The Induction of Cytokine Release in Monocytes by Electronegative Low-Density Lipoprotein (LDL) Is Related to Its Higher Ceramide Content than Native LDL



Abstract
:1. Introduction
2. Results and Discussion
2.1. Lipid and Apolipoprotein Composition
2.2. Phospholipolytic Activities
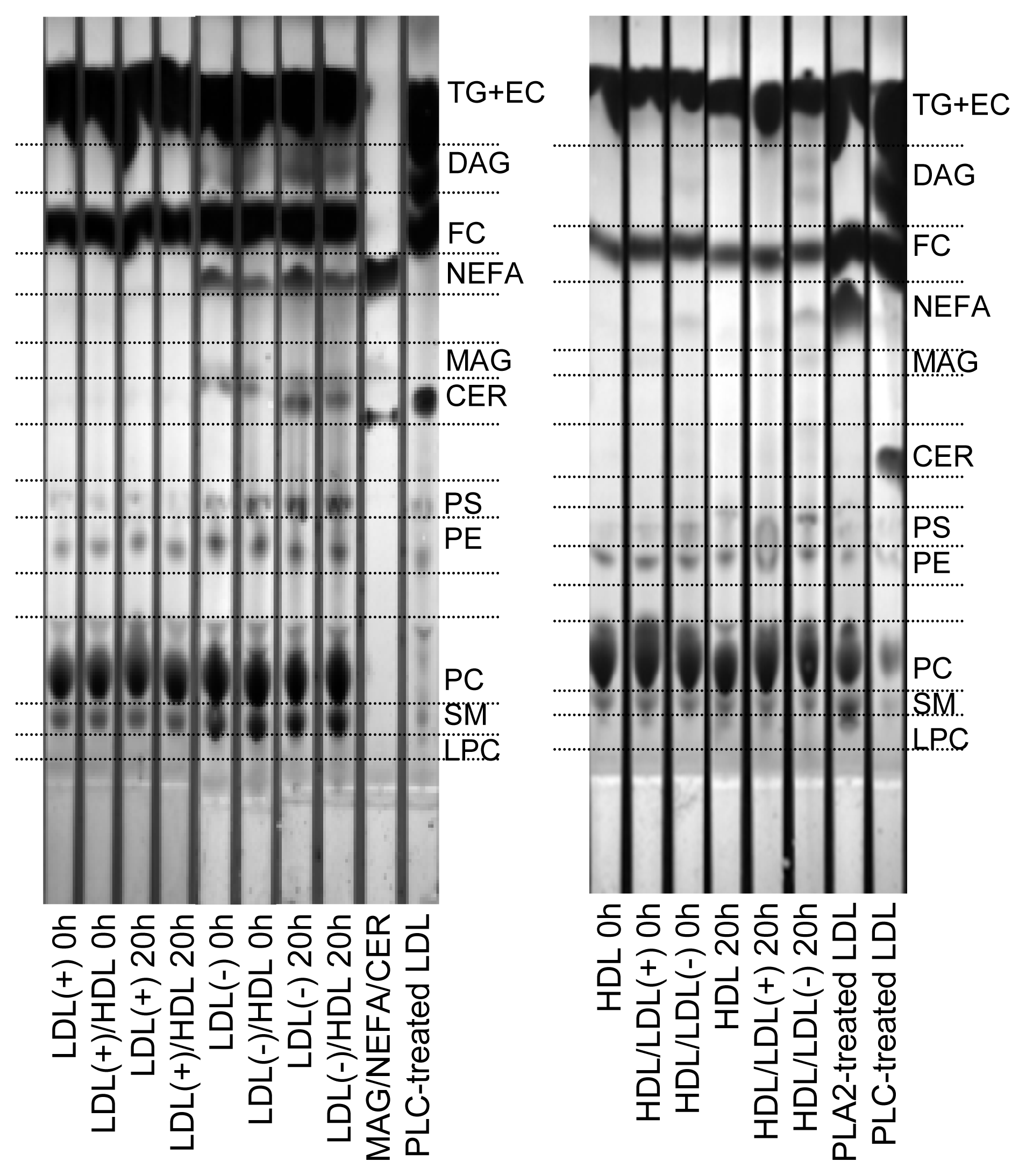
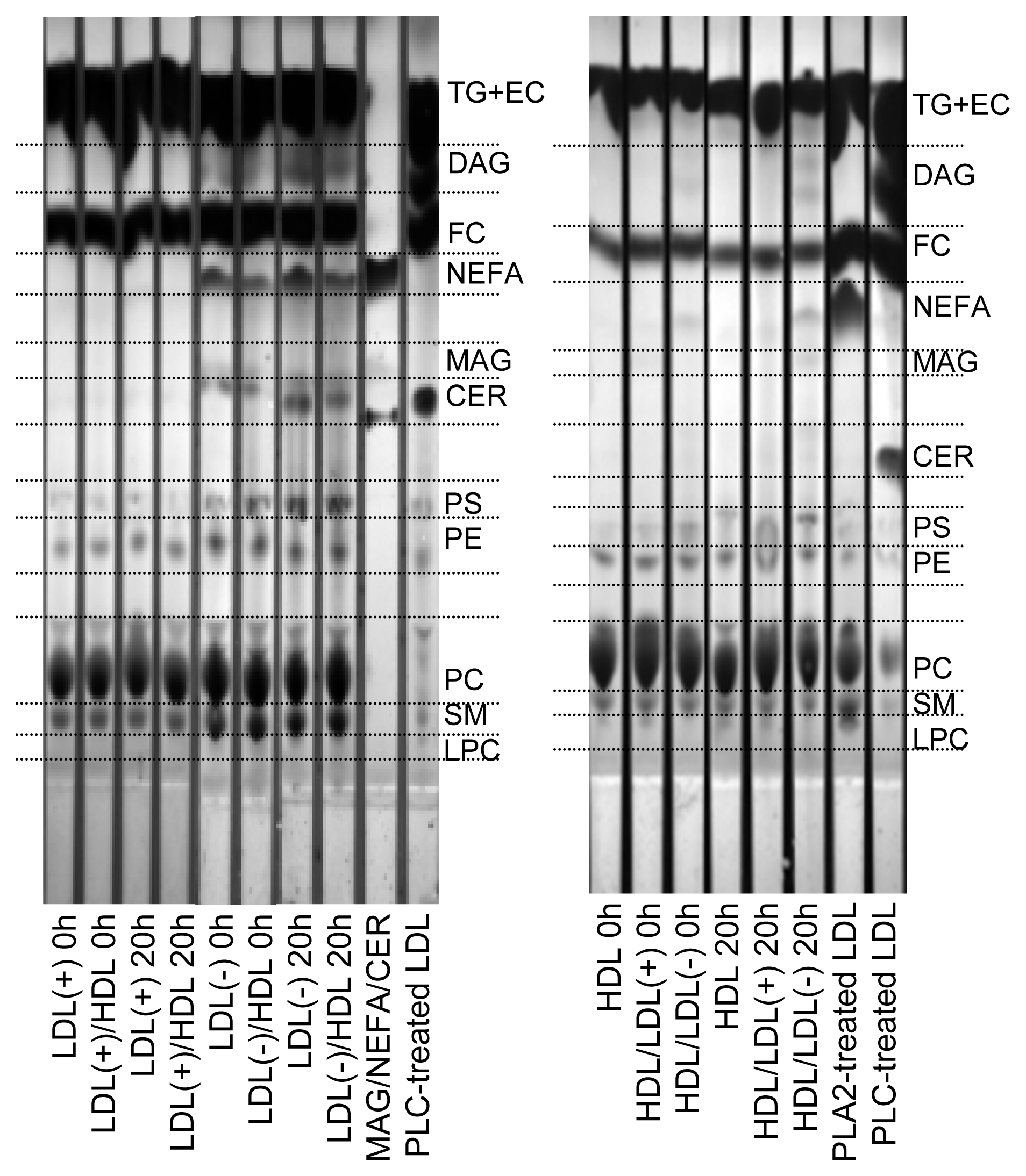
2.3. Products of PLC-Like Activity
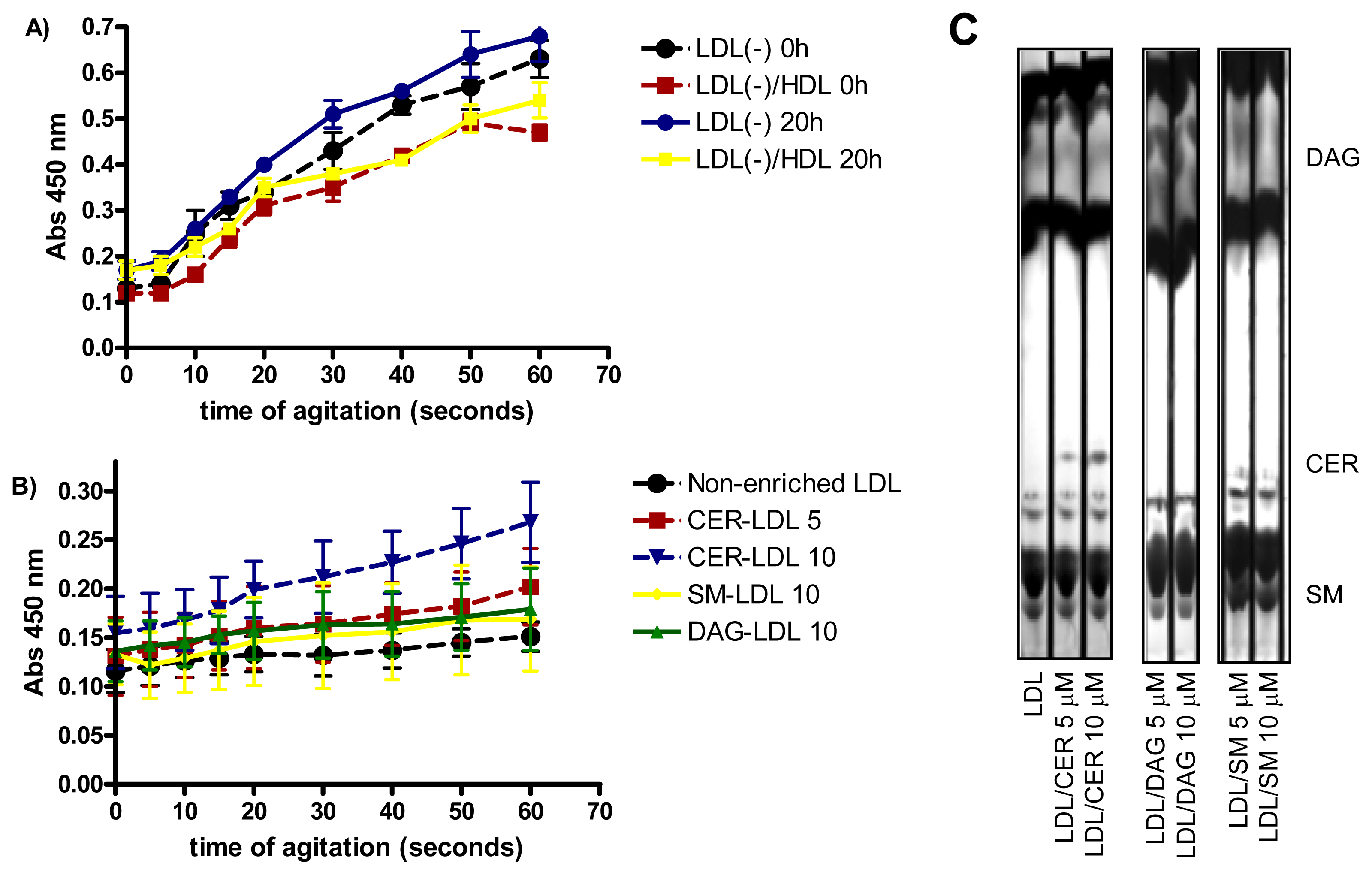
2.4. Effect of Incubation on LDL Aggregation
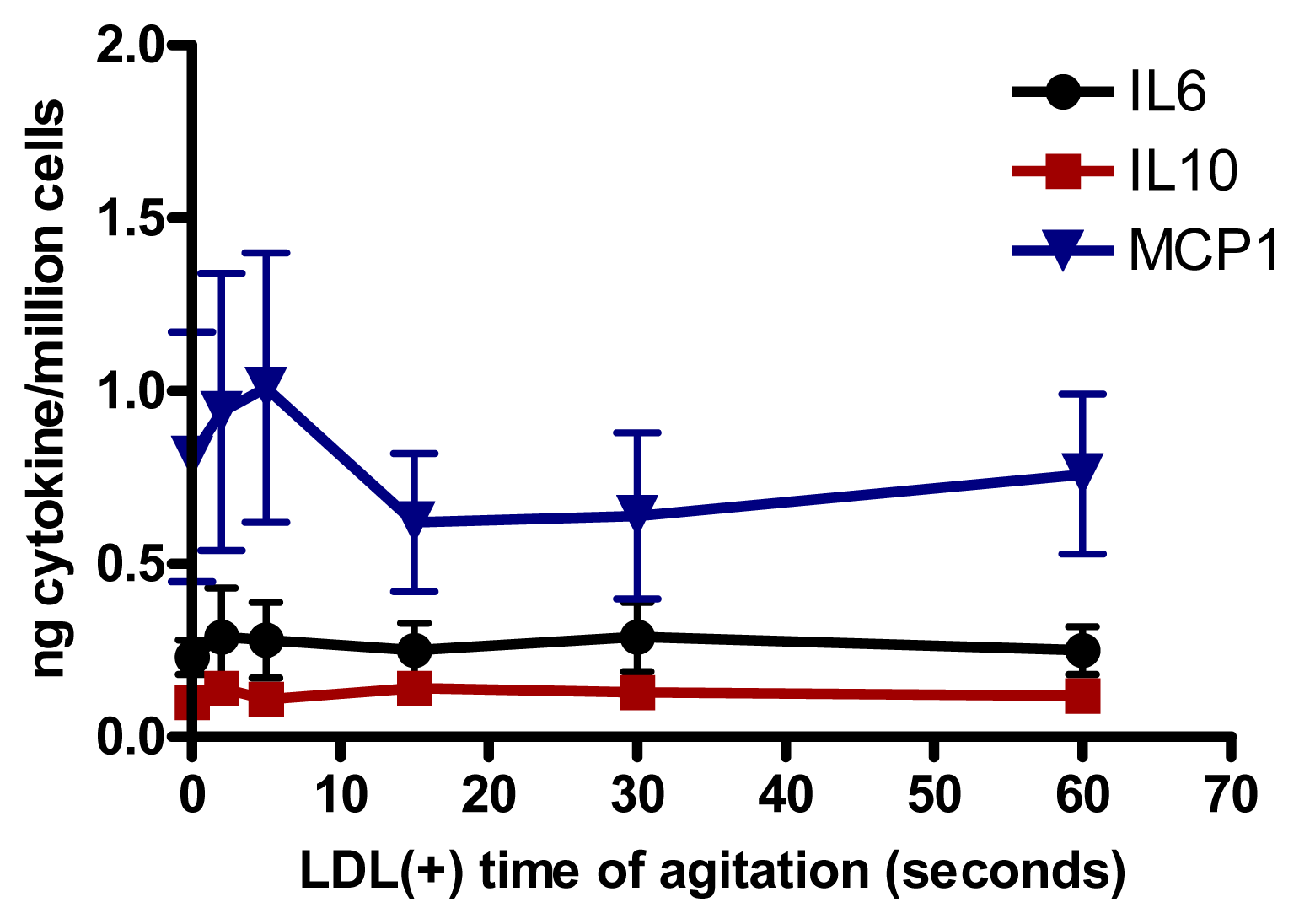
2.5. Relationship Between Aggregation of LDL(−) and Cytokine Release
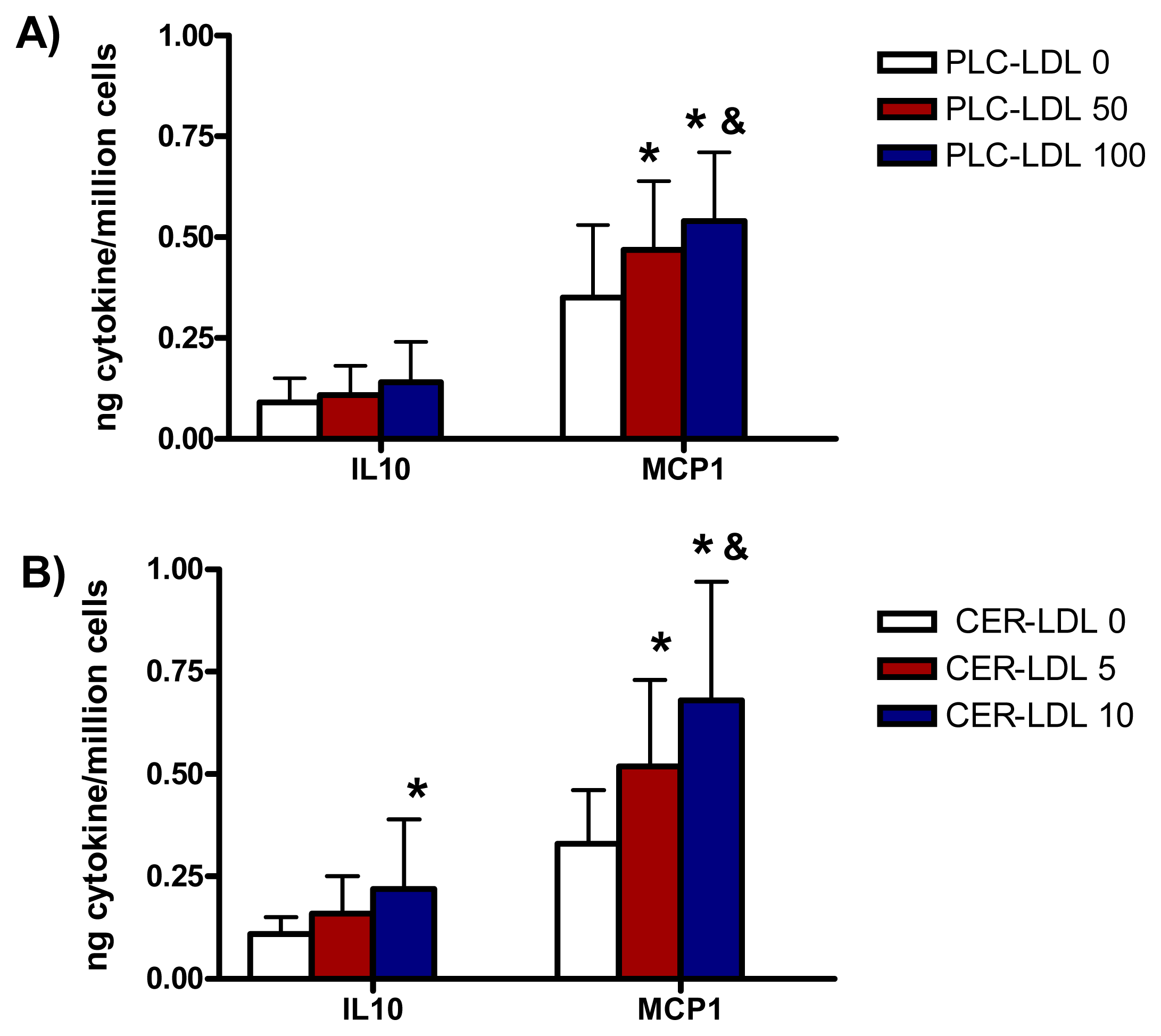
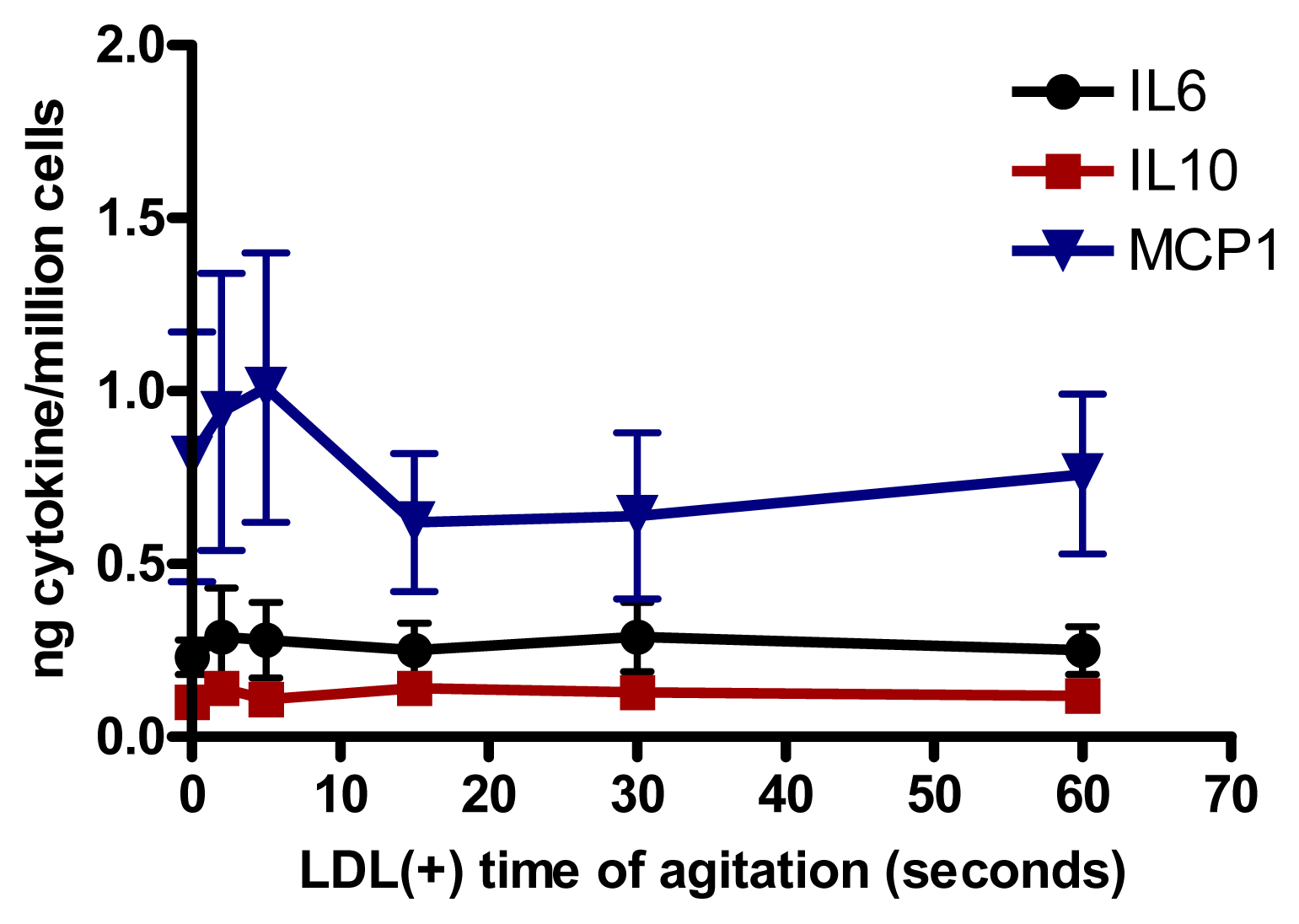
2.6. Involvement of PLC-Like Activity Products in Cytokine Release
3. Experimental Section
3.1. Lipoprotein Isolation and Separation of LDL Subfractions
3.2. LDL Incubation with HDL
3.3. LDL Modification
3.4. Lipid and Apoprotein Composition
3.5. Aggregation Level and Oxidation Tests
3.6. Enzymatic Activity Measurements
3.7. Cytokine Release Experiments in Monocytes
3.8. Statistical Analysis
4. Discussion
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Navab, M.; Berliner, J.A.; Watson, A.D.; Hama, S.Y.; Territo, M.C.; Lusis, A.J.; Shih, D.M.; van Lenten, B.J.; Frank, J.S.; Demer, L.L.; et al. The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler. Thromb. Vasc. Biol 1996, 16, 831–842. [Google Scholar]
- Sanchez-Quesada, J.L.; Benitez, S.; Ordonez-Llanos, J. Electronegative low-density lipoprotein. Curr. Opin. Lipidol 2004, 15, 329–335. [Google Scholar]
- Benitez, S.; Camacho, M.; Arcelus, R.; Vila, L.; Bancells, C.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Increased lysophosphatidylcholine and non-esterified fatty acid content in LDL induces chemokine release in endothelial cells. Relationship with electronegative LDL. Atherosclerosis 2004, 177, 299–305. [Google Scholar]
- Benitez, S.; Sanchez-Quesada, J.L.; Lucero, L.; Arcelus, R.; Ribas, V.; Jorba, O.; Castellvi, A.; Alonso, E.; Blanco-Vaca, F.; Ordonez-Llanos, J. Changes in low-density lipoprotein electronegativity and oxidizability after aerobic exercise are related to the increase in associated non-esterified fatty acids. Atherosclerosis 2002, 160, 223–232. [Google Scholar]
- Benitez, S.; Sanchez-Quesada, J.L.; Ribas, V.; Jorba, O.; Blanco-Vaca, F.; Gonzalez-Sastre, F.; Ordonez-Llanos, J. Platelet-activating factor acetylhydrolase is mainly associated with electronegative low-density lipoprotein subfraction. Circulation 2003, 108, 92–96. [Google Scholar]
- Bancells, C.; Benitez, S.; Villegas, S.; Jorba, O.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Novel phospholipolytic activities associated with electronegative low-density lipoprotein are involved in increased self-aggregation. Biochemistry 2008, 47, 8186–8194. [Google Scholar]
- Sanchez-Quesada, J.L.; Villegas, S.; Ordonez-Llanos, J. Electronegative LDL: A link between apoB misfolding, lipoprotein aggregation and proteoglycan binding. Curr. Opin. Lipidol 2012, 23, 479–486. [Google Scholar]
- Bancells, C.; Villegas, S.; Blanco, F.J.; Benitez, S.; Gallego, I.; Beloki, L.; Perez-Cuellar, M.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Aggregated electronegative low density lipoprotein in human plasma shows a high tendency toward phospholipolysis and particle fusion. J. Biol. Chem 2010, 285, 32425–32435. [Google Scholar]
- Bancells, C.; Benitez, S.; Ordonez-Llanos, J.; Oorni, K.; Kovanen, P.T.; Milne, R.W.; Sanchez-Quesada, J.L. Immunochemical analysis of the electronegative LDL subfraction shows that abnormal N-terminal apolipoprotein B conformation is involved in increased binding to proteoglycans. J. Biol. Chem 2011, 286, 1125–1133. [Google Scholar]
- Bancells, C.; Benitez, S.; Jauhiainen, M.; Ordonez-Llanos, J.; Kovanen, P.T.; Villegas, S.; Sanchez-Quesada, J.L.; Oorni, K. High binding affinity of electronegative LDL to human aortic proteoglycans depends on its aggregation level. J. Lipid Res 2009, 50, 446–455. [Google Scholar]
- Benitez, S.; Camacho, M.; Bancells, C.; Vila, L.; Sanchez-Quesada, J.L.; Ordonez-Llanos, J. Wide proinflammatory effect of electronegative low-density lipoprotein on human endothelial cells assayed by a protein array. Biochim. Biophys. Acta 2006, 1761, 1014–1021. [Google Scholar]
- Bancells, C.; Sanchez-Quesada, J.L.; Birkelund, R.; Ordonez-Llanos, J.; Benitez, S. HDL and electronegative LDL exchange anti- and pro-inflammatory properties. J. Lipid Res 2010, 51, 2947–2956. [Google Scholar]
- Benitez, S.; Bancells, C.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Pro-inflammatory action of LDL(−) on mononuclear cells is counteracted by increased IL10 production. Biochim. Biophys. Acta 2007, 1771, 613–622. [Google Scholar]
- Oorni, K.; Pentikainen, M.O.; Ala-Korpela, M.; Kovanen, P.T. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: Molecular mechanisms and effects on matrix interactions. J. Lipid Res 2000, 41, 1703–1714. [Google Scholar]
- Schissel, S.L.; Tweedie-Hardman, J.; Rapp, J.H.; Graham, G.; Williams, K.J.; Tabas, I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J. Clin. Invest 1996, 98, 1455–1464. [Google Scholar]
- Mathias, S.; Pena, L.A.; Kolesnick, R.N. Signal transduction of stress via ceramide. Biochem. J 1998, 335, 465–480. [Google Scholar]
- Laulederkind, S.J.; Bielawska, A.; Raghow, R.; Hannun, Y.A.; Ballou, L.R. Ceramide induces interleukin 6 gene expression in human fibroblasts. J. Exp. Med 1995, 182, 599–604. [Google Scholar]
- Ballou, L.R.; Laulederkind, S.J.; Rosloniec, E.F.; Raghow, R. Ceramide signalling and the immune response. Biochim. Biophys. Acta 1996, 1301, 273–287. [Google Scholar]
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gomez-Munoz, A. Ceramide and ceramide 1-phosphate in health and disease. Lipids Health Dis 2010, 9, 15. [Google Scholar]
- Hajjar, D.P.; Pomerantz, K.B. Signal transduction in atherosclerosis: Integration of cytokines and the eicosanoid network. FASEB J 1992, 6, 2933–2941. [Google Scholar]
- Sweet, M.J.; Hume, D.A. Endotoxin signal transduction in macrophages. J. Leukoc. Biol 1996, 60, 8–26. [Google Scholar]
- Kinscherf, R.; Claus, R.; Deigner, H.P.; Nauen, O.; Gehrke, C.; Hermetter, A.; Russwurm, S.; Daniel, V.; Hack, V.; Metz, J. Modified low density lipoprotein delivers substrate for ceramide formation and stimulates the sphingomyelin-ceramide pathway in human macrophages. FEBS Lett 1997, 405, 55–59. [Google Scholar]
- Grandl, M.; Bared, S.M.; Liebisch, G.; Werner, T.; Barlage, S.; Schmitz, G. E-LDL and Ox-LDL differentially regulate ceramide and cholesterol raft microdomains in human macrophages. Cytometry A 2006, 69, 189–191. [Google Scholar]
- Lightle, S.; Tosheva, R.; Lee, A.; Queen-Baker, J.; Boyanovsky, B.; Shedlofsky, S.; Nikolova-Karakashian, M. Elevation of ceramide in serum lipoproteins during acute phase response in humans and mice: Role of serine-palmitoyl transferase. Arch. Biochem. Biophys 2003, 419, 120–128. [Google Scholar]
- De Castellarnau, C.; Sanchez-Quesada, J.L.; Benitez, S.; Rosa, R.; Caveda, L.; Vila, L.; Ordonez-Llanos, J. Electronegative LDL from normolipemic subjects induces IL-8 and monocyte chemotactic protein secretion by human endothelial cells. Arterioscler. Thromb. Vasc. Biol 2000, 20, 2281–2287. [Google Scholar]
- Holopainen, J.M.; Medina, O.P.; Metso, A.J.; Kinnunen, P.K. Sphingomyelinase activity associated with human plasma low density lipoprotein. J. Biol. Chem 2000, 275, 16484–16489. [Google Scholar]
- Bancells, C.; Canals, F.; Benítez, S.; Colomé, N.; Julve, J.; Ordóñez-Llanos, J.; Sanchez-Quesada, J. Proteomic analysis of electronegative low density lipoprotein. J. Lipid Res 2010, 51, 3508–3515. [Google Scholar]
- Sevanian, A.; Bittolo-Bon, G.; Cazzolato, G.; Hodis, H.; Hwang, J.; Zamburlini, A.; Maiorino, M.; Ursini, F. LDL-is a lipid hydroperoxide-enriched circulating lipoprotein. J. Lipid Res 1997, 38, 419–428. [Google Scholar]
- Sanchez-Quesada, J.L.; Camacho, M.; Anton, R.; Benitez, S.; Vila, L.; Ordonez-Llanos, J. Electronegative LDL of FH subjects: Chemical characterization and induction of chemokine release from human endothelial cells. Atherosclerosis 2003, 166, 261–270. [Google Scholar]
- Boyanovsky, B.; Karakashian, A.; King, K.; Giltiay, N.; Nikolova-Karakashian, M. Uptake and metabolism of low density lipoproteins with elevated ceramide content by human microvascular endothelial cells: Implications for the regulation of apoptosis. J. Biol. Chem 2003, 278, 26992–26999. [Google Scholar]
- Auerbach, B.J.; Kiely, J.S.; Cornicelli, J.A. A spectrophotometric microtiter-based assay for the detection of hydroperoxy derivatives of linoleic acid. Anal. Biochem 1992, 201, 375–380. [Google Scholar]
- Serhan, C.N.; Haeggstrom, J.Z.; Leslie, C.C. Lipid mediator networks in cell signaling: Update and impact of cytokines. FASEB J 1996, 10, 1147–1158. [Google Scholar]
- Chatterjee, S. Sphingolipids in atherosclerosis and vascular biology. Arterioscler. Thromb. Vasc. Biol 1998, 18, 1523–1533. [Google Scholar]
- Estruch, M.; Sanchez-Quesada, J.L.; Bancells, C.; Beloki, L.; Ordonez-Llanos, J.; Benitez, S. Involvement of CD14 and TLR4 in the binding of electronegative LDL and consequent cytokine release in monocytes. Proceedings of the 80th European Atherosclerosis Congress, Milan, Italy, 25–28 May 2012.
- Pfeiffer, A.; Bottcher, A.; Orso, E.; Kapinsky, M.; Nagy, P.; Bodnar, A.; Spreitzer, I.; Liebisch, G.; Drobnik, W.; Gempel, K.; et al. Lipopolysaccharide and ceramide docking to CD14 provokes ligand-specific receptor clustering in rafts. Eur. J. Immunol 2001, 31, 3153–3164. [Google Scholar]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol 2007, 27, 84–91. [Google Scholar]
- Auge, N.; Nikolova-Karakashian, M.; Carpentier, S.; Parthasarathy, S.; Negre-Salvayre, A.; Salvayre, R.; Merrill, A.H., Jr; Levade, T. Role of sphingosine 1-phosphate in the mitogenesis induced by oxidized low density lipoprotein in smooth muscle cells via activation of sphingomyelinase, ceramidase and sphingosine kinase. J. Biol. Chem. 1999, 274, 21533–21538. [Google Scholar]
- Bancells, C.; Sánchez-Quesada, J.L.; Birkelund, R.; Ordóñez-Llanos, J.; Benítez, S. Electronegative LDL induces Fas and modifies gene expression in mononuclear cells. Front. Biosci 2010, 2, 78–86. [Google Scholar]
- Cifone, M.G.; de Maria, R.; Roncaioli, P.; Rippo, M.R.; Azuma, M.; Lanier, L.L.; Santoni, A.; Testi, R. Apoptotic signaling through CD95 (Fas/Apo-1) activates an acidic sphingomyelinase. J. Exp. Med 1994, 180, 1547–1552. [Google Scholar]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell. Biol 2003, 4, 397–407. [Google Scholar]
- Chandru, H.; Boggaram, V. The role of sphingosine 1-phosphate in the TNF-alpha induction of IL-8 gene expression in lung epithelial cells. Gene 2007, 391, 150–160. [Google Scholar]
- Lai, W.Q.; Irwan, A.W.; Goh, H.H.; Howe, H.S.; Yu, D.T.; Valle-Onate, R.; McInnes, I.B.; Melendez, A.J.; Leung, B.P. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. J. Immunol 2008, 181, 8010–8017. [Google Scholar]
- Mao, C.; Obeid, L.M. Ceramidases: Regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta 2008, 1781, 424–434. [Google Scholar]
- Barter, P.J.; Nicholls, S.; Rye, K.A.; Anantharamaiah, G.M.; Navab, M.; Fogelman, A.M. Antiinflammatory properties of HDL. Circ. Res 2004, 95, 764–772. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pre-incubation | Time of incubation | % CT | % TG | % PL | % apoB or apoAI | NEFA (mol/mol apo) | |
|---|---|---|---|---|---|---|---|
| LDL(+) | - | 0 h | 42.3 ± 2.4 | 7.0 ± 1.2 | 28.8 ± 3.4 | 21.9 ± 2.5 | 9.7 ± 4.7 |
| HDL | 0 h | 42.6 ± 1.7 | 6.8 ± 1.2 | 29.1 ± 2.7 | 21.4 ± 1.9 | 8.2 ± 5.2 | |
| - | 20 h | 42.3 ± 1.7 | 6.8 ± 1.1 | 28.1 ± 2.9 | 22.8 ± 2.7 | 11.7 ± 5.6 | |
| HDL | 20 h | 42.2 ± 1.8 | 7.2 ± 1.4 | 27.8 ± 3.2 | 22.8 ± 2.3 | 9.6 ± 4.1 | |
| LDL(−) | - | 0 h | 42.7 ± 2.3 | 10.6 ± 0.9 # | 26.4 ± 2.4 # | 20.3 ± 2.8 # | 32.2 ± 7.5 # |
| HDL | 0 h | 43.3 ± 2.2 | 10.6 ± 1.3 | 26.3 ± 2.2 | 19.9 ± 1.6 | 19.7 ± 3.4 | |
| - | 20 h | 42.6 ± 1.6 | 15.1 ± 1.5 #* | 22.0 ± 3.4 #* | 20.3 ± 2.7 # | 57.0 ± 17.4 #* | |
| HDL | 20 h | 42.4 ± 1.3 | 13.8 ± 1.3 * | 23.4 ± 2.4 * | 20.4 ± 2.7 | 42.3 ± 10.2 | |
| HDL | - | 0 h | 16.8 ± 2.7 | 4.1 ± 1.0 | 32.5 ± 4.6 | 46.0 ± 7.4 | 0.1 ± 0.1 |
| LDL(+) | 0 h | 18.0 ± 2.3 | 4.1 ± 0.8 | 33.3 ± 3.8 | 44.6 ± 6.0 | 0.2 ± 0.1 | |
| LDL(−) | 0 h | 20.1 ± 2.6 & | 5.4 ± 1.0 & | 31.8 ± 3.6 | 42.7 ± 5.6 | 0.8 ± 0.4 & | |
| - | 20 h | 16.3 ± 3.4 | 3.7 ± 1.2 | 31.3 ± 6.2 | 47.9 ± 10.5 | 0.2 ± 0.1 | |
| LDL(+) | 20 h | 17.8 ± 2.9 | 4.6 ± 1.2 | 31.7 ± 4.6 | 45.9 ± 7.9 | 0.3 ± 0.2 | |
| LDL(−) | 20 h | 20.0 ± 5.5 & | 9.7 ± 4.5 *& | 27.1 ± 4.7 *& | 43.2 ± 12.0 | 1.6 ± 0.9 *& |
| Pre-incubation | Time of incubation | PLC-like activity (mU equivalents/mg apo) | μmol peroxides/g apo | |
|---|---|---|---|---|
| LDL(+) | - | 0 h | 4.5 ± 8.7 | 4.88 ± 0.63 |
| HDL | 0 h | 0.6 ± 0.8 | 3.77 ± 1.16 | |
| - | 20 h | 6.0 ± 6.1 | 4.69 ± 1.18 | |
| HDL | 20 h | 5.1 ± 3.8 | 3.70 ± 1.28 | |
| LDL(−) | - | 0 h | 69.3 ± 15.6 # | 4.66 ± 1.01 |
| HDL | 0 h | 48.7 ± 9.8 & | 3.85 ± 0.20 | |
| - | 20 h | 201.5 ± 35.5 #* | 3.92 ± 0.51 | |
| HDL | 20 h | 134.9 ± 38.4 *& | 4.47 ± 0.67 | |
| HDL | - | 0 h | 0.2 ± 0.2 | 6.34 ± 2.47 |
| LDL(+) | 0 h | 0.3 ± 0.3 | 6.44 ± 2.55 | |
| LDL(−) | 0 h | 72.4 ± 16.9 & | 6.54 ± 2.30 | |
| - | 20 h | 0.2 ± 0.1 | 5.24 ± 0.81 | |
| LDL(+) | 20 h | 0.7 ± 0.7 | 5.14 ± 1.37 | |
| LDL(−) | 20 h | 165.5 ± 27.9 *& | 5.08 ± 1.22 |
| Pre-incubation | Time of incubation | Absorbance 450 nm (abs units) | |
|---|---|---|---|
| LDL(+) | - | 0 h | 0.092 ± 0.005 |
| HDL | 0 h | 0.088 ± 0.009 | |
| - | 20 h | 0.101 ± 0.008 | |
| HDL | 20 h | 0.101 ± 0.006 | |
| LDL(−) | - | 0 h | 0.101 ± 0.015 # |
| HDL | 0 h | 0.090 ± 0.006 & | |
| - | 20 h | 0.152 ± 0.017 #* | |
| HDL | 20 h | 0.130 ± 0.028 *#& | |
| HDL | - | 0 h | 0.046 ± 0.007 |
| LDL(+) | 0 h | 0.046 ± 0.005 | |
| LDL(−) | 0 h | 0.053 ± 0.008 & | |
| - | 20 h | 0.047 ± 0.008 | |
| LDL(+) | 20 h | 0.047 ± 0.010 | |
| LDL(−) | 20 h | 0.049 ± 0.008 |
| IL6 release (ng/million cells) | |
|---|---|
| LDL(+) | 0.30 ± 0.06 |
| LDL(−) | 0.64 ± 0.09 * |
| Non-enriched LDL | 0.33 ± 0.09 |
| PLC-LDL (50 U/L) | 0.38 ± 0.09 |
| PLC-LDL (100 U/L) | 0.56 ± 0.12 * |
| CER-LDL (5 μM) | 0.45 ± 0.12 * |
| CER-LDL (10 μM) | 0.55 ± 0.15 * |
| DAG-LDL (5 μM) | 0.32 ± 0.08 |
| DAG-LDL (10 μM) | 0.35 ± 0.12 |
| SM-LDL (5 μM) | 0.33 ± 0.04 |
| SM-LDL (10 μM) | 0.36 ± 0.10 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Estruch, M.; Sanchez-Quesada, J.L.; Beloki, L.; Ordoñez-Llanos, J.; Benitez, S. The Induction of Cytokine Release in Monocytes by Electronegative Low-Density Lipoprotein (LDL) Is Related to Its Higher Ceramide Content than Native LDL. Int. J. Mol. Sci. 2013, 14, 2601-2616. https://doi.org/10.3390/ijms14022601
Estruch M, Sanchez-Quesada JL, Beloki L, Ordoñez-Llanos J, Benitez S. The Induction of Cytokine Release in Monocytes by Electronegative Low-Density Lipoprotein (LDL) Is Related to Its Higher Ceramide Content than Native LDL. International Journal of Molecular Sciences. 2013; 14(2):2601-2616. https://doi.org/10.3390/ijms14022601
Chicago/Turabian StyleEstruch, Montserrat, Jose Luis Sanchez-Quesada, Lorea Beloki, Jordi Ordoñez-Llanos, and Sonia Benitez. 2013. "The Induction of Cytokine Release in Monocytes by Electronegative Low-Density Lipoprotein (LDL) Is Related to Its Higher Ceramide Content than Native LDL" International Journal of Molecular Sciences 14, no. 2: 2601-2616. https://doi.org/10.3390/ijms14022601