Parkinson’s Disease: A Complex Interplay of Mitochondrial DNA Alterations and Oxidative Stress

 and
and 
Abstract
: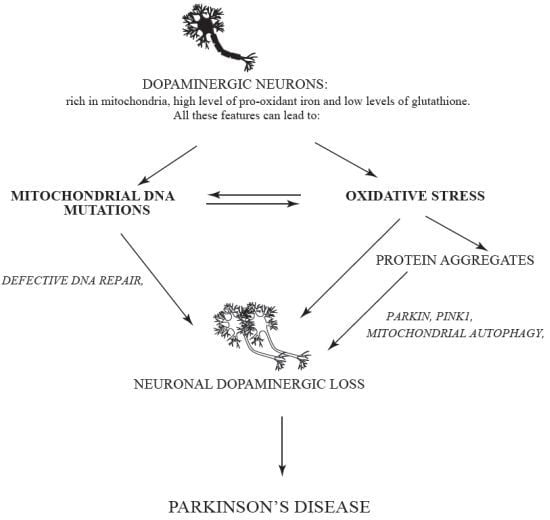
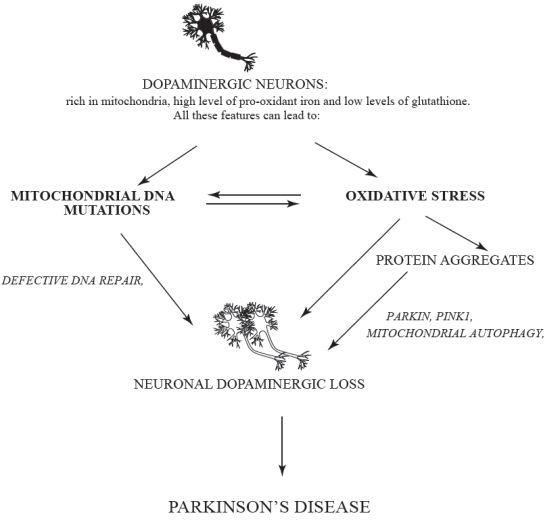{kind=link}
{kind=link}
{kind=link}
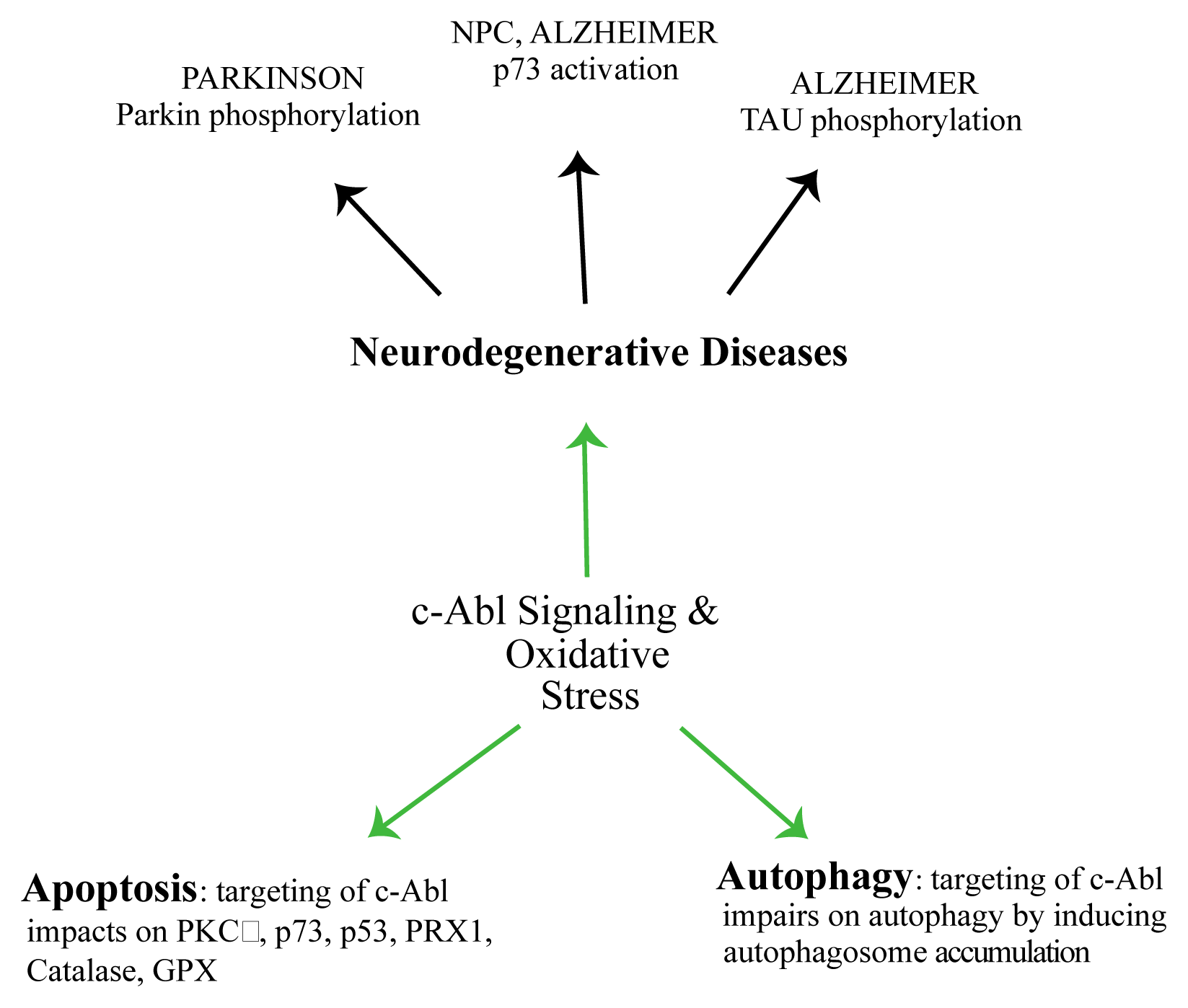{kind=link}
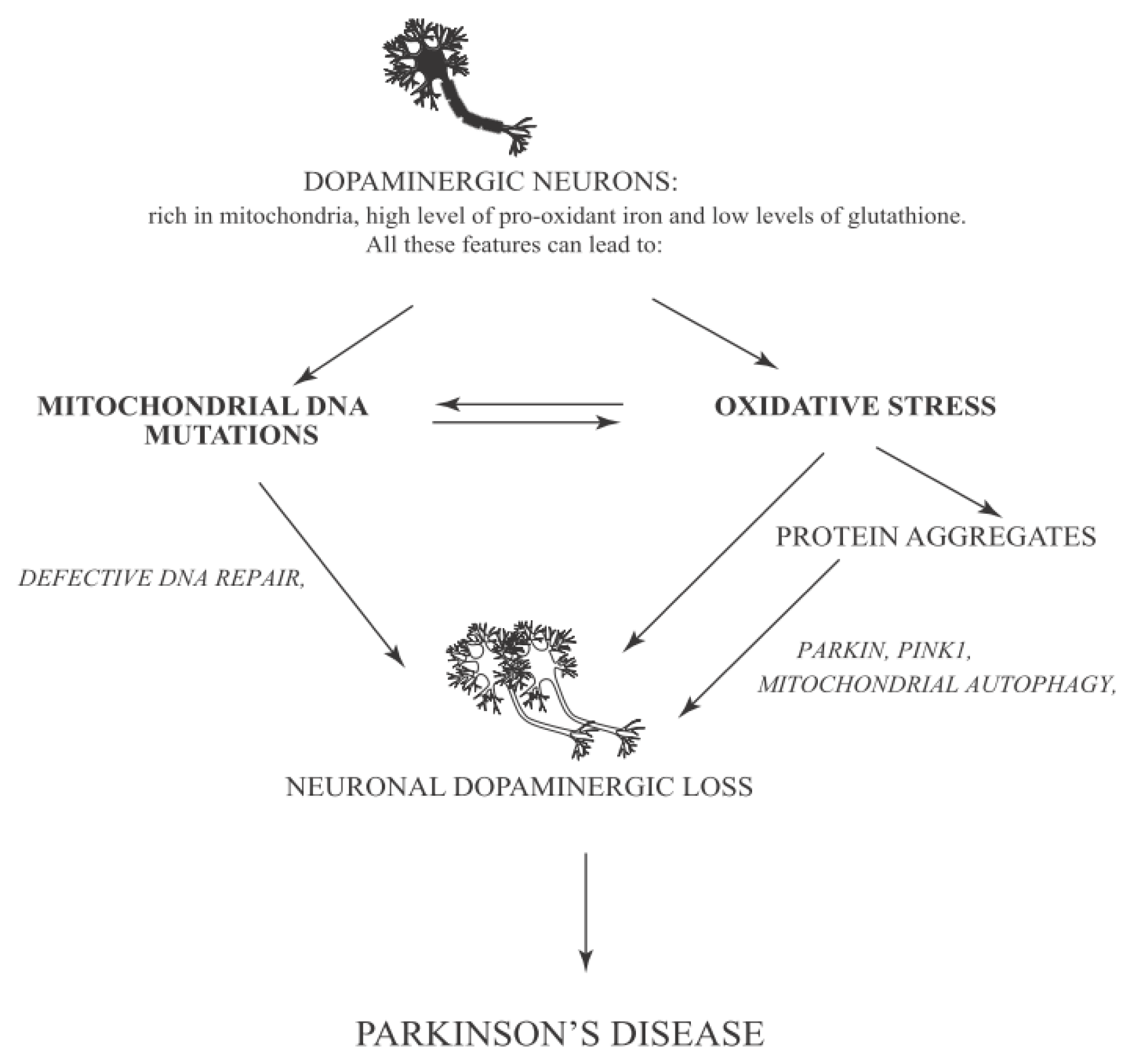{kind=link}
1. Introduction
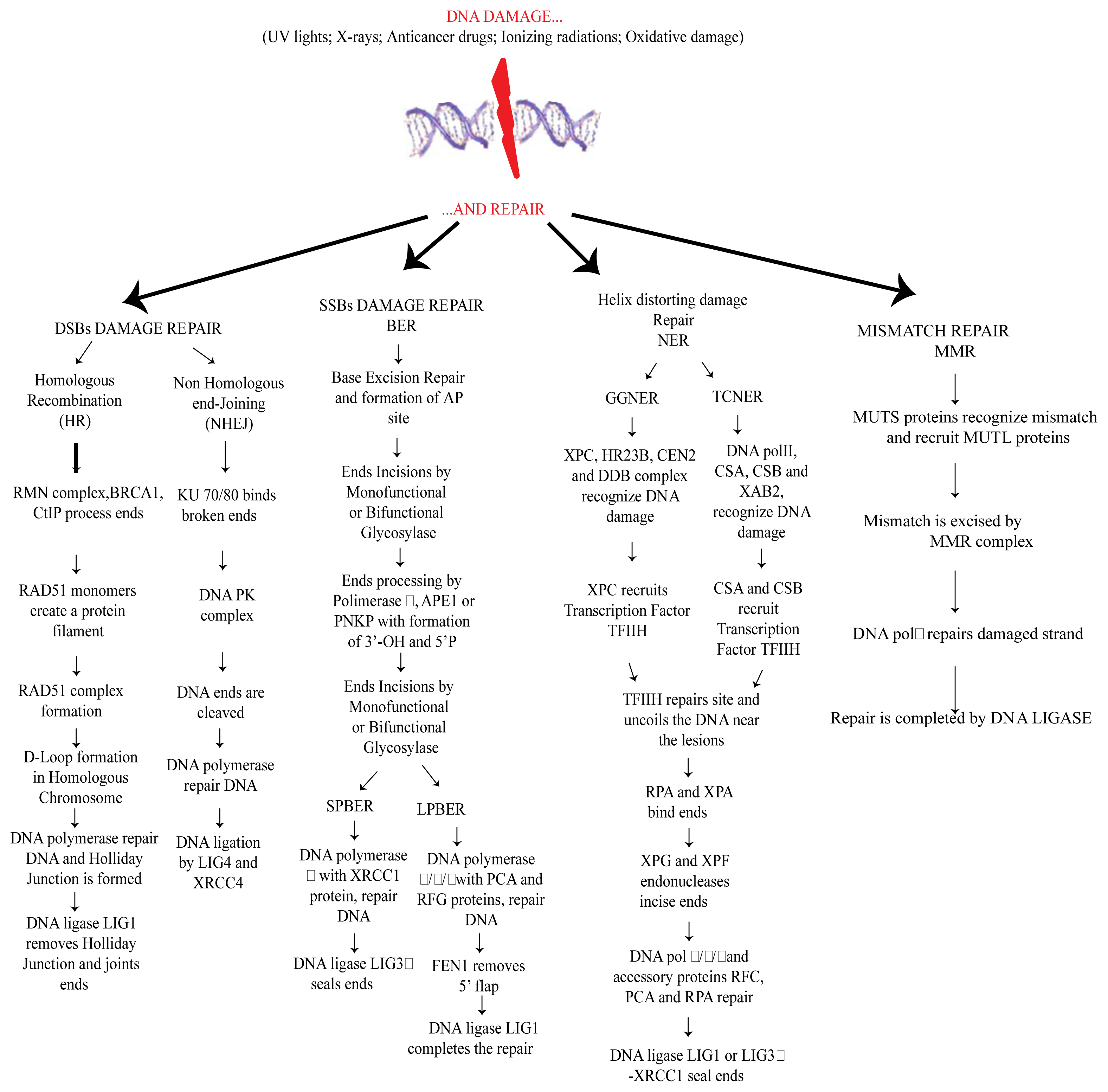
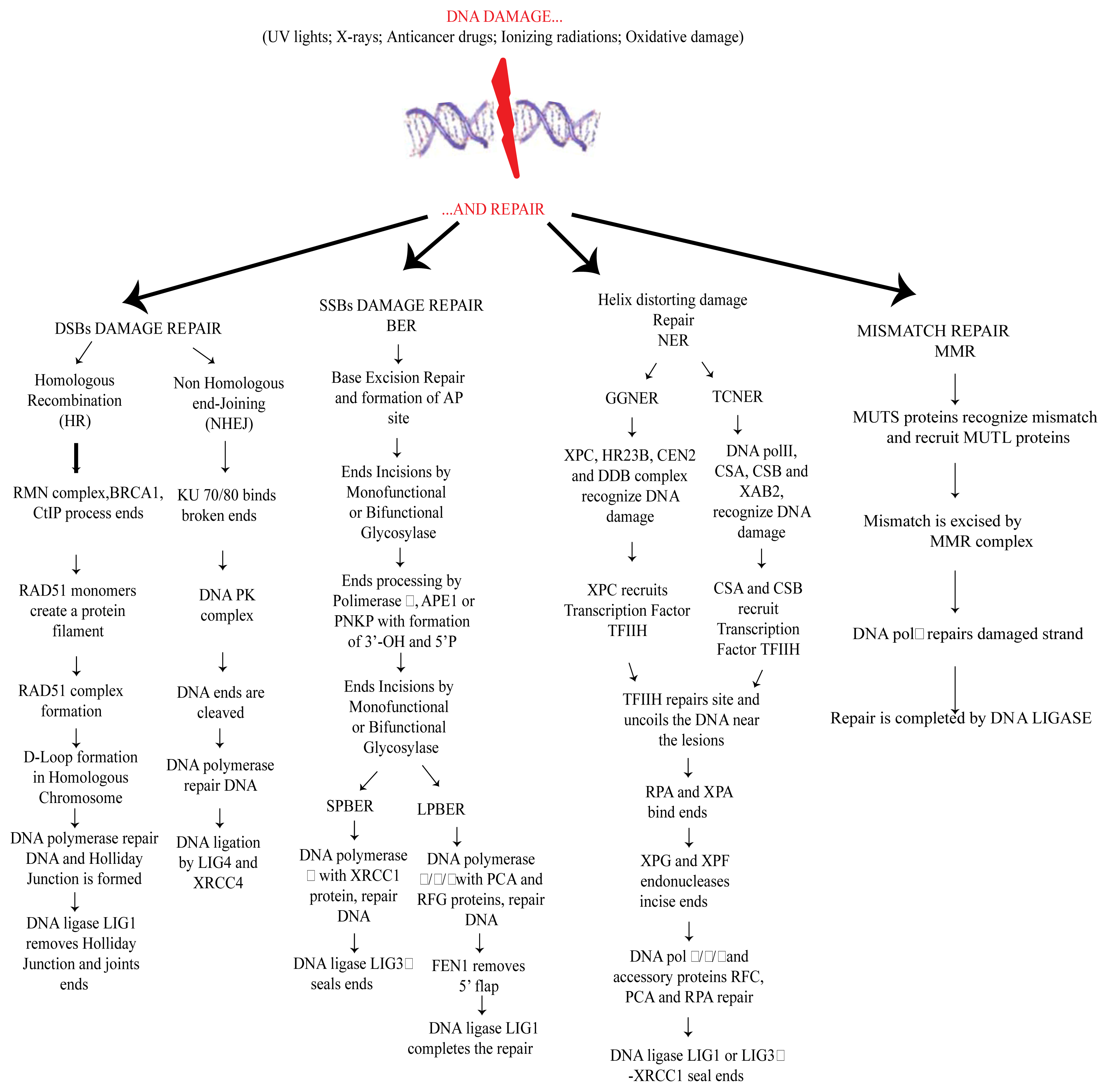
2. DNA Damage and Repair
3. Mitochondrial DNA Alterations in Diseases
4. Mitochondrial DNA Damage Repair
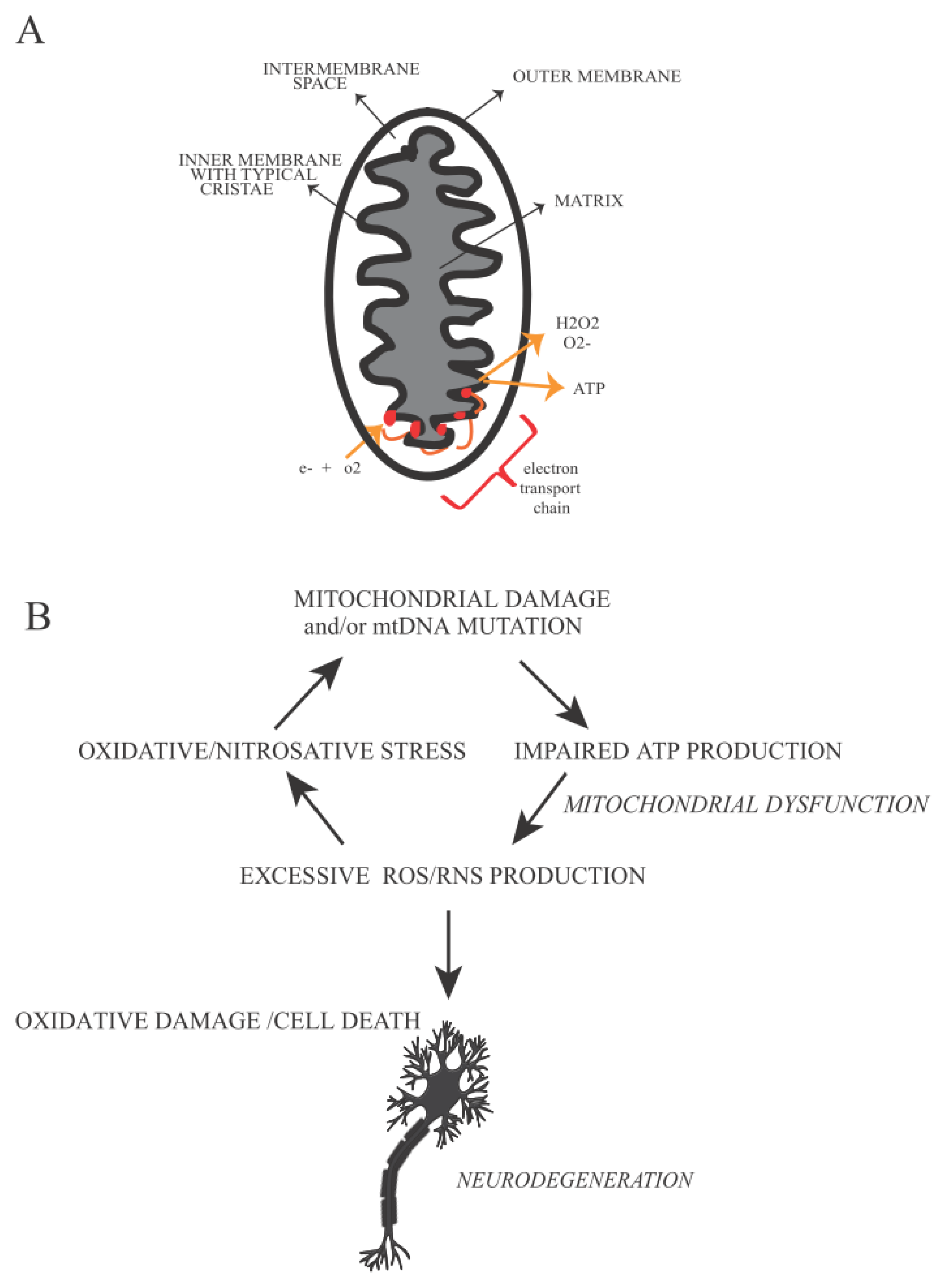
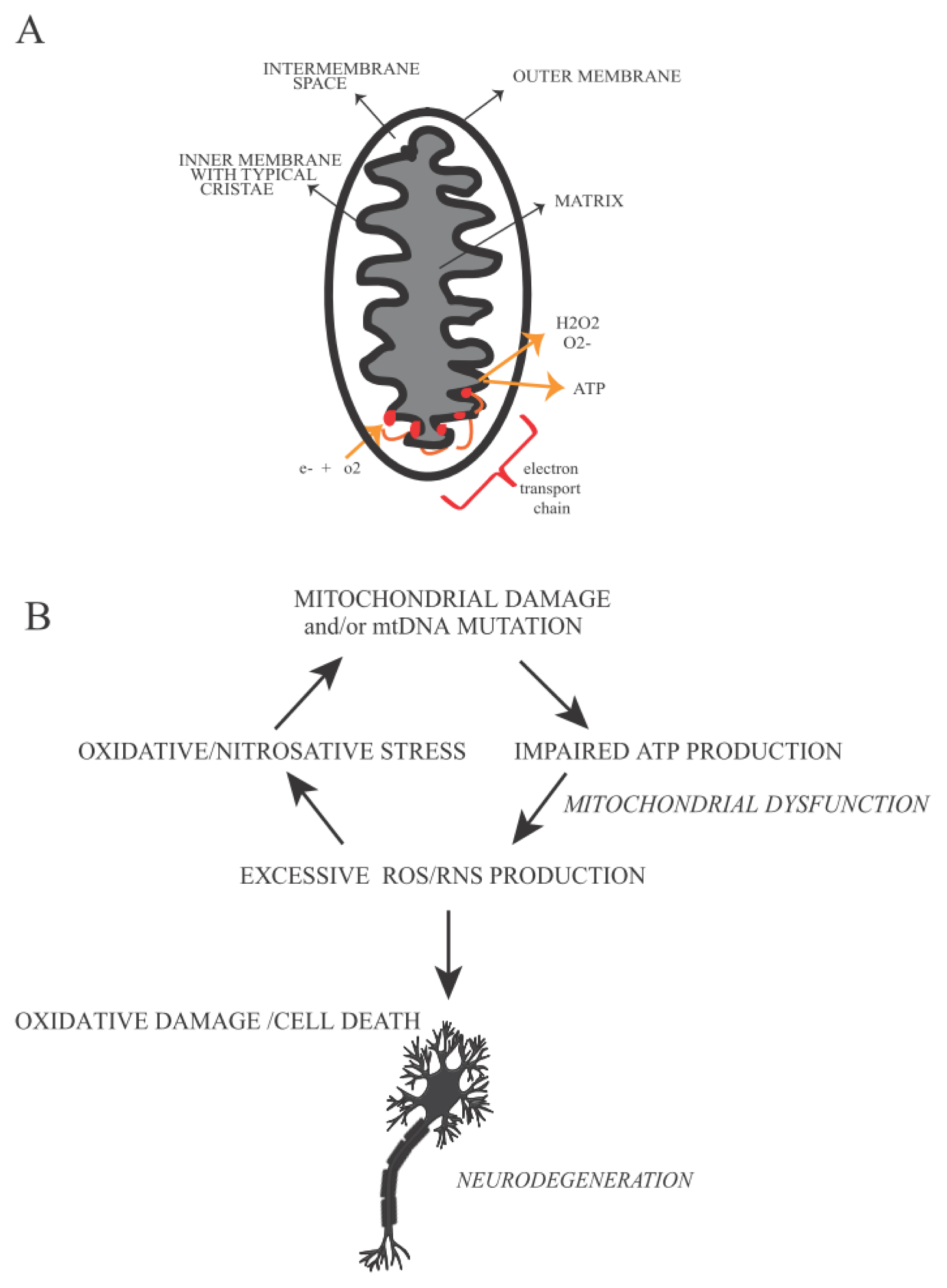
5. Mitochondrial Defects and Oxidative Stress
6. Parkinson’s Disease
6.1. Progression and Typical Symptoms
6.2. Oxidative Stress and Mitochondrial Mutations/Dysfunction in Parkinson Disease
7. Conclusions
Acknowledgements
Abbreviations
| AD | Alzheimer’s Disease |
| BER | Base Excision Repair |
| c-Abl | Abelson tyrosine kinase |
| CDK5 | Cyclin-Dependent kinase 5 |
| DSB | Double Strand Break |
| HR | Homologous Recombination |
| JNK | c-Jun N-terminal kinase |
| LBs | Lewy Bodies |
| LNs | Lewy Neurites |
| mtDNA | mitochondrial DNA |
| nDNA | nuclear DNA |
| NER | Nucleotide Excision Repair |
| NHEJ | Non Homologous End Joins |
| PD | Parkinson’s Disease |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxidative Species |
| SN | Substantia Nigra |
| SSB | Single Strand Break |
| α-syn | α-synuclein |
Conflict of Interest
References
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol 2011, 94, 166–200. [Google Scholar]
- Hegde, M.L.; Mantha, A.K.; Hazra, T.K.; Bhakat, K.K.; Mitra, S.; Szczesny, B. Oxidative genome damage and its repair: Implications in aging and neurodegenerative diseases. Mech. Ageing Dev 2012, 133, 157–168. [Google Scholar]
- Bohgaki, T.; Bohgaki, M.; Hakem, R. DNA double-strand break signaling and human disorders. Genome Integr 2010, 1, 15. [Google Scholar]
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2012. [Google Scholar] [CrossRef]
- Freitas, A.A.; de Magalhaes, J.P. A review and appraisal of the DNA damage theory of ageing. Mut. Res 2011, 728, 12–22. [Google Scholar]
- Polidori, M.C.; Griffiths, H.R.; Mariani, E.; Mecocci, P. Hallmarks of protein oxidative damage in neurodegenerative diseases: Focus on Alzheimer’s disease. Amino Acids 2007, 32, 553–559. [Google Scholar]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2012. [Google Scholar] [CrossRef]
- Hsieh, R.H.; Hou, J.H.; Hsu, H.S.; Wei, Y.H. Age-dependent respiratory function decline and DNA deletions in human muscle mitochondria. Biochem. Mol. Biol. Int 1994, 32, 1009–1022. [Google Scholar]
- Lezza, A.M.; Boffoli, D.; Scacco, S.; Cantatore, P.; Gadaleta, M.N. Correlation between mitochondrial DNA 4977-bp deletion and respiratory chain enzyme activities in aging human skeletal muscles. Biochem. Biophys. Res. Commun 1994, 205, 772–779. [Google Scholar]
- Lax, N.Z.; Turnbull, D.M.; Reeve, A.K. Mitochondrial mutations: Newly discovered players in neuronal degeneration. Neuroscientist 2011, 17, 645–658. [Google Scholar]
- Lee, H.C.; Pang, C.Y.; Hsu, H.S.; Wei, Y.H. Differential accumulations of 4,977 bp deletion in mitochondrial DNA of various tissues in human ageing. Biochim. Biophys. Acta 1994, 1226, 37–43. [Google Scholar]
- Hayashi, M.; Miyata, R.; Tanuma, N. Oxidative stress in developmental brain disorders. Adv. Exp. Med. Biol 2012, 724, 278–290. [Google Scholar]
- Mecocci, P. Oxidative stress in mild cognitive impairment and Alzheimer disease: A continuum. J. Alzheimer’s Dis 2004, 6, 159–163. [Google Scholar]
- Schlatterer, S.D.; Acker, C.M.; Davies, P. c-Abl in neurodegenerative disease. J. Mol. Neurosci 2011, 45, 445–452. [Google Scholar]
- Gonfloni, S. DNA damage stress response in germ cells: Role of c-Abl and clinical implications. Oncogene 2010, 29, 6193–6202. [Google Scholar]
- Colicelli, J. ABL tyrosine kinases: Evolution of function, regulation, and specificity. Sci. Signal. 2010, 3, re6. [Google Scholar]
- Shaul, Y.; Ben-Yehoyada, M. Role of c-Abl in the DNA damage stress response. Cell Res 2005, 15, 33–35. [Google Scholar]
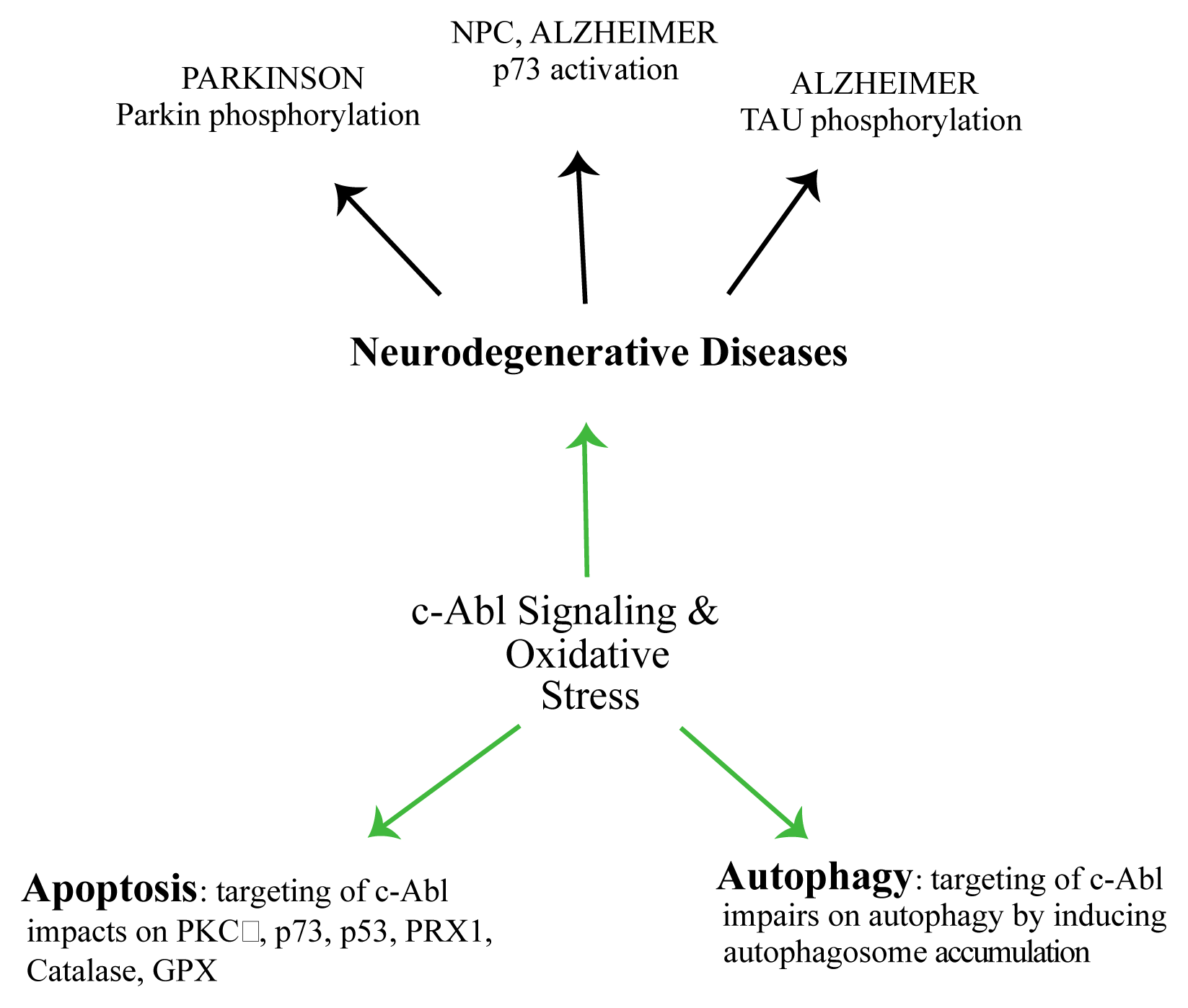
- Gonfloni, S.; Maiani, E.; Di Bartolomeo, C.; Diederich, M.; Cesareni, G. Oxidative stress, DNA damage, and c-Abl signaling: At the crossroad in neurodegenerative diseases? Int. J. Cell Biol 2012, 2012, 683097. [Google Scholar]
- Schlatterer, S.D.; Tremblay, M.A.; Acker, C.M.; Davies, P. Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. J. Alzheimer’s Dis 2011, 25, 119–133. [Google Scholar]
- Devine, M.J.; Plun-Favreau, H.; Wood, N.W. Parkinson’s disease and cancer: Two wars, one front. Nat. Rev. Cancer 2011, 11, 812–823. [Google Scholar]
- Nakamura, J.; Swenberg, J.A. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999, 59, 2522–2526. [Google Scholar]
- Friedberg, E.C.; Aguilera, A.; Gellert, M.; Hanawalt, P.C.; Hays, J.B.; Lehmann, A.R.; Lindahl, T.; Lowndes, N.; Sarasin, A.; Wood, R.D. DNA repair: From molecular mechanism to human disease. DNA Repair 2006, 5, 986–996. [Google Scholar]
- Liu, Y.; Wilson, S.H. DNA base excision repair: A mechanism of trinucleotide repeat expansion. Trends Biochem. Sci 2012, 37, 162–172. [Google Scholar]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet 2008, 9, 619–631. [Google Scholar]
- Cahill, D.; Connor, B.; Carney, J.P. Mechanisms of eukaryotic DNA double strand break repair. Front. Biosci 2006, 11, 1958–1976. [Google Scholar]
- Engels, W.R.; Johnson-Schlitz, D.; Flores, C.; White, L.; Preston, C.R. A third link connecting aging with double strand break repair. Cell Cycle 2007, 6, 131–135. [Google Scholar]
- Chen, B.P.; Li, M.; Asaithamby, A. New insights into the roles of ATM and DNA-PKcs in the cellular response to oxidative stress. Cancer Lett 2012, 327, 103–110. [Google Scholar]
- Korwek, Z.; Sewastianik, T.; Bielak-Zmijewska, A.; Mosieniak, G.; Alster, O.; Moreno-Villaneuva, M.; Burkle, A.; Sikora, E. Inhibition of ATM blocks the etoposide-induced DNA damage response and apoptosis of resting human T cells. DNA Repair 2012, 11, 864–873. [Google Scholar]
- Aziz, K.; Nowsheen, S.; Pantelias, G.; Iliakis, G.; Gorgoulis, V.G.; Georgakilas, A.G. Targeting DNA damage and repair: Embracing the pharmacological era for successful cancer therapy. Pharmacol. Ther 2012, 133, 334–350. [Google Scholar]
- Vyjayanti, V.N.; Rao, K.S. DNA double strand break repair in brain: Reduced NHEJ activity in aging rat neurons. Neurosci. Lett 2006, 393, 18–22. [Google Scholar]
- Niedernhofer, L.J. Tissue-specific accelerated aging in nucleotide excision repair deficiency. Mechanisms of ageing and development. Mech. Ageing Dev 2008, 129, 408–415. [Google Scholar]
- Fousteri, M.; Mullenders, L.H. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res 2008, 18, 73–84. [Google Scholar]
- Shuck, S.C.; Short, E.A.; Turchi, J.J. Eukaryotic nucleotide excision repair: From understanding mechanisms to influencing biology. Cell Res 2008, 18, 64–72. [Google Scholar]
- Fishel, M.L.; Vasko, M.R.; Kelley, M.R. DNA repair in neurons: So if they don’t divide what’s to repair? Mut. Res 2007, 614, 24–36. [Google Scholar]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol 2006, 7, 335–346. [Google Scholar]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Ann. Rev. Biochem. 2005, 74, 681–710. [Google Scholar]
- Hong, Z.; Jiang, J.; Hashiguchi, K.; Hoshi, M.; Lan, L.; Yasui, A. Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J. Cell Sci 2008, 121, 3146–3154. [Google Scholar]
- Conde-Perezprina, J.C.; Leon-Galvan, M.A.; Konigsberg, M. DNA mismatch repair system: Repercussions in cellular homeostasis and relationship with aging. Oxid. Med. Cell. Longevity 2012, 2012, 728430. [Google Scholar]
- Lujan, S.A.; Williams, J.S.; Pursell, Z.F.; Abdulovic-Cui, A.A.; Clark, A.B.; Nick McElhinny, S.A.; Kunkel, T.A. Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genet 2012, 8, e1003016. [Google Scholar]
- Lenaz, G. The mitochondrial production of reactive oxygen species: Mechanisms and implications in human pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab 2012, 26, 711–723. [Google Scholar]
- Sorolla, M.A.; Rodriguez-Colman, M.J.; Vall-llaura, N.; Tamarit, J.; Ros, J.; Cabiscol, E. Protein oxidation in Huntington disease. Biofactors 2012, 38, 173–185. [Google Scholar]
- Zadori, D.; Klivenyi, P.; Szalardy, L.; Fulop, F.; Toldi, J.; Vecsei, L. Mitochondrial disturbances, excitotoxicity, neuroinflammation and kynurenines: Novel therapeutic strategies for neurodegenerative disorders. J. Neurol. Sci 2012, 322, 187–191. [Google Scholar]
- Gasparre, G.; Romeo, G.; Rugolo, M.; Porcelli, A.M. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim. Biophys. Acta 2011, 1807, 633–642. [Google Scholar]
- Karbownik-Lewi Ska, M.G.; St Pniak, J.; Lewi Ski, A. High level of oxidized nucleosides in thyroid mitochondrial DNA; damaging effects of Fenton reaction substrates. Thyroid Res 2012, 5, 24. [Google Scholar]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen 2010, 51, 440–450. [Google Scholar]
- Zhao, D.; Wang, Z.; Hong, D.; Zhang, W.; Yuan, Y. Chronic progressive external ophthalmoplegia coexistent with motor neuron disease in a patient with a novel large-scale mitochondrial DNA deletion. Clin. Neurol. Neurosurg. 2012. [Google Scholar] [CrossRef]
- Comte, C.; Tonin, Y.; Heckel-Mager, A.M.; Boucheham, A.; Smirnov, A.; Aure, K.; Lornbes, A.; Martin, R.P.; Entelis, N.; Tarassov, I. Mitochondrial targeting of recombinant RNAs modulates the level of a heteroplasmic mutation in human mitochondrial DNA associated with Kearns Sayre Syndrome. Nucleic Acids Res 2013, 41, 418–433. [Google Scholar]
- Caporali, L.; Ghelli, A.M.; Iommarini, L.; Maresca, A.; Valentino, M.L.; La Morgia, C.; Liguori, R.; Zanna, C.; Barboni, P.; De Nardo, V.; et al. Cybrid studies establish the causal link between the mtDNA m.3890G>A/MT-ND1 mutation and optic atrophy with bilateral brainstem lesions. Biochim. Biophys. Acta 2012, 1832, 445–452. [Google Scholar]
- Tschampa, H.J.; Urbach, H.; Greschus, S.; Kunz, W.S.; Kornblum, C. Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.3243A>G MTTL1 mutation. J. Neurol. 2012. [Google Scholar] [CrossRef]
- Lopez-Gallardo, E.; Solano, A.; Herrero-Martin, M.D.; Martinez-Romero, I.; Castano-Perez, M.D.; Andreu, A.L.; Herrera, A.; López-Pérez, M.J.; Ruiz-Pesini, E.; Montoya, J. NARP syndrome in a patient harbouring an insertion in the MT-ATP6 gene that results in a truncated protein. J. Med. Genet 2009, 46, 64–67. [Google Scholar]
- Goto, Y.; Nonaka, I.; Horai, S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990, 61, 931–937. [Google Scholar]
- Monden, Y.; Mori, M.; Kuwajima, M.; Goto, T.; Yamagata, T.; Momoi, M.Y. Late-onset Leigh syndrome with myoclonic epilepsy with ragged-red fibers. Brain Dev. 2012. [Google Scholar] [CrossRef]
- Barja, G.; Herrero, A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J 2000, 14, 312–318. [Google Scholar]
- Mecocci, P.; MacGarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, D.C.; Beal, M.F. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol 1993, 34, 609–616. [Google Scholar]
- Wei, Y.H.; Lee, H.C. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp. Biol. Med. (Maywood) 2002, 227, 671–682. [Google Scholar]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar]
- Albers, D.S.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J. Neural Transm. Suppl 2000, 59, 133–154. [Google Scholar]
- Lee, C.K.; Weindruch, R.; Prolla, T.A. Gene-expression profile of the ageing brain in mice. Nat. Genet 2000, 25, 294–297. [Google Scholar]
- Greaves, L.C.; Elson, J.L.; Nooteboom, M.; Grady, J.P.; Taylor, G.A.; Taylor, R.W.; Mathers, J.C.; Kirkwood, T.B.; Turnbull, D.M. Comparison of mitochondrial mutation spectra in ageing human colonic epithelium and disease: Absence of evidence for purifying selection in somatic mitochondrial DNA point mutations. PLoS Genet 2012, 8, e1003082. [Google Scholar]
- Lee, H.C.; Chang, C.M.; Chi, C.W. Somatic mutations of mitochondrial DNA in aging and cancer progression. Ageing Res. Rev 2010, 9, S47–S58. [Google Scholar]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2012, 20, 31–42. [Google Scholar]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009, 8, 704–719. [Google Scholar]
- Gredilla, R. DNA damage and base excision repair in mitochondria and their role in aging. J. Aging Res 2010, 2011, 257093. [Google Scholar]
- Lauritzen, K.H.; Moldestad, O.; Eide, L.; Carlsen, H.; Nesse, G.; Storm, J.F.; Mansuy, I.M.; Bergersen, L.H.; Klungland, A. Mitochondrial DNA toxicity in forebrain neurons causes apoptosis, neurodegeneration, and impaired behavior. Mol. Cell. Biol 2010, 30, 1357–1367. [Google Scholar]
- Mitra, S.; Izumi, T.; Boldogh, I.; Bhakat, K.K.; Chattopadhyay, R.; Szczesny, B. Intracellular trafficking and regulation of mammalian AP-endonuclease 1 (APE1), an essential DNA repair protein. DNA Repair 2007, 6, 461–469. [Google Scholar]
- Martin, L.J. Biology of mitochondria in neurodegenerative diseases. Prog. Mol. Biol. Transl. Sci 2012, 107, 355–415. [Google Scholar]
- Larsen, N.B.; Rasmussen, M.; Rasmussen, L.J. Nuclear and mitochondrial DNA repair: Similar pathways? Mitochondrion 2005, 5, 89–108. [Google Scholar]
- Gredilla, R.; Bohr, V.A.; Stevnsner, T. Mitochondrial DNA repair and association with aging—An update. Exp. Gerontol 2010, 45, 478–488. [Google Scholar]
- Chakrabarti, S.; Munshi, S.; Banerjee, K.; Thakurta, I.G.; Sinha, M.; Bagh, M.B. Mitochondrial dysfunction during brain aging: Role of oxidative stress and modulation by antioxidant supplementation. Aging Dis 2011, 2, 242–256. [Google Scholar]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci 2012, 322, 254–262. [Google Scholar]
- Filosto, M.; Scarpelli, M.; Cotelli, M.S.; Vielmi, V.; Todeschini, A.; Gregorelli, V.; Tonin, P.; Tomelleri, G.; Padovani, A. The role of mitochondria in neurodegenerative diseases. J. Neurol 2011, 258, 1763–1774. [Google Scholar]
- Loeb, L.A.; Wallace, D.C.; Martin, G.M. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 18769–18770. [Google Scholar]
- Nicholls, D.G. Oxidative stress and energy crises in neuronal dysfunction. Ann. N. Y. Acad. Sci 2008, 1147, 53–60. [Google Scholar]
- Lee, H.C.; Wei, Y.H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol 2005, 37, 822–834. [Google Scholar]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev 2008, 88, 611–638. [Google Scholar]
- Cheng, X.; Siow, R.C.; Mann, G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: A role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid. Redox Signal 2011, 14, 469–487. [Google Scholar]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Ann. Rev. Pharmacol. Toxicol 2007, 47, 89–116. [Google Scholar]
- Niture, S.K.; Kaspar, J.W.; Shen, J.; Jaiswal, A.K. Nrf2 signaling and cell survival. Toxicol. Appl. Pharmacol 2010, 244, 37–42. [Google Scholar]
- Ozanne, B.; Wheeler, T.; Zack, J.; Smith, G.; Dale, B. Transforming gene of a human leukaemia cell is unrelated to the expressed tumour virus related gene of the cell. Nature 1982, 299, 744–747. [Google Scholar]
- Maiani, E.; Diederich, M.; Gonfloni, S. DNA damage response: The emerging role of c-Abl as a regulatory switch? Biochem. Pharmacol 2011, 82, 1269–1266. [Google Scholar]
- Herrup, K. Reimagining Alzheimer’s disease—An age-based hypothesis. J. Neurosci 2010, 30, 16755–16762. [Google Scholar]
- Jing, Z.; Caltagarone, J.; Bowser, R. Altered subcellular distribution of c-Abl in Alzheimer’s disease. J. Alzheimer’s Dis 2009, 17, 409–422. [Google Scholar]
- Kharbanda, S.; Ren, R.; Pandey, P.; Shafman, T.D.; Feller, S.M.; Weichselbaum, R.R.; Kufe, D.W. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature 1995, 376, 785–788. [Google Scholar]
- Cancino, G.I.; Toledo, E.M.; Leal, N.R.; Hernandez, D.E.; Yévenes, L.F.; Inestrosa, N.C.; Alvarez, A.R. STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer’s beta-amyloid deposits. Brain 2008, 131, 2425–2442. [Google Scholar]
- Ko, H.S.; Lee, Y.; Shin, J.H.; Karuppagounder, S.S.; Gadad, B.S.; Koleske, A.J.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc. Natl Acad. Sci. USA 2010, 107, 16691–16696. [Google Scholar]
- Cancino, G.I.; Perez de Arce, K.; Castro, P.U.; Toledo, E.M.; von Bernhardi, R.; Alvarez, A.R. c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol. Aging 2011, 32, 1249–1261. [Google Scholar]
- Cacciatore, I.; Baldassarre, L.; Fornasari, E.; Mollica, A.; Pinnen, F. Recent advances in the treatment of neurodegenerative diseases based on GSH delivery systems. Oxid. Med. Cell. Longevity 2012, 2012, 240146. [Google Scholar]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev 1997, 25, 335–358. [Google Scholar]
- Garrido, M.; Tereshchenko, Y.; Zhevtsova, Z.; Taschenberger, G.; Bahr, M.; Kugler, S. Glutathione depletion and overproduction both initiate degeneration of nigral dopaminergic neurons. Acta Neuropathol 2011, 121, 475–485. [Google Scholar]
- Drake, J.; Kanski, J.; Varadarajan, S.; Tsoras, M.; Butterfield, D.A. Elevation of brain glutathione by gamma-glutamylcysteine ethyl ester protects against peroxynitrite-induced oxidative stress. J. Neurosci. Res 2002, 68, 776–784. [Google Scholar]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Ann. Rev. pharmacol. Toxicol 2005, 45, 51–88. [Google Scholar]
- Wilce, M.C.; Parker, M.W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta 1994, 1205, 1–18. [Google Scholar]
- Nuccetelli, M.; Mazzetti, A.P.; Rossjohn, J.; Parker, M.W.; Board, P.; Caccuri, A.M.; Federici, G.; Ricci, G.; Lo Bello, M. Shifting substrate specificity of human glutathione transferase (from class Pi to class alpha) by a single point mutation. Biochem. Biophys. Res. Commun 1998, 252, 184–189. [Google Scholar]
- Fabrini, R.; de Luca, A.; Stella, L.; Mei, G.; Orioni, B.; Ciccone, S.; Federici, G.; Lo Bello, M.; Ricci, G. Monomer-dimer equilibrium in glutathione transferases: A critical re-examination. Biochemistry 2009, 48, 10473–10482. [Google Scholar]
- Chen, J.; Liou, A.; Zhang, L.; Weng, Z.; Gao, Y.; Cao, G.; Zigmond, M.J.; Chen, J. GST P1, a novel downstream regulator of LRRK2, G2019S-induced neuronal cell death. Front. Biosci 2012, 4, 2365–2377. [Google Scholar]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar]
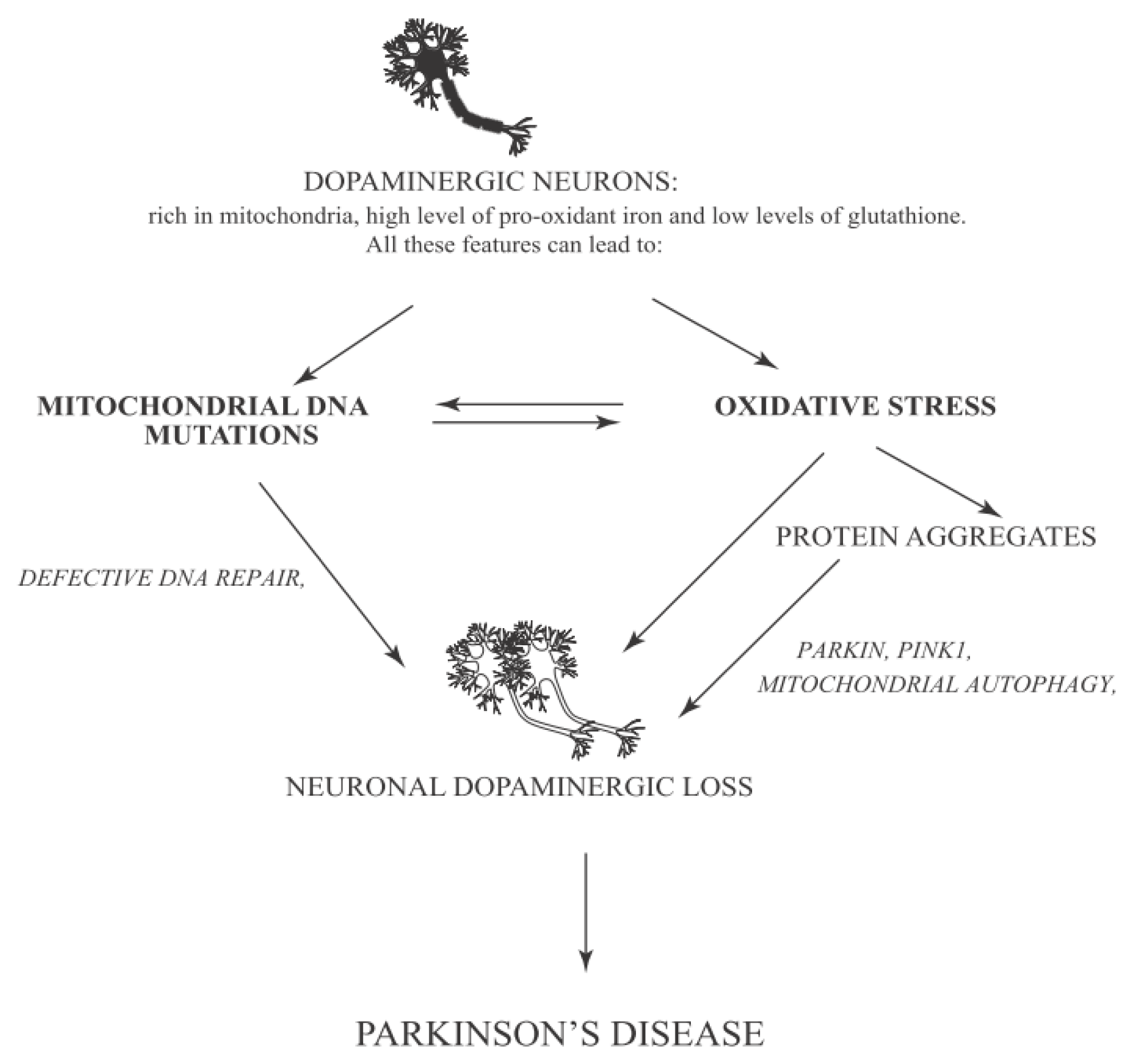
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Ann. N. Y. Acad. Sci 2008, 1147, 93–104. [Google Scholar]
- Chinta, S.J.; Andersen, J.K. Redox imbalance in Parkinson’s disease. Biochim. Biophys. Acta 2008, 1780, 1362–1367. [Google Scholar]
- Hirsch, E.C.; Brandel, J.P.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: An X-ray microanalysis. J. Neurochem 1991, 56, 446–451. [Google Scholar]
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: Implications for Lewy-body formation in Parkinson disease. Nat. Med 2001, 7, 1144–1150. [Google Scholar]
- Chung, K.K.; Thomas, B.; Li, X.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, V.L.; Dawson, T.M. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 2004, 304, 1328–1331. [Google Scholar]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Ann. Rev. Neurosci 2005, 28, 57–87. [Google Scholar]
- Blalock, E.M.; Chen, K.C.; Sharrow, K.; Herman, J.P.; Porter, N.M.; Foster, T.C.; Landfield, P.W. Gene microarrays in hippocampal aging: Statistical profiling identifies novel processes correlated with cognitive impairment. J. Neurosci 2003, 23, 3807–3819. [Google Scholar]
- Shulman, L.M.; Singer, C.; Bean, J.A.; Weiner, W.J. Internal tremor in patients with Parkinson’s disease. Mov. Disord 1996, 11, 3–7. [Google Scholar]
- Villarejo, A.; Camacho, A.; García-Ramos, R.; Moreno, T.; Penas, M.; Juntas, R.; Ruiz, J. Cholinergic-dopaminergic imbalance in Pisa syndrome. Clin. Neuropharmacol 2003, 26, 119–121. [Google Scholar]
- Berardelli, A.; Rothwell, J.C.; Thompson, P.D.; Hallett, M. Pathophysiology of bradykinesia in Parkinson’s disease. Brain 2001, 124, 2131–2146. [Google Scholar]
- Stefani, A.; Lozano, A.M.; Peppe, A.; Stanzione, P.; Galati, S.; Tropepi, D.; Pierantozzi, M.; Brusa, L.; Scarnati, E.; Mazzone, P. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson’s disease. Brain 2007, 130, 1596–1607. [Google Scholar]
- Zesiewicz, T.A.; Sullivan, K.L.; Hauser, R.A. Nonmotor symptoms of Parkinson’s disease. Expert Rev. Neurother 2006, 6, 1811–1822. [Google Scholar]
- Zhu, M.; Li, W.; Lu, C. Role of alpha-synuclein protein levels in mitochondrial morphology and cell survival in cell lines. PLoS One 2012, 7, e36377. [Google Scholar]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar]
- Elkon, H.; Don, J.; Melamed, E.; Ziv, I.; Shirvan, A.; Offen, D. Mutant and wild-type alpha-synuclein interact with mitochondrial cytochrome C oxidase. J. Mol. Neurosci 2002, 18, 229–238. [Google Scholar]
- Braak, H.; Braak, E. Pathoanatomy of Parkinson’s disease. J. Neurol. 2000, 247, II3–10. [Google Scholar]
- Arduino, D.M.; Esteves, A.R.; Cardoso, S.M. Mitochondria drive autophagy pathology via microtubule disassembly: A new hypothesis for Parkinson disease. Autophagy 2012, 9, 112–114. [Google Scholar]
- Pienaar, I.S.; Chinnery, P.F. Existing and emerging mitochondrial-targeting therapies for altering Parkinson’s disease severity and progression. Pharmacol. Ther. 2012. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 2012, 13, 378–385. [Google Scholar]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.S.; Li, L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol 2007, 5, e172. [Google Scholar]
- Heeman, B.; Van den Haute, C.; Aelvoet, S.A.; Valsecchi, F.; Rodenburg, R.J.; Reumers, V.; Debyser, Z.; Callewaert, G.; Koopman, W.J.; Willems, P.H.; et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell Sci. 2011, 124, 1115–1125. [Google Scholar]
- Zhou, C.; Huang, Y.; Shao, Y.; May, J.; Prou, D.; Perier, C.; Dauer, W.; Schon, E.A.; Przedborski, S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc. Natl. Acad. Sci. USA 2008, 105, 12022–1207. [Google Scholar]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.Y.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; Deas, E.; et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS One 2008, 3, e2455. [Google Scholar]
- Marongiu, R.; Spencer, B.; Crews, L.; Adame, A.; Patrick, C.; Trejo, M.; Dallapiccola, B.; Valente, E.M.; Masliah, E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J. Neurochem 2009, 108, 1561–1574. [Google Scholar]
- Shimura, H.; Hattori, N.; Kubo, S.i.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet 2000, 25, 302–305. [Google Scholar]
- Shimura, H.; Hattori, N.; Kubo, S.; Yoshikawa, M.; Kitada, T.; Matsumine, H.; Asakawa, S.; Minoshima, S.; Yamamura, Y.; Shimizu, N.; et al. Immunohistochemical and subcellular localization of Parkin protein: Absence of protein in autosomal recessive juvenile parkinsonism patients. Ann. Neurol. 1999, 45, 668–672. [Google Scholar]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010, 8, e1000298. [Google Scholar]
- Jenner, P.; Olanow, C.W. Understanding cell death in Parkinson’s disease. Ann. Neurol 1998, 44, S72–S84. [Google Scholar]
- Pickrell, A.M.; Pinto, M.; Hida, A.; Moraes, C.T. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci 2011, 31, 17649–17658. [Google Scholar]
- Grespi, F.; Melino, G. P73 and age-related diseases: Is there any link with Parkinson Disease? Aging. 2012. Available online: http://www.impactaging.com/papers/v4/n12/full/100515.html (accessed on 5 December 2012).
- Imam, S.Z.; Zhou, Q.; Yamamoto, A.; Valente, A.J.; Ali, S.F.; Bains, M.; Roberts, J.L.; Kahle, P.J.; Clark, R.A.; Li, S. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: Implications for Parkinson’s disease. J. Neurosci 2011, 31, 157–163. [Google Scholar]
- Xiao, L.; Chen, D.; Hu, P.; Wu, J.; Liu, W.; Zhao, Y.; Cao, M.; Fang, Y.; Bi, W.; Zheng, Z.; et al. The c-Abl-MST1 signaling pathway mediates oxidative stress-induced neuronal cell death. J. Neurosci 2011, 31, 9611–9619. [Google Scholar]
- Zhang, Y.; Dawson, V.L.; Dawson, T.M. Oxidative stress and genetics in the pathogenesis of Parkinson’s disease. Neurobiol. Dis 2000, 7, 240–250. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ciccone, S.; Maiani, E.; Bellusci, G.; Diederich, M.; Gonfloni, S. Parkinson’s Disease: A Complex Interplay of Mitochondrial DNA Alterations and Oxidative Stress. Int. J. Mol. Sci. 2013, 14, 2388-2409. https://doi.org/10.3390/ijms14022388
Ciccone S, Maiani E, Bellusci G, Diederich M, Gonfloni S. Parkinson’s Disease: A Complex Interplay of Mitochondrial DNA Alterations and Oxidative Stress. International Journal of Molecular Sciences. 2013; 14(2):2388-2409. https://doi.org/10.3390/ijms14022388
Chicago/Turabian StyleCiccone, Sarah, Emiliano Maiani, Giovanna Bellusci, Marc Diederich, and Stefania Gonfloni. 2013. "Parkinson’s Disease: A Complex Interplay of Mitochondrial DNA Alterations and Oxidative Stress" International Journal of Molecular Sciences 14, no. 2: 2388-2409. https://doi.org/10.3390/ijms14022388