βArrestins in Cardiac G Protein-Coupled Receptor Signaling and Function: Partners in Crime or “Good Cop, Bad Cop”?


{kind=link}
Abstract
:1. General Considerations of βarrs
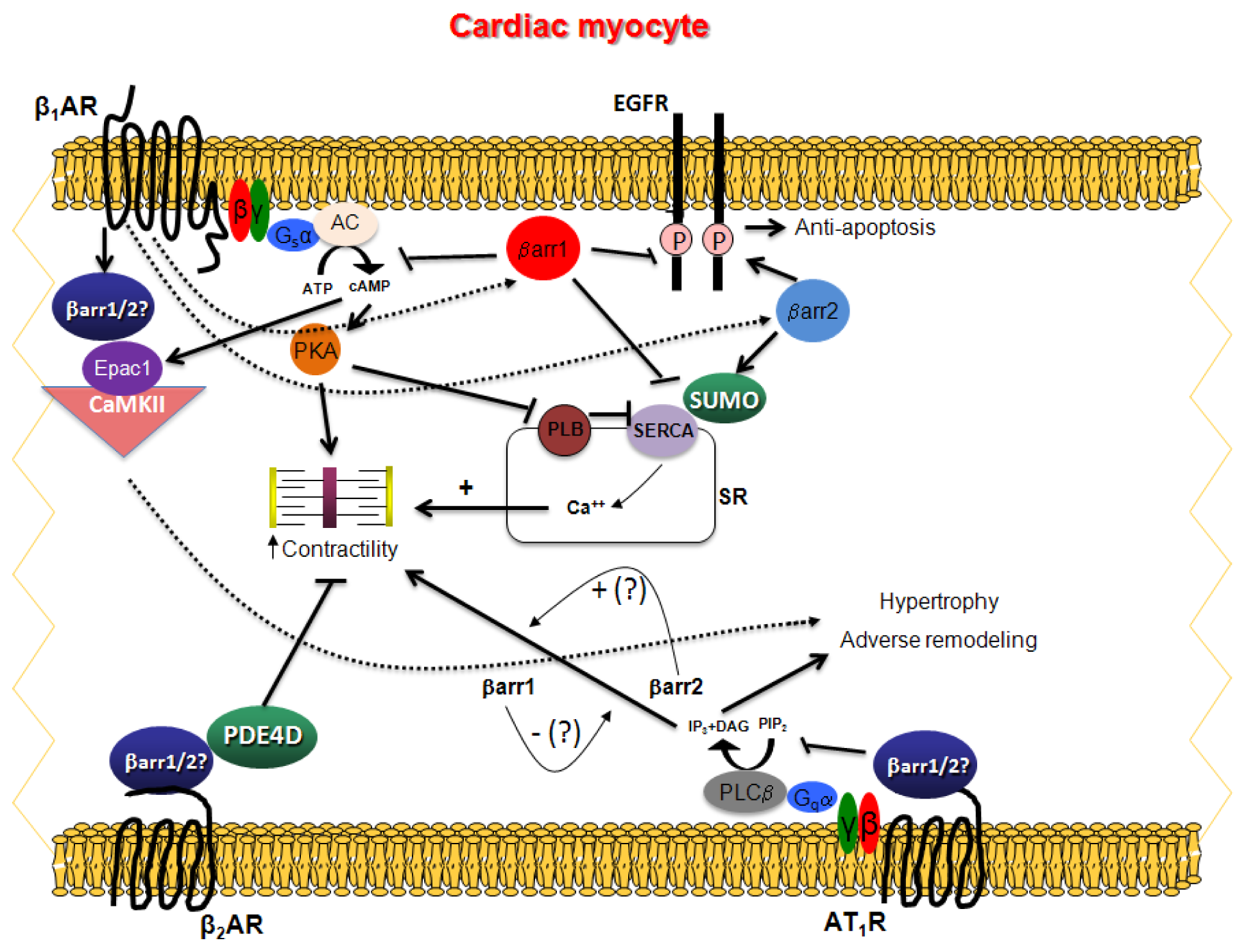
2. Cardiac β1AR Signaling and βarrs
3. Cardiac β2AR Signaling and βarrs
4. Cardiac AT1R Signaling and βarrs
5. Other Cardiac GPCRs and βarrs
6. Unanswered Questions on Cardiac βarrs
7. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Lymperopoulos, A.; Bathgate, A. Arrestins in the cardiovascular system. Prog. Mol. Biol. Transl. Sci 2013, 118, 297–334. [Google Scholar]
- Kenakin, T. Making receptors a reality: The 2012 Nobel Prize in Chemistry. Trends Pharmacol. Sci 2013, 34, 2–5. [Google Scholar]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res 2013, 113, 739–753. [Google Scholar]
- Siryk-Bathgate, A.; Dabul, S.; Lymperopoulos, A. Current and future G protein-coupled receptor signaling targets for heart failure therapy. Drug Des. Dev. Ther 2013, 7, 1209–1222. [Google Scholar]
- Lymperopoulos, A. Physiology and pharmacology of the cardiovascular adrenergic system. Front. Physiol 2013, 4, 240. [Google Scholar]
- Lymperopoulos, A. β-arrestin biased agonism/antagonism at cardiovascular seven transmembrane-spanning receptors. Curr. Pharm. Des 2012, 18, 192–198. [Google Scholar]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. GRK2 inhibition in heart failure: Something old, something new. Curr. Pharm. Des 2012, 18, 186–191. [Google Scholar]
- Gurevich, V.V.; Gurevich, E.V. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol. Ther 2006, 110, 465–502. [Google Scholar]
- Shukla, A.K.; Violin, J.D.; Whalen, E.J.; Gesty-Palmer, D.; Shenoy, S.K.; Lefkowitz, R.J. Distinct conformational changes in β-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 9988–9993. [Google Scholar]
- Gurevich, V.V.; Pals-Rylaarsdam, R.; Benovic, J.L.; Hosey, M.M.; Onorato, J.J. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J. Biol. Chem 1997, 272, 28849–28852. [Google Scholar]
- Luttrell, L.M.; Gesty-Palmer, D. Beyond desensitization: Physiological relevance of arrestin dependent signaling. Pharmacol. Rev 2010, 62, 305–330. [Google Scholar]
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev 2001, 53, 1–24. [Google Scholar]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Caron, M.G.; Barak, L.S. Differential affinities of visual arrestin, βarrestin1, and βarrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem 2000, 275, 17201–17210. [Google Scholar]
- Charest, P.G.; Terrillon, S.; Bouvier, M. Monitoring agonist-promoted conformational changes of β-arrestin in living cells by intramolecular BRET. EMBO Rep 2005, 6, 334–340. [Google Scholar] [Green Version]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91. [Google Scholar]
- Communal, C.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Opposing effects of β1- and β2-aadrenergic receptors on cardiac myocyte apoptosis: Role of a pertussis toxin-sensitive G protein. Circulation 1999, 100, 2210–2212. [Google Scholar]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The β2-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through Gidependent coupling to phosphatidylinositol 3-kinase. Circ. Res 2000, 87, 1172–1179. [Google Scholar]
- Zhu, W.Z.; Zheng, M.; Koch, W.J.; Lefkowitz, R.J.; Kobilka, B.K.; Xiao, R.P. Dual modulation of cell survival and cell death by β2-adrenergic signalling in adult mouse cardiomyocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 1607–1612. [Google Scholar]
- Dorn, G.W.; Tepe, N.M.; Lorenz, J.N.; Koch, W.J.; Liggett, S.B. Low- and high-level transgenic expression of β2-adrenergic receptors differentially affect cardiac hypertrophy and function in Gαq-overexpressing mice. Proc. Natl. Acad. Sci. USA 1999, 96, 6400–6405. [Google Scholar]
- Conner, D.A.; Mathier,, M.A.; Mortensen, R.M.; Christe, M.; Vatner, S.F.; Seidman, C.E.; Seidman, J.G. β-Arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to β-adrenergic stimulation. Circ. Res 1997, 81, 1021–1026. [Google Scholar]
- Xiang, Y.; Kobilka, B.K. Myocyte adrenoceptor signaling pathways. Science 2003, 300, 1530–1532. [Google Scholar]
- Bristow, M.R. β-adrenergic receptor blockade in chronic heart failure. Circulation 2000, 101, 558–569. [Google Scholar]
- Packer, M.; Bristow, M.R.; Cohn, J.N.; Colucci, W.S.; Fowler, M.B.; Gilbert, E.M.; Shusterman, N.H. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N. Engl. J. Med 1996, 334, 1349–1355. [Google Scholar]
- Noma, T.; Lemaire, A.; Naga Prasad, S.V.; Barki-Harrington, L.; Tilley, D.G.; Chen, J.; le Corvoisier, P.; Violin, J.D.; Wei, H.; Lefkowitz, R.J.; et al. β-arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Invest 2007, 117, 2445–2458. [Google Scholar]
- Tilley, D.G. G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ. Res 2011, 109, 217–230. [Google Scholar]
- Noor, N.; Patel, C.B.; Rockman, H.A. β-arrestin: A signaling molecule and potential therapeutic target for heart failure. J. Mol. Cell. Cardiol 2011, 51, 534–41. [Google Scholar]
- Bathgate-Siryk, A.; Dabul, S.; Pandya, K.; Walklett, K.; Rengo, G.; Cannavo, A.; de Lucia, C.; Liccardo, D.; Gao, E.; Leosco, D.; et al. Negative impact of β-arrestin-1 on post-myocardial infarction heart failure via cardiac and adrenal-dependent neurohormonal mechanisms. Hypertension 2013. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol 2008, 70, 23–49. [Google Scholar]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar]
- Lymperopoulos, A. Nova Southeastern University, Unpublished Observation; 2013. [Google Scholar]
- Mangmool, S.; Shukla, A.K.; Rockman, H.A. β-Arrestin-dependent activation of Ca2+/calmodulin kinase II after β1-adrenergic receptor stimulation. J. Cell Biol 2010, 189, 573–587. [Google Scholar]
- Anderson, M.E. CaMKII and a failing strategy for growth in heart. J. Clin. Invest 2009, 119, 1082–1085. [Google Scholar]
- Xiao, R.P.; Zhu, W.; Zheng, M.; Chakir, K.; Bond, R.; Lakatta, E.G.; Cheng, H. Subtype-specific β-adrenoceptor signaling pathways in the heart and their potential clinical implications. Trends Pharmacol. Sci 2004, 25, 358–365. [Google Scholar]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar]
- Witherow, D.S.; Garrison, T.R.; Miller, W.E.; Lefkowitz, R.J. β-Arrestin inhibits NF-κB activity by means of its interaction with the NF-κB inhibitor IkappaBalpha. Proc. Natl. Acad. Sci. USA 2004, 101, 8603–8607. [Google Scholar]
- Gao, H.; Sun, Y.; Wu, Y.; Luan, B.; Wang, Y.; Qu, B.; Pei, G. Identification of β-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol. Cell 2004, 14, 303–317. [Google Scholar]
- Richter, W.; Day, P.; Agrawal, R.; Bruss, M.D.; Granier, S.; Wang, Y.L.; Rasmussen, S.G.; Horner, K.; Wang, P.; Lei, T.; et al. Signaling from β1- and β2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J 2008, 27, 384–393. [Google Scholar]
- Xiang, Y.; Naro, F.; Zoudilova, M.; Jin, S.L.; Conti, M.; Kobilka, B. Phosphodiesterase 4D is required for β2-adrenoceptor subtype-specific signaling in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 909–914. [Google Scholar]
- Salazar, N.C.; Vallejos, X.; Siryk, A.; Rengo, G.; Cannavo, A.; Liccardo, D.; de Lucia, C.; Gao, E.; Leosco, D.; Koch, W.J.; et al. GRK2 blockade with βARKct is essential for cardiac β2-adrenergic receptor signaling towards increased contractility. Cell Commun. Signal 2013, 11, 64. [Google Scholar]
- Houslay, M.D.; Baillie, G.S.; Maurice, D.H. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: A molecular toolbox for generating compartmentalized cAMP signaling. Circ. Res 2007, 100, 950–966. [Google Scholar]
- Drake, M.T.; Violin, J.D.; Whalen, E.J.; Wisler, J.W.; Shenoy, S.K.; Lefkowitz, R.J. β-arrestin-biased agonism at the β2-adrenergic receptor. J. Biol. Chem 2008, 283, 5669–5676. [Google Scholar]
- Wisler, J.W.; DeWire, S.M.; Whalen, E.J.; Violin, J.D.; Drake, M.T.; Ahn, S.; Shenoy, S.K.; Lefkowitz, R.J. A unique mechanism of β-blocker action: Carvedilol stimulates β-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar]
- Billet, S.; Aguilar, F.; Baudry, C.; Clauser, E. Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney Int 2008, 74, 1379–1384. [Google Scholar]
- Balmforth, A.J.; Shepherd, F.H.; Warburton, P.; Ball, S.G. Evidence of an important and direct role for protein kinase C in agonist-induced phosphorylation leading to desensitization of the angiotensin AT1A receptor. Br. J. Pharmacol 1997, 122, 1469–1477. [Google Scholar]
- Olivares-Reyes, J.A.; Smith, R.D.; Hunyady, L.; Shah, B.H.; Catt, K.J. Agonist-induced signaling, desensitization, and internalization of a phosphorylation-deficient AT1A angiotensin receptor. J. Biol. Chem 2001, 276, 37761–37768. [Google Scholar]
- Thomas, W.G.; Thekkumkara, T.J.; Baker, K.M. Cardiac effects of AII. AT1A receptor signaling, desensitization, and internalization. Adv. Exp. Med. Biol 1996, 396, 59–69. [Google Scholar]
- Zhai, P.; Yamamoto, M.; Galeotti, J.; Liu, J.; Masurekar, M.; Thaisz, J.; Irie, K.; Holle, E.; Yu, X.; Kupershmidt, S.; et al. Cardiac-specific overexpression of AT1 receptor mutant lacking G αq/G α i coupling causes hypertrophy and bradycardia in transgenic mice. J. Clin. Invest 2005, 115, 3045–3056. [Google Scholar]
- Holloway, A.C.; Qian, H.; Pipolo, L.; Ziogas, J.; Miura, S.; Karnik, S.; Southwell, B.R.; Lew, M.J.; Thomas, W.G. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol. Pharmacol 2002, 61, 768–777. [Google Scholar]
- Rajagopal, K.; Whalen, E.J.; Violin, J.D.; Stiber, J.A.; Rosenberg, P.B.; Premont, R.T.; Coffman, T.M.; Rockman, H.A.; Lefkowitz, R.J. β-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 16284–16289. [Google Scholar]
- Mishra, S.; Ling, H.; Grimm, M.; Zhang, T.; Bers, D.M.; Brown, J.H. Cardiac hypertrophy and heart failure development through Gq and CaM kinase II signaling. J. Cardiovasc. Pharmacol 2010, 56, 598–603. [Google Scholar]
- Dorn, G.W. II Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Invest 2005, 115, 527–537. [Google Scholar]
- Kukkonen, J.P. Regulation of receptor-coupling to (multiple) G proteins. A challenge for basic research and drug discovery. Recept. Channels 2004, 10, 167–183. [Google Scholar]
- Zidar, D.A.; Violin, J.D.; Whalen, E.J.; Lefkowitz, R.J. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 9649–9654. [Google Scholar]
- Aplin, M.; Christensen, G.L.; Schneider, M.; Heydorn, A.; Gammeltoft, S.; Kjølbye, A.L.; Sheikh, S.P.; Hansen, J.L. Differential extracellular signal-regulated kinases 1 and 2 activation by the angiotensin type 1 receptor supports distinct phenotypes of cardiac myocytes. Basic Clin. Pharmacol. Toxicol 2007, 100, 296–301. [Google Scholar]
- Boerrigter, G.; Lark, M.W.; Whalen, E.J.; Soergel, D.G.; Violin, J.D.; Burnett, J.C., Jr. Cardiorenal actions of TRV120027, a novel β-arrestin-biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: A novel therapeutic strategy for acute heart failure. Circ. Heart Fail 2011, 4, 770–778. [Google Scholar]
- Rakesh, K.; Yoo, B.; Kim, I.M.; Salazar, N.; Kim, K.S.; Rockman, H.A. β-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci. Signal 2010, 3, ra46. [Google Scholar]
- Lymperopoulos, A.; Rengo, G.; Funakoshi, H.; Eckhart, A.D.; Koch, W.J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med 2007, 13, 315–323. [Google Scholar]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Soltys, S.; Koch, W.J. An adrenal β-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5825–5830. [Google Scholar]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Koch, W.J. Adrenal β-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J. Am. Coll. Cardiol 2011, 57, 356–365. [Google Scholar]
- Hasseldine, A.R.; Harper, E.A.; Black, J.W. Cardiac-specific overexpression of human β2 adrenoceptors in mice exposes coupling to both Gs and Gi proteins. Br. J. Pharmacol 2003, 138, 1358–1366. [Google Scholar]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. β-arrestins and cell signaling. Annu. Rev. Physiol 2007, 69, 483–510. [Google Scholar]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar]
- Kim, J.; Ahn, S.; Ren, X.R.; Whalen, E.J.; Reiter, E.; Wei, H.; Lefkowitz, R.J. Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 1442–1447. [Google Scholar]
- DeWire, S.M.; Violin, J.D. Biased ligands for better cardiovascular drugs: Dissecting G-protein-coupled receptor pharmacology. Circ. Res 2011, 109, 205–216. [Google Scholar]
- Violin, J.D.; Soergel, D.G.; Boerrigter, G.; Burnett, J.C., Jr.; Lark, M.W. GPCR biased ligands as novel heart failure therapeutics. Trends Cardiovasc. Med 2013, 23, 242–249. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lymperopoulos, A.; Negussie, S. βArrestins in Cardiac G Protein-Coupled Receptor Signaling and Function: Partners in Crime or “Good Cop, Bad Cop”? Int. J. Mol. Sci. 2013, 14, 24726-24741. https://doi.org/10.3390/ijms141224726
Lymperopoulos A, Negussie S. βArrestins in Cardiac G Protein-Coupled Receptor Signaling and Function: Partners in Crime or “Good Cop, Bad Cop”? International Journal of Molecular Sciences. 2013; 14(12):24726-24741. https://doi.org/10.3390/ijms141224726
Chicago/Turabian StyleLymperopoulos, Anastasios, and Shmuel Negussie. 2013. "βArrestins in Cardiac G Protein-Coupled Receptor Signaling and Function: Partners in Crime or “Good Cop, Bad Cop”?" International Journal of Molecular Sciences 14, no. 12: 24726-24741. https://doi.org/10.3390/ijms141224726