The Antidiabetic Drug Metformin Inhibits the Proliferation of Bladder Cancer Cells in Vitro and in Vivo

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
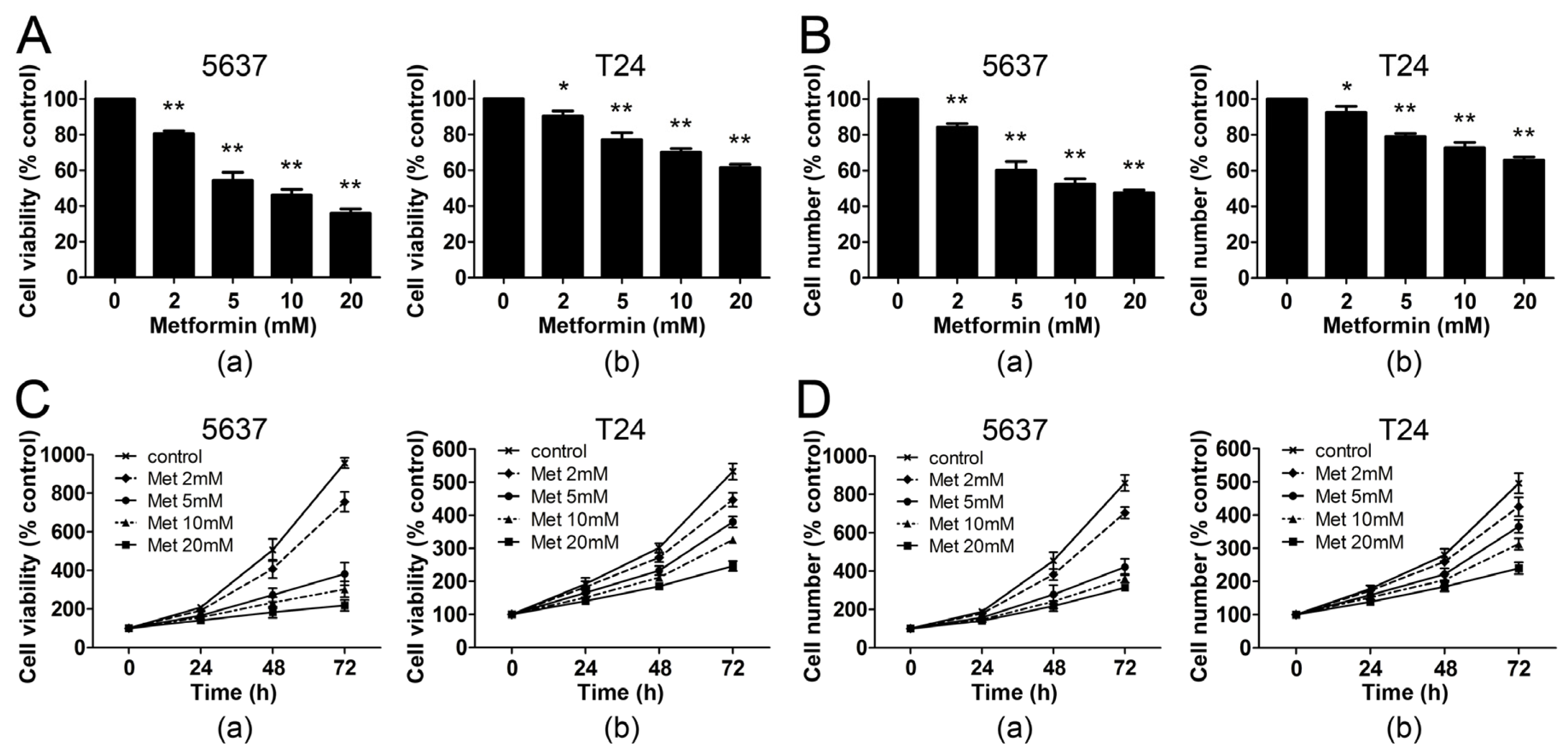
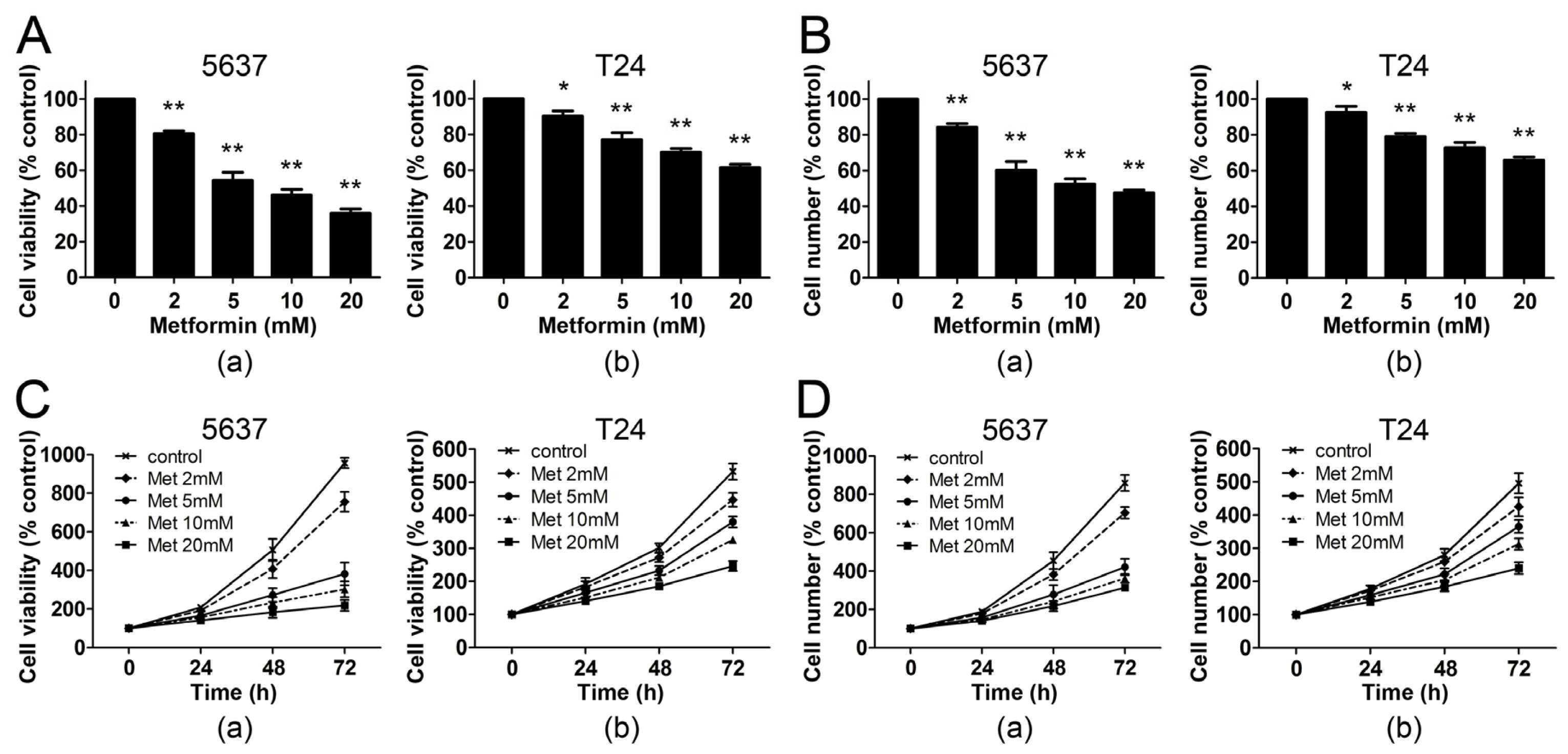
2.1. Metformin Inhibits the Proliferation of Human Bladder Cancer Cells
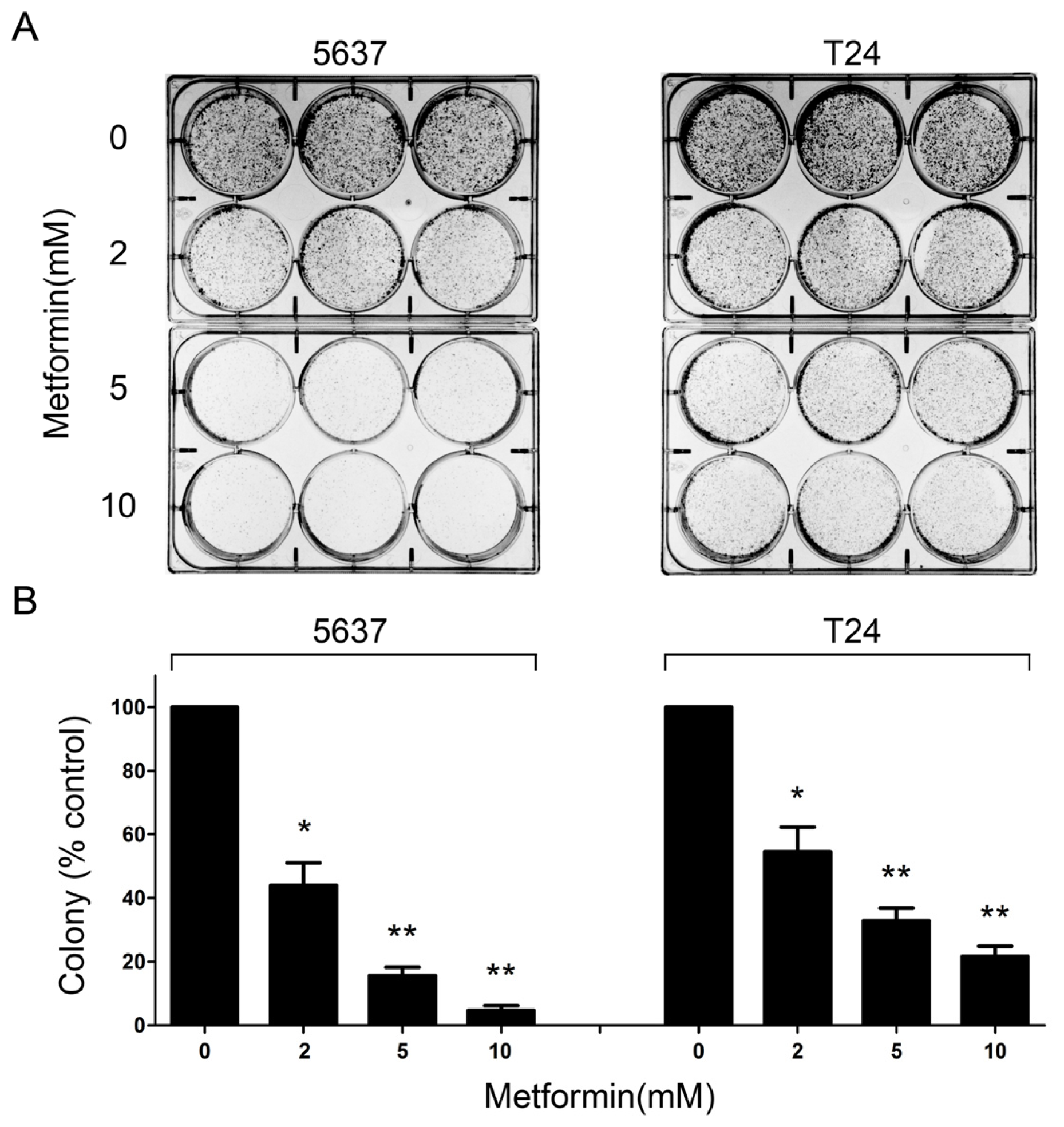
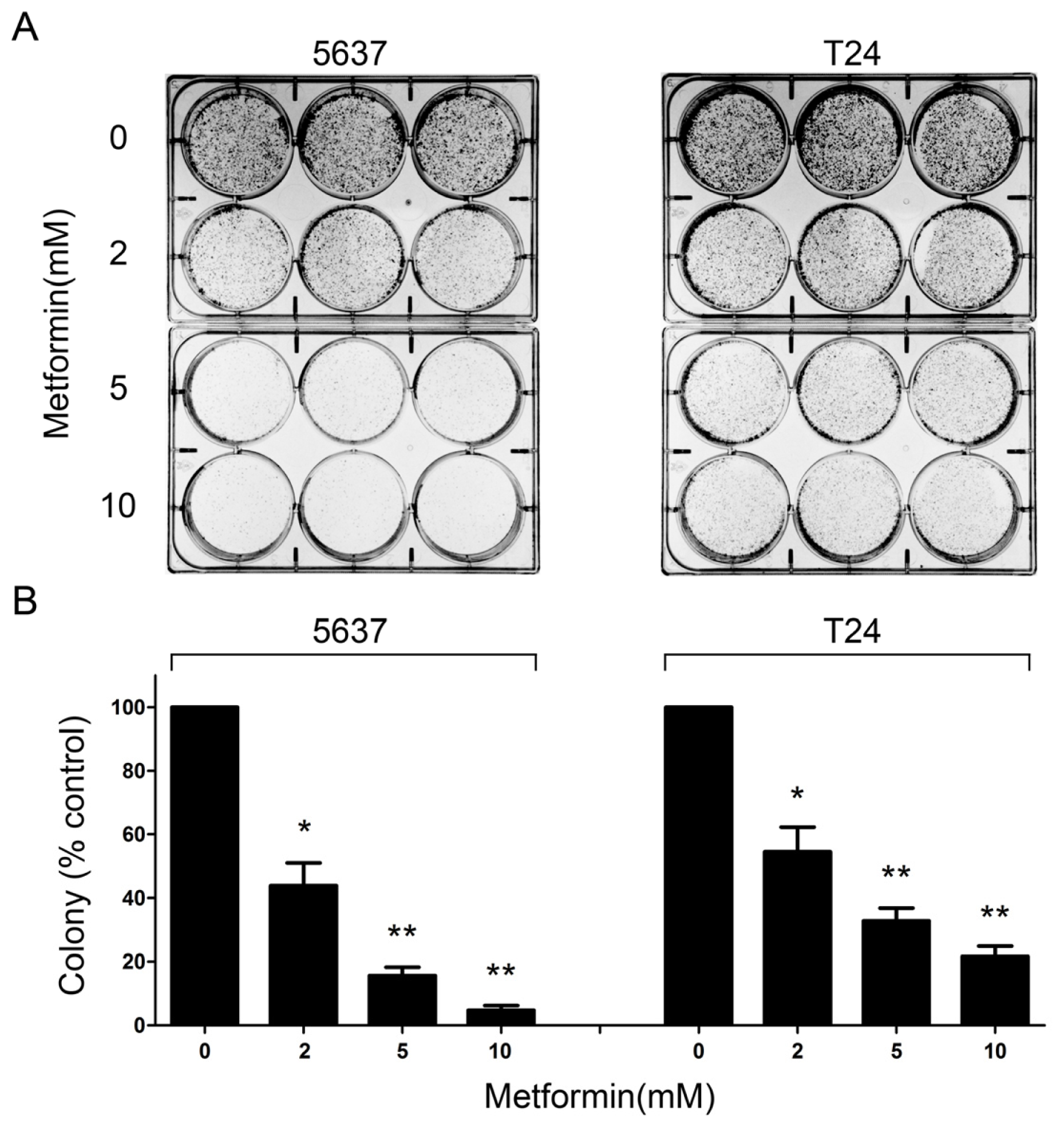
2.2. Metformin Reduces Colony Formation of Bladder Cancer Cells in Vitro
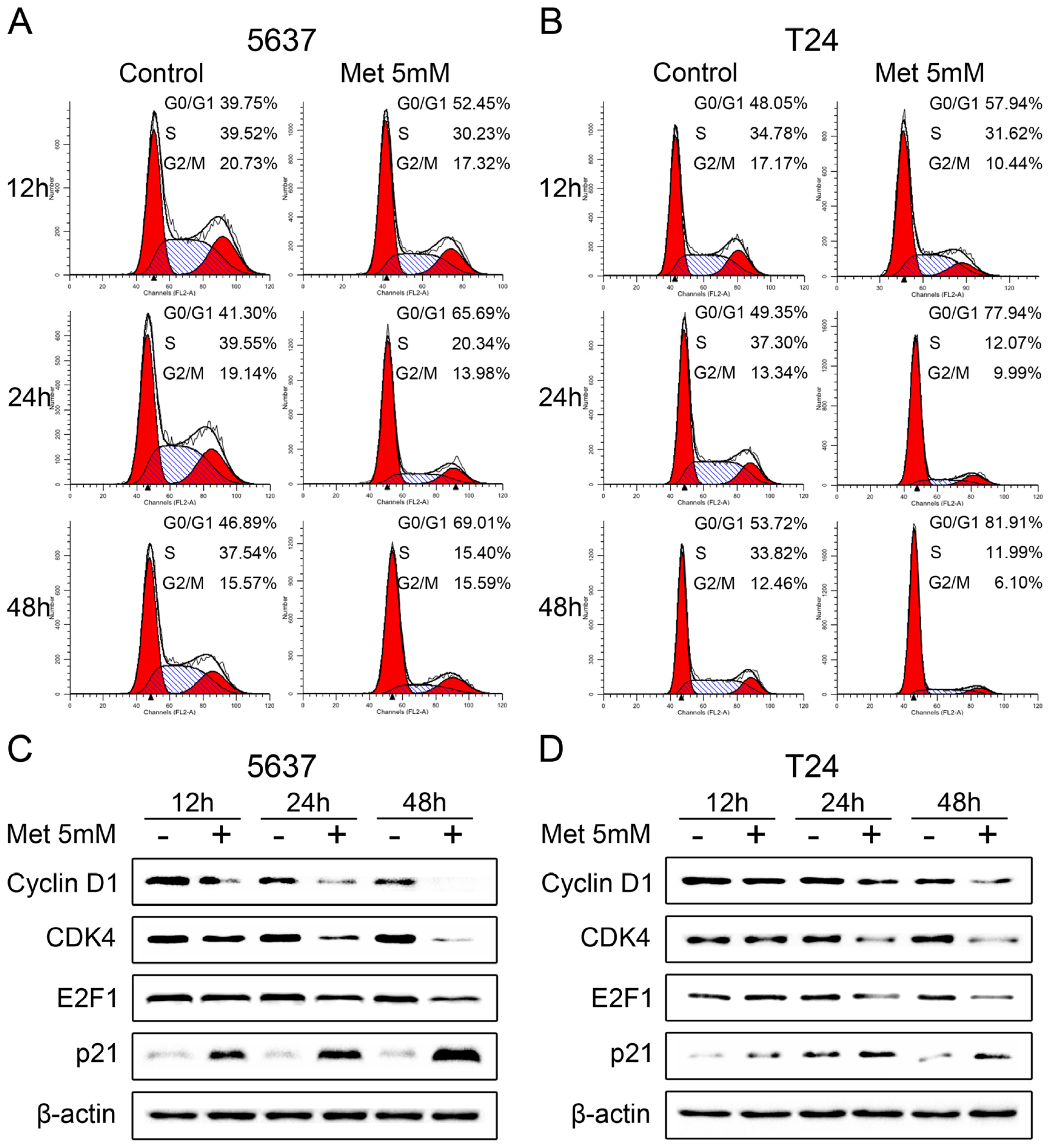
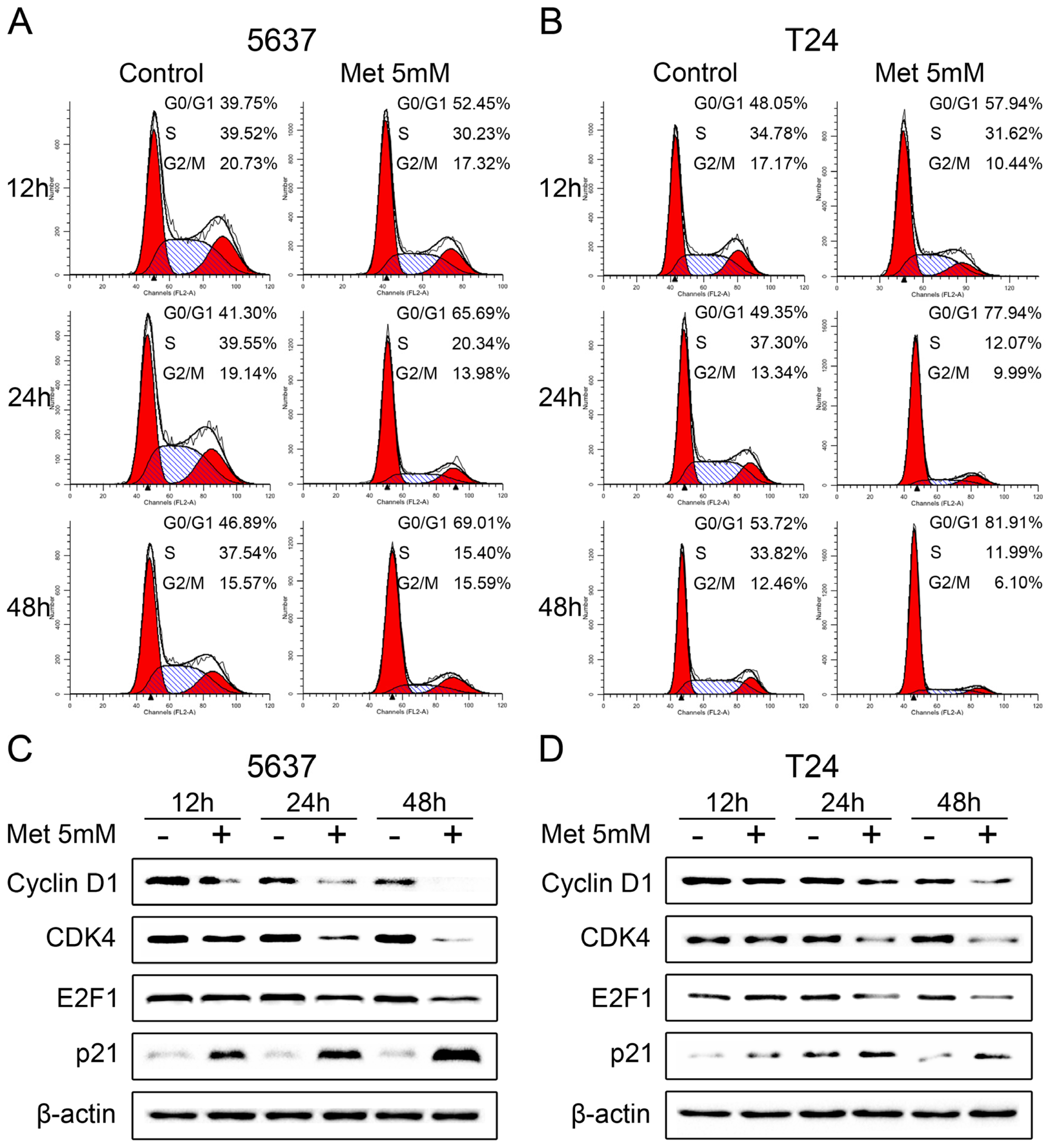
2.3. Metformin Induces Cell Cycle Arrest in G0/G1 Phases
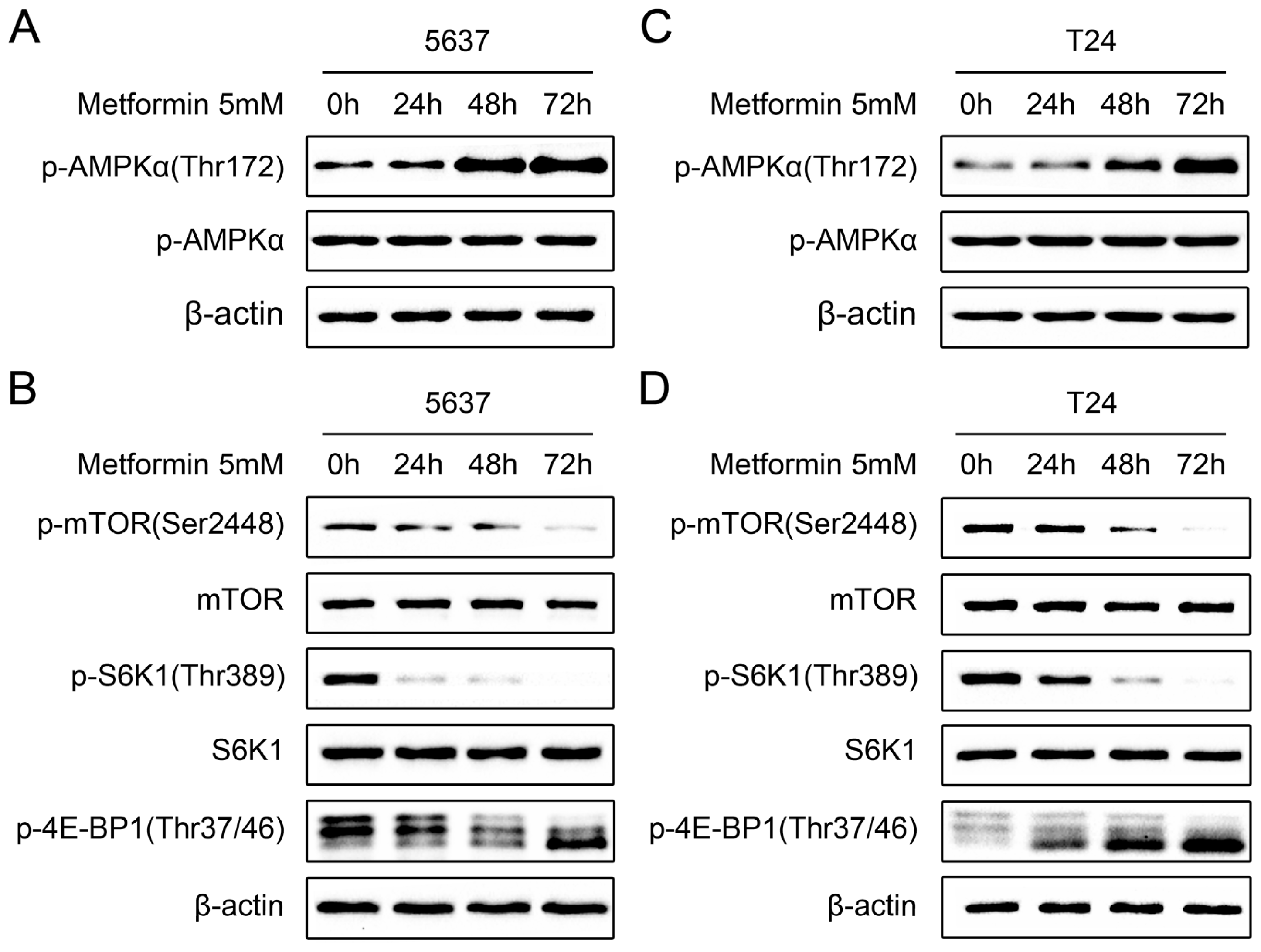
2.4. Metformin Activates AMPK and Inhibits mTOR Signaling in Bladder Cancer Cells
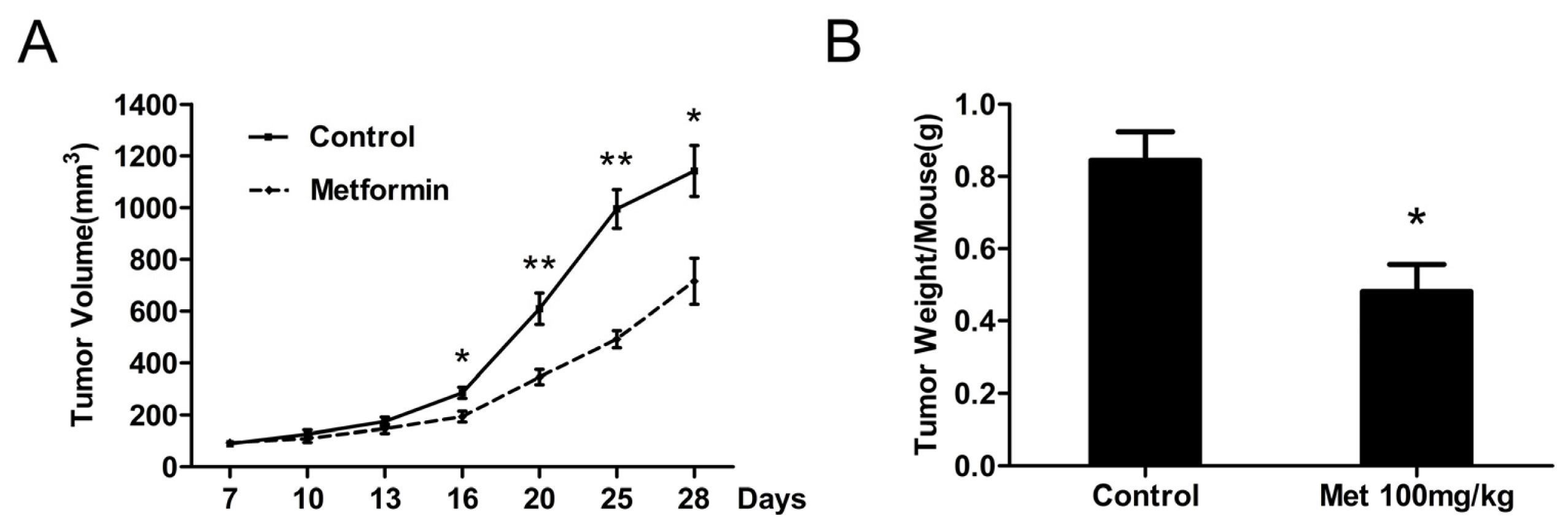
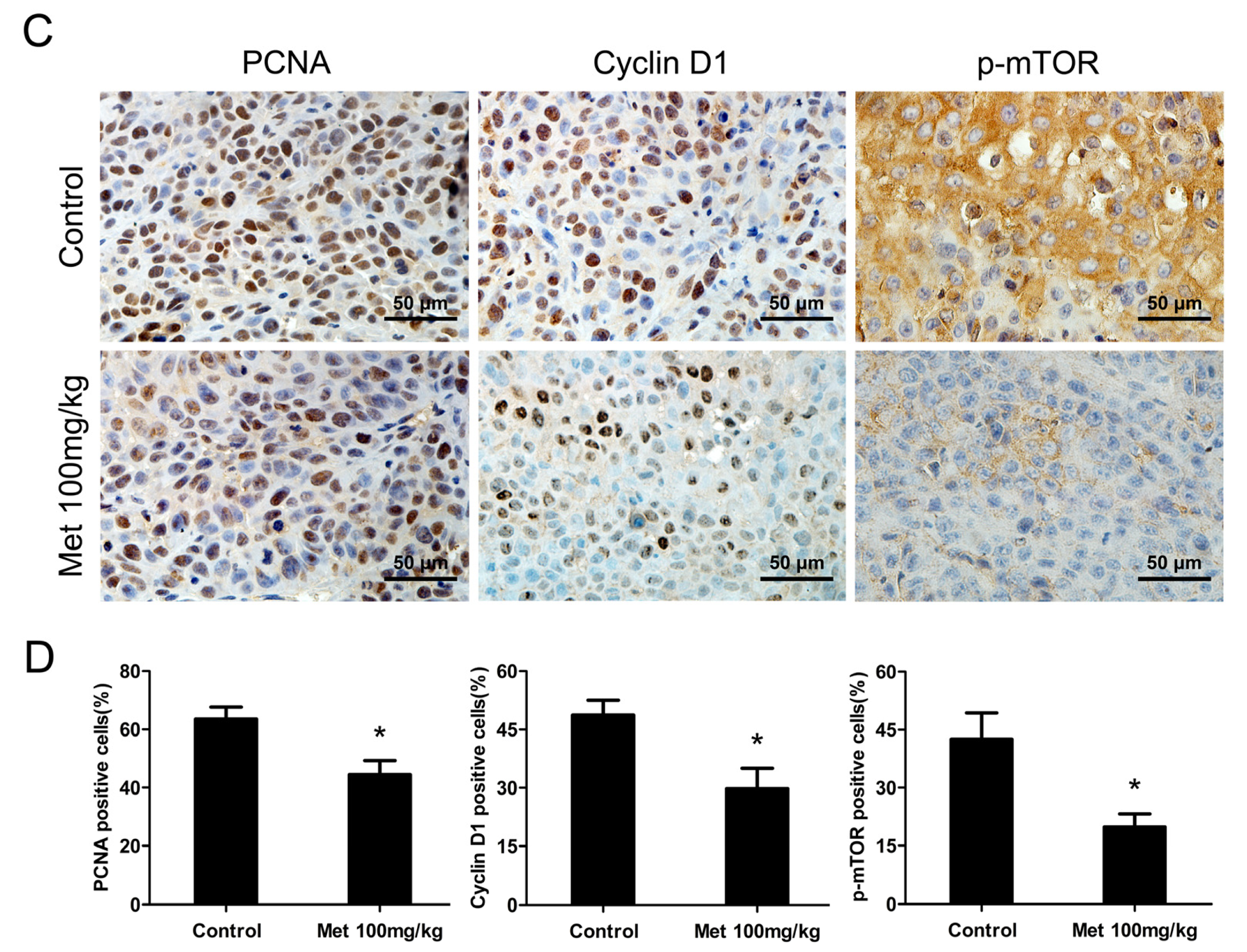
2.5. Metformin Inhibits the Growth of Human Bladder Tumor Xenografts in Nude Mice
3. Discussion
4. Experimental Section
4.1. Reagents
4.2. Cell Lines and Culture Conditions
4.3. Cell Viability Assay
4.4. Cell Count Assay
4.5. Colony Formation Assay
4.6. Flow Cytometric Analysis
4.7. Protein Extraction and Western Blot Analysis
4.8. Tumor Xenograft Model
4.9. Immunohistochemistry
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin 2011, 61, 212–236. [Google Scholar]
- Bischoff, C.J.; Clark, P.E. Bladder cancer. Curr. Opin. Oncol 2009, 21, 272–277. [Google Scholar]
- Schenk-Braat, E.A.; Bangma, C.H. Immunotherapy for superficial bladder cancer. Cancer Immunol. Immunother 2005, 54, 414–423. [Google Scholar]
- Herr, H.W.; Dotan, Z.; Donat, S.M.; Bajorin, D.F. Defining optimal therapy for muscle invasive bladder cancer. J. Urol 2007, 177, 437–443. [Google Scholar]
- Choueiri, T.K.; Raghavan, D. Chemotherapy for muscle-invasive bladder cancer treated with definitive radiotherapy: Persisting uncertainties. Nat. Clin. Pract. Oncol 2008, 5, 444–454. [Google Scholar]
- Witters, L.A. The blooming of the French lilac. J. Clin. Invest 2001, 108, 1105–1107. [Google Scholar]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest 2001, 108, 1167–1174. [Google Scholar]
- Boyle, J.G.; Salt, I.P.; McKay, G.A. Metformin action on AMP-activated protein kinase: A translational research approach to understanding a potential new therapeutic target. Diabet. Med 2010, 27, 1097–1106. [Google Scholar]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 2006, 66, 10269–10273. [Google Scholar]
- Anisimov, V.N.; Berstein, L.M.; Egormin, P.A.; Piskunova, T.S.; Popovich, I.G.; Zabezhinski, M.A.; Kovalenko, I.G.; Poroshina, T.E.; Semenchenko, A.V.; Provinciali, M.; et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp. Gerontol 2005, 40, 685–693. [Google Scholar]
- Rattan, R.; Giri, S.; Hartmann, L.C.; Shridhar, V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J. Cell. Mol. Med 2011, 15, 166–178. [Google Scholar]
- Rattan, R.; Graham, R.P.; Maguire, J.L.; Giri, S.; Shridhar, V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 2011, 13, 483–491. [Google Scholar]
- Ben, S.I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar]
- Kato, K.; Gong, J.; Iwama, H.; Kitanaka, A.; Tani, J.; Miyoshi, H.; Nomura, K.; Mimura, S.; Kobayashi, M.; Aritomo, Y.; et al. The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol. Cancer Ther 2012, 11, 549–560. [Google Scholar]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS One 2012, 7, e33411. [Google Scholar]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar]
- Rieken, M.; Xylinas, E.; Kluth, L.; Crivelli, J.J.; Chrystal, J.; Faison, T.; Lotan, Y.; Karakiewicz, P.I.; Fajkovic, H.; Babjuk, M.; et al. Association of diabetes mellitus and metformin use with oncological outcomes of patients with non-muscle invasive bladder cancer. BJU Int 2013, 112, 1105–1112. [Google Scholar]
- Kimball, S.R. Interaction between the AMP-activated protein kinase and mTOR signaling pathways. Med. Sci. Sports Exerc 2006, 38, 1958–1964. [Google Scholar]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar]
- Bringuier, P.P.; Tamimi, Y.; Schuuring, E.; Schalken, J. Expression of cyclin D1 and EMS1 in bladder tumours; relationship with chromosome 11q13 amplification. Oncogene 1996, 12, 1747–1753. [Google Scholar]
- Schuuring, E.; Verhoeven, E.; Mooi, W.J.; Michalides, R.J. Identification and cloning of two overexpressed genes, U21B31/PRAD1 and EMS1, within the amplified chromosome 11q13 region in human carcinomas. Oncogene 1992, 7, 355–361. [Google Scholar]
- Shin, K.Y.; Kong, G.; Kim, W.S.; Lee, T.Y.; Woo, Y.N.; Lee, J.D. Overexpression of cyclin D1 correlates with early recurrence in superficial bladder cancers. Br. J. Cancer 1997, 75, 1788–1792. [Google Scholar]
- Yuan, L.; Gu, X.; Shao, J.; Wang, M.; Wang, M.; Zhu, Q.; Zhang, Z. Cyclin D1 G870A polymorphism is associated with risk and clinicopathologic characteristics of bladder cancer. DNA Cell Biol 2010, 29, 611–617. [Google Scholar]
- Lin, H.H.; Ke, H.L.; Hsiao, K.H.; Tsai, C.W.; Wu, W.J.; Bau, D.T.; Chang, L.L. Potential role of CCND1 G870A genotype as a predictor for urothelial carcinoma susceptibility and muscle-invasiveness in Taiwan. Chin. J. Physiol 2011, 54, 196–202. [Google Scholar]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 2005, 1, 15–25. [Google Scholar]
- Hardie, D.G. AMPK: A key regulator of energy balance in the single cell and the whole organism. Int. J. Obes 2008, 32, S7–S12. [Google Scholar]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res 2007, 100, 328–341. [Google Scholar]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol 2010, 6, 457–470. [Google Scholar]
- Zakikhani, M.; Dowling, R.J.; Sonenberg, N.; Pollak, M.N. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev. Res 2008, 1, 369–375. [Google Scholar]
- Wysocki, P.J.; Wierusz-Wysocka, B. Obesity, hyperinsulinemia and breast cancer: Novel targets and a novel role for metformin. Expert. Rev. Mol. Diagn 2010, 10, 509–519. [Google Scholar]
- Hansel, D.E.; Platt, E.; Orloff, M.; Harwalker, J.; Sethu, S.; Hicks, J.L.; de Marzo, A.; Steinle, R.E.; Hsi, E.D.; Theodorescu, D.; et al. Mammalian target of rapamycin (mTOR) regulates cellular proliferation and tumor growth in urothelial carcinoma. Am. J. Pathol 2010, 176, 3062–3072. [Google Scholar]
- Mansure, J.J.; Nassim, R.; Chevalier, S.; Rocha, J.; Scarlata, E.; Kassouf, W. Inhibition of mammalian target of rapamycin as a therapeutic strategy in the management of bladder cancer. Cancer Biol. Ther 2009, 8, 2339–2347. [Google Scholar]
- Seager, C.M.; Puzio-Kuter, A.M.; Patel, T.; Jain, S.; Cordon-Cardo, C.; Mc, K.J.; Abate-Shen, C. Intravesical delivery of rapamycin suppresses tumorigenesis in a mouse model of progressive bladder cancer. Cancer Prev. Res 2009, 2, 1008–1014. [Google Scholar]
- Pinto-Leite, R.; Botelho, P.; Ribeiro, E.; Oliveira, P.A.; Santos, L. Effect of sirolimus on urinary bladder cancer T24 cell line. J. Exp. Clin. Cancer Res 2009, 28, 3. [Google Scholar]
- Morgillo, F.; Sasso, F.C.; Della, C.C.; Vitagliano, D.; D’Aiuto, E.; Troiani, T.; Martinelli, E.; de Vita, F.; Orditura, M.; de Palma, R.; et al. Synergistic effects of metformin treatment in combination with gefitinib, a selective EGFR tyrosine kinase inhibitor, in LKB1 wild-type NSCLC cell lines. Clin. Cancer Res 2013, 19, 3508–3519. [Google Scholar]
- Platt, F.M.; Hurst, C.D.; Taylor, C.F.; Gregory, W.M.; Harnden, P.; Knowles, M.A. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin. Cancer Res 2009, 15, 6008–6017. [Google Scholar]
- Guo, Y.; Chekaluk, Y.; Zhang, J.; Du, J.; Gray, N.S.; Wu, C.L.; Kwiatkowski, D.J. TSC1 involvement in bladder cancer: Diverse effects and therapeutic implications. J. Pathol 2013, 230, 17–27. [Google Scholar]
- Martin-Castillo, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle 2010, 9, 1057–1064. [Google Scholar]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J 2000, 348, 607–614. [Google Scholar]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar]
- National Research Council, Guide for the Care and Use of Laboratory Animals, 7th ed; National Academy Press: Washington, DC, USA, 1996.



© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, T.; Guo, P.; Zhang, Y.; Xiong, H.; Yu, X.; Xu, S.; Wang, X.; He, D.; Jin, X. The Antidiabetic Drug Metformin Inhibits the Proliferation of Bladder Cancer Cells in Vitro and in Vivo. Int. J. Mol. Sci. 2013, 14, 24603-24618. https://doi.org/10.3390/ijms141224603
Zhang T, Guo P, Zhang Y, Xiong H, Yu X, Xu S, Wang X, He D, Jin X. The Antidiabetic Drug Metformin Inhibits the Proliferation of Bladder Cancer Cells in Vitro and in Vivo. International Journal of Molecular Sciences. 2013; 14(12):24603-24618. https://doi.org/10.3390/ijms141224603
Chicago/Turabian StyleZhang, Tao, Peng Guo, Yinan Zhang, Hui Xiong, Xiao Yu, Shan Xu, Xinyang Wang, Dalin He, and Xunbo Jin. 2013. "The Antidiabetic Drug Metformin Inhibits the Proliferation of Bladder Cancer Cells in Vitro and in Vivo" International Journal of Molecular Sciences 14, no. 12: 24603-24618. https://doi.org/10.3390/ijms141224603