.; Sultmann, H. Circulating miRNAs are correlated with tumor progression in prostate cancer. Int. J. Cancer 2011, 128, 608–616. [Google Scholar]
- Nishikawa, E.; Osada, H.; Okazaki, Y.; Arima, C.; Tomida, S.; Tatematsu, Y.; Taguchi, A.; Shimada, Y.; Yanagisawa, K.; Yatabe, Y.; et al. miR-375 is activated by ASH1 and inhibits YAP1 in a lineage-dependent manner in lung cancer. Cancer Res 2011, 71, 6165–6173. [Google Scholar]
- Bierkens, M.; Krijgsman, O.; Wilting, S.M.; Bosch, L.; Jaspers, A.; Meijer, G.A.; Meijer, C.J.; Snijders, P.J.; Ylstra, B.; Steenbergen, R.D. Focal aberrations indicate EYA2 and hsa-miR-375 as oncogene and tumor suppressor in cervical carcinogenesis. Genes Chromosomes Cancer 2013, 52, 56–68. [Google Scholar]
- Mahn, R.; Heukamp, L.C.; Rogenhofer, S.; von Ruecker, A.; Muller, S.C.; Ellinger, J. Circulating microRNAs (miRNA) in serum of patients with prostate cancer. Urology 2011, 77, 1265.e9–1265.e16. [Google Scholar]
- Bryant, R.J.; Pawlowski, T.; Catto, J.W.; Marsden, G.; Vessella, R.L.; Rhees, B.; Kuslich, C.; Visakorpi, T.; Hamdy, F.C. Changes in circulating microRNA levels associated with prostate cancer. Br. J. Cancer 2012, 106, 768–774. [Google Scholar]
- Catalona, W.J.; Smith, D.S.; Ratliff, T.L.; Dodds, K.M.; Coplen, D.E.; Yuan, J.J.; Petros, J.A.; Andriole, G.L. Measurement of prostate-specific antigen in serum as a screening test for prostate cancer. N. Engl. J. Med 1991, 324, 1156–1161. [Google Scholar]
- Cooner, W.H. Prostate-specific antigen and transrectal ultrasound of the prostate in detection of prostate cancer. Clin. Investig. Med 1993, 16, 471–474. [Google Scholar]
- Stamey, T.A.; Kabalin, J.N.; McNeal, J.E.; Johnstone, I.M.; Freiha, F.; Redwine, E.A.; Yang, N. Prostate specific antigen in the diagnosis and treatment of adenocarcinoma of the prostate. II. Radical prostatectomy treated patients. J. Urol 1989, 141, 1076–1083. [Google Scholar]
- Prostate-Specific Antigen (PSA) Test. Available online: http://www.cancer.gov/cancertopics/factsheet/detection/PSA accessed on 22 May 2011.
- Smith, D.S.; Humphrey, P.A.; Catalona, W.J. The early detection of prostate carcinoma with prostate specific antigen: The Washington University experience. Cancer 1997, 80, 1852–1856. [Google Scholar]
- Schmid, H.P.; Prikler, L.; Semjonow, A. Problems with prostate-specific antigen screening: A critical review. Recent Results Cancer Res. 2003, 163, 226–231, discussion 264–226. [Google Scholar]
- Heneghan, H.M.; Miller, N.; Lowery, A.J.; Sweeney, K.J.; Newell, J.; Kerin, M.J. Circulating microRNAs as novel minimally invasive biomarkers for breast cancer. Ann. Surg 2010, 251, 499–505. [Google Scholar]
- Yan, L.X.; Huang, X.F.; Shao, Q.; Huang, M.Y.; Deng, L.; Wu, Q.L.; Zeng, Y.X.; Shao, J.Y. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA 2008, 14, 2348–2360. [Google Scholar]
- Asaga, S.; Kuo, C.; Nguyen, T.; Terpenning, M.; Giuliano, A.E.; Hoon, D.S. Direct serum assay for microRNA-21 concentrations in early and advanced breast cancer. Clin. Chem 2011, 57, 84–91. [Google Scholar]
- Zhu, W.; Qin, W.; Atasoy, U.; Sauter, E.R. Circulating microRNAs in breast cancer and healthy subjects. BMC Res. Notes 2009, 2, 89. [Google Scholar]
- Roth, C.; Rack, B.; Muller, V.; Janni, W.; Pantel, K.; Schwarzenbach, H. Circulating microRNAs as blood-based markers for patients with primary and metastatic breast cancer. Breast Cancer Res 2010, 12, R90. [Google Scholar]
- Zhao, H.; Shen, J.; Medico, L.; Wang, D.; Ambrosone, C.B.; Liu, S. A pilot study of circulating miRNAs as potential biomarkers of early stage breast cancer. PLoS One 2010, 5, e13735. [Google Scholar]
- Schrauder, M.G.; Strick, R.; Schulz-Wendtland, R.; Strissel, P.L.; Kahmann, L.; Loehberg, C.R.; Lux, M.P.; Jud, S.M.; Hartmann, A.; Hein, A.; et al. Circulating micro-RNAs as potential blood-based markers for early stage breast cancer detection. PLoS One 2012, 7, e29770. [Google Scholar]
- Wei, J.; Gao, W.; Zhu, C.J.; Liu, Y.Q.; Mei, Z.; Cheng, T.; Shu, Y.Q. Identification of plasma microRNA-21 as a biomarker for early detection and chemosensitivity of non-small cell lung cancer. Chin. J. Cancer 2011, 30, 407–414. [Google Scholar]
- Foss, K.M.; Sima, C.; Ugolini, D.; Neri, M.; Allen, K.E.; Weiss, G.J. miR-1254 and miR-574-5p: Serum-based microRNA biomarkers for early-stage non-small cell lung cancer. J. Thorac. Oncol 2011, 6, 482–488. [Google Scholar]
- Zheng, D.; Haddadin, S.; Wang, Y.; Gu, L.Q.; Perry, M.C.; Freter, C.E.; Wang, M.X. Plasma microRNAs as novel biomarkers for early detection of lung cancer. Int. J. Clin. Exp. Pathol 2011, 4, 575–586. [Google Scholar]
- Silva, J.; Garcia, V.; Zaballos, A.; Provencio, M.; Lombardia, L.; Almonacid, L.; Garcia, J.M.; Dominguez, G.; Pena, C.; Diaz, R.; et al. Vesicle-related microRNAs in plasma of nonsmall cell lung cancer patients and correlation with survival. Eur. Respir. J 2010, 37, 617–623. [Google Scholar]
- Hu, Z.; Chen, X.; Zhao, Y.; Tian, T.; Jin, G.; Shu, Y.; Chen, Y.; Xu, L.; Zen, K.; Zhang, C.; et al. Serum microRNA signatures identified in a genome-wide serum microRNA expression profiling predict survival of non-small-cell lung cancer. J. Clin. Oncol 2010, 28, 1721–1726. [Google Scholar]
- Bianchi, F.; Nicassio, F.; Marzi, M.; Belloni, E.; Dall’olio, V.; Bernard, L.; Pelosi, G.; Maisonneuve, P.; Veronesi, G.; Di Fiore, P.P. A serum circulating miRNA diagnostic test to identify asymptomatic high-risk individuals with early stage lung cancer. EMBO Mol. Med 2011, 3, 495–503. [Google Scholar]
- Boeri, M.; Verri, C.; Conte, D.; Roz, L.; Modena, P.; Facchinetti, F.; Calabro, E.; Croce, C.M.; Pastorino, U.; Sozzi, G. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 3713–3718. [Google Scholar]
- Ng, E.K.; Chong, W.W.; Jin, H.; Lam, E.K.; Shin, V.Y.; Yu, J.; Poon, T.C.; Ng, S.S.; Sung, J.J. Differential expression of microRNAs in plasma of patients with colorectal cancer: A potential marker for colorectal cancer screening. Gut 2009, 58, 1375–1381. [Google Scholar]
- Kanaan, Z.; Rai, S.N.; Eichenberger, M.R.; Roberts, H.; Keskey, B.; Pan, J.; Galandiuk, S. Plasma miR-21: A potential diagnostic marker of colorectal cancer. Ann. Surg. 2012, 256, 544–551. [Google Scholar]
- Huang, Z.; Huang, D.; Ni, S.; Peng, Z.; Sheng, W.; Du, X. Plasma microRNAs are promising novel biomarkers for early detection of colorectal cancer. Int. J. Cancer 2010, 127, 118–126. [Google Scholar]
- Wang, L.G.; Gu, J. Serum microRNA-29a is a promising novel marker for early detection of colorectal liver metastasis. Cancer Epidemiol 2012, 36, e61–e67. [Google Scholar]
- Pu, X.X.; Huang, G.L.; Guo, H.Q.; Guo, C.C.; Li, H.; Ye, S.; Ling, S.; Jiang, L.; Tian, Y.; Lin, T.Y. Circulating miR-221 directly amplified from plasma is a potential diagnostic and prognostic marker of colorectal cancer and is correlated with p53 expression. J. Gastroenterol. Hepatol 2010, 25, 1674–1680. [Google Scholar]
- Nelson, P.T.; Wang, W.X.; Wilfred, B.R.; Tang, G. Technical variables in high-throughput miRNA expression profiling: Much work remains to be done. Biochim. Biophys. Acta 2008, 1779, 758–765. [Google Scholar]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar]
- Chen, Y.; Gelfond, J.A.; McManus, L.M.; Shireman, P.K. Reproducibility of quantitative RT-PCR array in miRNA expression profiling and comparison with microarray analysis. BMC Genomics 2009, 10, 407. [Google Scholar]
- Creighton, C.J.; Reid, J.G.; Gunaratne, P.H. Expression profiling of microRNAs by deep sequencing. Brief. Bioinf 2009, 10, 490–497. [Google Scholar]
- Kong, W.; Zhao, J.J.; He, L.; Cheng, J.Q. Strategies for profiling microRNA expression. J. Cell. Physiol 2009, 218, 22–25. [Google Scholar]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet 2009, 10, 704–714. [Google Scholar]
- Peltier, H.J.; Latham, G.J. Normalization of microRNA expression levels in quantitative RT-PCR assays: Identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 2008, 14, 844–852. [Google Scholar]
- Linsen, S.E.; de Wit, E.; Janssens, G.; Heater, S.; Chapman, L.; Parkin, R.K.; Fritz, B.; Wyman, S.K.; de Bruijn, E.; Voest, E.E.; et al. Limitations and possibilities of small RNA digital gene expression profiling. Nat. Methods 2009, 6, 474–476. [Google Scholar]
- Wark, A.W.; Lee, H.J.; Corn, R.M. Multiplexed detection methods for profiling microRNA expression in biological samples. Angew. Chem. Int. Ed. Engl 2008, 47, 644–652. [Google Scholar]
- Castoldi, M.; Schmidt, S.; Benes, V.; Hentze, M.W.; Muckenthaler, M.U. miChip: An array-based method for microRNA expression profiling using locked nucleic acid capture probes. Nat. Protoc 2008, 3, 321–329. [Google Scholar]
- Baskerville, S.; Bartel, D.P. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. Rna 2005, 11, 241–247. [Google Scholar]
- Li, W.; Ruan, K. MicroRNA detection by microarray. Anal. Bioanal. Chem 2009, 394, 1117–1124. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res 1996, 6, 986–994. [Google Scholar]
- Jiang, J.; Lee, E.J.; Gusev, Y.; Schmittgen, T.D. Real-time expression profiling of microRNA precursors in human cancer cell lines. Nucleic Acids Res 2005, 33, 5394–5403. [Google Scholar]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 2005, 33, e179. [Google Scholar]
- Guo, J.; Dong, Q.; Fang, Z.; Chen, X.; Lu, H.; Wang, K.; Yin, Y.; Cai, X.; Zhao, N.; Chen, J.; et al. Identification of miRNAs that are associated with tumor metastasis in neuroblastoma. Cancer Biol. Ther 2010, 9, 446–452. [Google Scholar]
- Kikkawa, N.; Hanazawa, T.; Fujimura, L.; Nohata, N.; Suzuki, H.; Chazono, H.; Sakurai, D.; Horiguchi, S.; Okamoto, Y.; Seki, N. miR-489 is a tumour-suppressive miRNA target PTPN11 in hypopharyngeal squamous cell carcinoma (HSCC). Br. J. Cancer 2010, 103, 877–884. [Google Scholar]
- Li, J.; Yao, B.; Huang, H.; Wang, Z.; Sun, C.; Fan, Y.; Chang, Q.; Li, S.; Wang, X.; Xi, J. Real-time polymerase chain reaction microRNA detection based on enzymatic stem-loop probes ligation. Anal. Chem 2009, 81, 5446–5451. [Google Scholar]
- Zhi, F.; Chen, X.; Wang, S.; Xia, X.; Shi, Y.; Guan, W.; Shao, N.; Qu, H.; Yang, C.; Zhang, Y.; et al. The use of hsa-miR-21, hsa-miR-181b and hsa-miR-106a as prognostic indicators of astrocytoma. Eur. J. Cancer 2010, 46, 1640–1649. [Google Scholar]
- Raymond, C.K.; Roberts, B.S.; Garrett-Engele, P.; Lim, L.P.; Johnson, J.M. Simple, quantitative primer-extension PCR assay for direct monitoring of microRNAs and short-interfering RNAs. Rna 2005, 11, 1737–1744. [Google Scholar]
- Sharbati-Tehrani, S.; Kutz-Lohroff, B.; Bergbauer, R.; Scholven, J.; Einspanier, R. miR-Q: A novel quantitative RT-PCR approach for the expression profiling of small RNA molecules such as miRNAs in a complex sample. BMC Mol. Biol 2008, 9, 34. [Google Scholar]
- Castro, F.O.; Sharbati, S.; Rodriguez-Alvarez, L.L.; Cox, J.F.; Hultschig, C.; Einspanier, R. MicroRNA expression profiling of elongated cloned and in vitro-fertilized bovine embryos. Theriogenology 2010, 73, 71–85. [Google Scholar]
- Fu, H.J.; Zhu, J.; Yang, M.; Zhang, Z.Y.; Tie, Y.; Jiang, H.; Sun, Z.X.; Zheng, X.F. A novel method to monitor the expression of microRNAs. Mol. Biotechnol 2006, 32, 197–204. [Google Scholar]
- Ro, S.; Park, C.; Jin, J.; Sanders, K.M.; Yan, W. A PCR-based method for detection and quantification of small RNAs. Biochem. Biophys. Res. Commun 2006, 351, 756–763. [Google Scholar]
- Marras, S.A.; Tyagi, S.; Kramer, F.R. Real-time assays with molecular beacons and other fluorescent nucleic acid hybridization probes. Clin. Chim. Acta 2006, 363, 48–60. [Google Scholar]
- Tyagi, S.; Kramer, F.R. Molecular beacons: Probes that fluoresce upon hybridization. Nat. Biotechnol 1996, 14, 303–308. [Google Scholar]
- Paiboonskuwong, K.; Kato, Y. Detection of the mature, but not precursor, RNA using a fluorescent DNA probe. Nucleic Acids Symp. Ser. (Oxf. ) 2006, 50, 327–328. [Google Scholar]
- Kang, W.J.; Cho, Y.L.; Chae, J.R.; Lee, J.D.; Choi, K.J.; Kim, S. Molecular beacon-based bioimaging of multiple microRNAs during myogenesis. Biomaterials 2011, 32, 1915–1922. [Google Scholar]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol 2008, 26, 1135–1145. [Google Scholar]
- Ansorge, W.J. Next-generation DNA sequencing techniques. N. Biotechnol 2009, 25, 195–203. [Google Scholar]
- Mardis, E.R. Anticipating the 1,000 dollar genome. Genome Biol 2006, 7, 112. [Google Scholar]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar]
- Gu, L.Q.; Wanunu, M.; Wang, M.X.; McReynolds, L.; Wang, Y. Detection of miRNAs with a nanopore single-molecule counter. Expert Rev. Mol. Diagn 2012, 12, 573–584. [Google Scholar]
- Neely, L.A.; Patel, S.; Garver, J.; Gallo, M.; Hackett, M.; McLaughlin, S.; Nadel, M.; Harris, J.; Gullans, S.; Rooke, J. A single-molecule method for the quantitation of microRNA gene expression. Nat. Methods 2006, 3, 41–46. [Google Scholar]
- Kapanidis, A.N.; Lee, N.K.; Laurence, T.A.; Doose, S.; Margeat, E.; Weiss, S. Fluorescence-aided molecule sorting: Analysis of structure and interactions by alternating-laser excitation of single molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 8936–8941. [Google Scholar]
- Lee, N.K.; Kapanidis, A.N.; Wang, Y.; Michalet, X.; Mukhopadhyay, J.; Ebright, R.H.; Weiss, S. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J 2005, 88, 2939–2953. [Google Scholar]
- Kapanidis, A.N.; Laurence, T.A.; Lee, N.K.; Margeat, E.; Kong, X.; Weiss, S. Alternating-Laser Excitation of Single Molecules. Acc. Chem. Res 2005, 38, 523–533. [Google Scholar]
- Kapanidis, A.N.; Margeat, E.; Laurence, T.A.; Doose, S.; Ho, S.O.; Mukhopadhyay, J.; Kortkhonjia, E.; Mekler, V.; Ebright, R.H.; Weiss, S. Retention of transcription initiation factor sigma(70) in transcription elongation: Single-molecule analysis. Mol. Cell 2005, 20, 347–356. [Google Scholar]
- Laurence, T.A.; Kong, X.; Jager, M.; Weiss, S. Probing structural heterogeneities and fluctuations of nucleic acids and denatured proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 17348–17353. [Google Scholar]
- Jager, M.; Michalet, X.; Weiss, S. Protein-protein interactions as a tool for site-specific labeling of proteins. Protein Sci 2005, 14, 2059–2068. [Google Scholar]
- Jager, M.; Nir, E.; Weiss, S. Site-specific labeling of proteins for single-molecule FRET by combining chemical and enzymatic modification. Protein Sci 2006, 15, 640–646. [Google Scholar]
- Margeat, E.; Kapanidis, A.N.; Tinnefeld, P.; Wang, Y.; Mukhopadhyay, J.; Ebright, R.H.; Weiss, S. Direct observation of abortive initiation and promoter escape within single immobilized transcription complexes. Biophys. J 2006, 90, 1419–1431. [Google Scholar]
- Kapanidis, A.N.; Margeat, E.; Ho, S.O.; Kortkhonjia, E.; Weiss, S.; Ebright, R.H. Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science 2006, 314, 1144–1147. [Google Scholar]
- Nir, E.; Michalet, X.; Hamadani, K.M.; Laurence, T.A.; Neuhauser, D.; Kovchegov, Y.; Weiss, S. Shot-noise limited single-molecule FRET histograms: Comparison between theory and experiments. J. Phys. Chem. B 2006, 110, 22103–22124. [Google Scholar]
- Lee, N.K.; Kapanidis, A.N.; Koh, H.R.; Korlann, Y.; Ho, S.O.; Kim, Y.; Gassman, N.; Kim, S.K.; Weiss, S. Three-color alternating-laser excitation of single molecules: Monitoring multiple interactions and distances. Biophys. J. 2007, 92, 303–312. [Google Scholar]
- Yim, S.W.; Kim, T.; Laurence, T.A.; Partono, S.; Kim, D.; Kim, Y.; Weiss, S.; Reitmair, A. Four-color alternating laser excitation single-molecule fluorescence spectroscopy for next-generation biodetection assays. Clin. Chem 2012, 58, 707–716. [Google Scholar]
- Kim, S.; Streets, A.M.; Lin, R.R.; Quake, S.R.; Weiss, S.; Majumdar, D.S. High-throughput single-molecule optofluidic analysis. Nat. Methods 2011, 8, 242–245. [Google Scholar]
- Crawford, E.D. PSA testing: What is the use? Lancet 2005, 365, 1447–1449. [Google Scholar]
- Du, X.-L.; Duan, D.-M.; Cao, R.; Jin, G.; Li, J. Enhancing DNA detection sensitivity through a two-step enrichment method with magnetic beads and droplet evaporation. Anal. Lett 2010, 43, 1525–1533. [Google Scholar]
- Colyer, R.A.; Scalia, G.; Kim, T.; Rech, I.; Resnatic, D.; Marangonic, S.; Ghionic, M.; Cova, S.; Weiss, S.; Michalet, X. High-throughput multispot single-molecule spectroscopy. Proc. SPIE 2010, 7571. [Google Scholar] [CrossRef]
- Colyer, R.A.; Scalia, G.; Rech, I.; Gulinatti, A.; Ghioni, M.; Cova, S.; Weiss, S.; Michalet, X. High-throughput FCS using an LCOS spatial light modulator and an 8 × 1 SPAD array. Biomed. Opt. Express 2010, 1, 1408–1431. [Google Scholar]
- Baker, M. MicroRNA profiling: Separating signal from noise. Nat. Methods 2010, 7, 687–692. [Google Scholar]


{kind=link}
{kind=link}
| Type | Class | Symbol | Characteristic | Cancer/biological function associations |
|---|---|---|---|---|
| Long non-coding RNAs (lncRNAs, ≥200 nt) | Long intergenic non-coding RNAs | lincRNAs | ranging from several hundreds to tens of thousands nts; lie within the genomic intervals between two genes; transcriptional | involved in tumorigenesis and cancer metastasis/involved in diverse biological processes such as dosage compensation and/or imprinting |
| Long intronic non-coding RNAs | cis-regulation of neighbouring genes lie within introns; evolutionary conserved; tissue-specific expression patterns | aberrantly expressed in human cancers/possible link with posttranscriptional gene silencing | ||
| Telomere-associated ncRNAs | TERRAs | 100 bp >9 kb; conserved among eukaryotes; synthesized from C-rich strand; polyadenylated; form intermolecular G-quadruplex structure with single-stranded telomeric DNA | possible impact on telomere-associated diseases including many cancers/negative regulation of telomere length and activity through inhibition of telomerase | |
| Long non-coding RNAs with dual functions | both protein-coding and functionally regulatory RNA capacity | deregulation has been described in breast and ovarian tumors/modulate gene expression through diverse mechanisms | ||
| Pseudogene RNAs | gene copies that have lost the ability to code for a protein; potential to regulate their protein-coding cousin; created via retrotrans-positions; tissue-specific | often deregulated during tumorigenesis and cancer progression/regulation of tumor suppressors and oncogenes by acting as microRNA decoys | ||
| Transcribed-ultraconserved regions | T-UCRs | longer than 200 bp; absolutely conserved between orthologous regions of human, rat, and mouse; located in both intra- and intergenic regions | expression is often altered in some cancers; possible involvement in tumorigenesis; antisense inhibitors for protein-coding genes or other ncRNAs | |
| Antisense RNAs | aRNAs | complementary antisense transcripts | antisense transcripts appear to be a pervasive feature of human cells, which suggests that they are a fundamental component of gene regulation | |
| Long stress-induced non-coding transcripts | LSINCTs | longer than 300 nucleotides; expression is increased in response to the DNA damage-inducing tobacco carcinogen 4-(methylnitrosamino)-1-(3- pyridyl)-1-butanone (NNK) | increased expression in a number of cancer-derived cell lines | |
| Small non-coding RNAs (<200 nt) | MicroRNAs | miRNAs | 18–25 nt; account 1%–2% of the human genome; control the 50% of protein-coding genes; guide suppression of translation; Drosha- and Dicer-dependent small ncRNAs | initiation of various disorders including many, if not all, cancers/regulation of proliferation, differentiation, and apoptosis; involved in human development |
| Small nucleolar RNAs | snoRNAs | 60–300 nt; enriched in the nucleolus; excised from pre-mRNA introns in vertebrates; bind snoRNP proteins | association with development of some cancers/important function in the maturation of other non-coding RNAs, above all, rRNAs and snRNAs; miRNA-like snoRNAs regulate mRNAs | |
| Pyknons | subset of patterns of variable length; form mosaics in untranslated and protein-coding regions; more frequently in 3′ UTRs | expected association with cancer biology/possible link with posttranscriptional silencing of genes, mainly involved in cell communication, regulation of transcription, signaling, transport, etc. | ||
| Sample type | Method | Patients (n) | HDs (n) | Normalization procedure based on | Candidate miRNAs (n) | Differentially expressed miRNAs | Prognostic | Reference |
|---|---|---|---|---|---|---|---|---|
| Prostate cancer | ||||||||
| Serum | qRT-PCR | 25 | 25 | Spiked-in miRNA | 6 | miR-141 | No | Mitchell et al.[118] |
| Serum | qRT-PCR array † | 21 | None | Spiked-in miRNA | 667 | 69 miRs up ††, 0 miRs down †† | Brase et al.[126] | |
| qRT-PCR | 45 | None | Spiked-in miRNA | 5 | miR-9†, miR-516a-3p | No | ||
| 116 | None | Spiked-in miRNA | miR-141, miR-375 | Yes | ||||
| miR-200b | No | |||||||
| Serum | qRT-PCR | 35 | 20 | 4 | miR-26a, miR-32, miR-195, let-7i (only in benign prostate hyperplasia) | Mahn et al.[129] | ||
| Plasma | qRT-PCR | 78 | 28 | 742 | 16 miRs including miR-141, miR-200b, miR-375 | Bryant et al.[130] | ||
| Breast cancer | ||||||||
| Whole blood | qRT-PCR | 148 | 44 | miR-16 | 7 | miR-195 let-7a | No | Heneghan et al.[137] |
| Serum | qRT-PCR | 102 | 20 | miR-16 | 1 | miR-21 | No | Asaga et al.[139] |
| Serum | qRT-PCR | 13 | 8 | cel-miR-39, has-miR-145 | miR-155 | Zhu et al.[140] | ||
| Serum | qRT-PCR | 89 | 29 | miR-16 | 4 | miR-10b, miR-155 miR-34a | No | Roth et al.[141] |
| Plasma | Illumina microarray † | 10CA | 10 CA | Quantile normalization algorithm | 1145 | 17 miRs up, 14 miRs down | No | Zhao et al.[142] |
| 10AA | 10 AA | 9 miRs up, 9 miRs down | ||||||
| Whole blood | microarray | 48 | 57 | 1100 | 13 miRs up, 46miRs down | Schrauder et al.[143] | ||
| NSCLC | ||||||||
| Plasma | qRT-PCR | 63 | 30 | miR-21 up-regulation | Wei et al.[144] | |||
| Serum | qRT-PCR | 11 (profiling) 31 (validation) | 11 (profiling) 22 (validation) | miR-1254, miR-574-5p up-regulation | Foss et al.[145] | |||
| Plasma | qRT-PCR | 74 | 68 | miR-155, miR-197, miR-182 | Zheng et al.[146] | |||
| Serum | Solexa sequencing † | 11 ‡ | 21 ‡ | Total RNA | 190 | 63 miRs up, 28 miRs down | Chen et al.[111] | |
| qRT-PCR | 152 | 75 | Average of HDs | 3 | let-7a, miR-223 miR-25 | No | ||
| Exosomes | qRT-PCR array after EpCAM-based enrichment step † | 28 | 20 | miR-142-3p; miR-30b | 365 | 0 miRs up, 10 miRs down | Silva et al.[147] | |
| qRT-PCR | miR-142-3p; miR-30b | 5 | let-7f, miR-30e-3p | Yes | ||||
| miR-20b | No | |||||||
| Serum | Solexa sequencing † | 2 × 30 ‡ | None | Spiked-in miRNA | 101/109 § | 3 miRs up §, 8 miRs down § | Hu et al.[148] | |
| qRT-PCR | 303 | 1 | One HD | 11 | miR-486, miR-1# miR-30d, miR-499# | Yes | ||
| Serum | qRT-PCR array | 59 | 69 | 34 miRs | Bianchi et al.[149] | |||
| Tissues and Plasma | Microarray followed by qRT-PCR | 19 (training set), 22 (validation set) | 13 miRs | Boeri et al.[150] | ||||
| Colorectal cancer | ||||||||
| Serum | Solexa sequencing † | 11 ‡ | 21 ‡ | Total RNA | 190 | 69miRs when compared to healthy controls | Chen et al.[111] | |
| qRT-PCR | 152 | 75 | Average of HDs | 3 | 14 miRs when compared to a lung cancer group | |||
| Plasma | qRT-PCR array † | 25 | 20 | U6 | 95 | miR-17-3p, miR-135b miR-92, miR-222 miR-95 | Ng et al.[151] | |
| qRT-PCR | 90 | 50 | U6 | 5 | miR-17-3p miR-92 | No | ||
| Tissues and Plasma | Microfluidic array | 20 | 20 | 380 | 90% of 19 miRs dysregulated in colorectal cancer patient plasma, especially miR-21 | Kanaan et al.[152] | ||
| Plasma | qRT-PCR | 100 | 59 | miR-16 | 12 | miR-29a miR-92a | No | Huang et al.[153] |
| Serum | qRT-PCR | 74 (40 for validation) | 3 | miR-29a | Wang et al.[154] | |||
| Plasma | qRT-PCR | 103 | 37 | Standard curve | 3 | miR-221 | Yes | Pu et al.[155] |
| Plasma | qRT-PCR | 102 | miR-141 | Cheng et al.[125] | ||||
| Characteristic | qPCR * | Microarray * | Sequencing * | 4c-ALEX (96 wells; current prototype) | Multifoci (64) 4c-ALEX (384 well optofluidics chip; in development) |
|---|---|---|---|---|---|
| Throughput time | ~6 h | ~2 days | 1–2 weeks | ~2.5 days (each well takes 30 min acquisition time) to ~1 weeks (triplicates) | ~3 h (64 times faster data acquisition) to 9 h (triplicates) |
| Total RNA required | 500 ng | 100–1000 ng | 500–5000 ng | ~50 ng | ~50 ng |
| Estimated cost per sample, including reagents and supplies | $400 (754 human microRNAs queried per sample) | $250–$350 (at least 950 microRNAs queried per sample) | $1000–$1300 (theoretically, all microRNAs queried per sample) | $10 (576 microRNAs queried per sample) | $10 (theoretically, all microRNAs queried per sample) |
| Dynamic range detected | Six orders of magnitude | Four orders of magnitude | Five or more orders of magnitude | Four orders of magnitude | Four orders of magnitude |
| Ease-of-use | Easy | Moderate | Difficult | Easy | Easy |
| Infrastructure and technical requirements | Few | Moderate | Substantial | Few | Moderate |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, T.; Reitmair, A. Non-Coding RNAs: Functional Aspects and Diagnostic Utility in Oncology. Int. J. Mol. Sci. 2013, 14, 4934-4968. https://doi.org/10.3390/ijms14034934
Kim T, Reitmair A. Non-Coding RNAs: Functional Aspects and Diagnostic Utility in Oncology. International Journal of Molecular Sciences. 2013; 14(3):4934-4968. https://doi.org/10.3390/ijms14034934
Chicago/Turabian StyleKim, Taiho, and Armin Reitmair. 2013. "Non-Coding RNAs: Functional Aspects and Diagnostic Utility in Oncology" International Journal of Molecular Sciences 14, no. 3: 4934-4968. https://doi.org/10.3390/ijms14034934