The Effects of H2S on the Activities of CYP2B6, CYP2D6, CYP3A4, CYP2C19 and CYP2C9 in Vivo in Rat

Abstract
:1. Introduction
2. Results and Discussion
2.1. Method Validation
2.2. Pharmacokinetic Study
3. Material and Methods
3.1. Chemicals and Reagents
3.2. Animals
3.3. Instrumentation and Conditions
3.4. Preparation of Standard Solutions
3.5. Pharmacokinetic Study
4. Conclusion
Acknowledgments
Conflicts of Interest
References
- Reiffenstein, R.J.; Hulbert, W.C.; Roth, S.H. Toxicology of hydrogen sulfide. Annu. Rev. Pharmacol. Toxicol 1992, 32, 109–134. [Google Scholar]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J 2002, 16, 1792–1798. [Google Scholar]
- Kamoun, P. Endogenous production of hydrogen sulfide in mammals. Amino Acids 2004, 26, 243–254. [Google Scholar]
- Zhou, C.F.; Tang, X.Q. Hydrogen sulfide and nervous system regulation. Chin. Med. J 2011, 124, 3576–3582. [Google Scholar]
- Lowicka, E.; Beltowski, J. Hydrogen sulfide (H2S)—The third gas of interest for pharmacologists. Pharmacol. Rep 2007, 59, 4–24. [Google Scholar]
- Wang, R. Hydrogen sulfide: The third gasotransmitter in biology and medicine. Antioxid. Redox Signal 2010, 12, 1061–1064. [Google Scholar]
- Lefer, D.J. A new gaseous signaling molecule emerges: Cardioprotective role of hydrogen sulfide. Proc. Natl. Acad. Sci. USA 2007, 104, 17907–17908. [Google Scholar]
- Kimura, H. Hydrogen sulfide as a neuromodulator. Mol. Neurobiol 2002, 26, 13–19. [Google Scholar]
- Benavides, G.A.; Squadrito, G.L.; Mills, R.W.; Patel, H.D.; Isbell, T.S.; Patel, R.P.; Darley-Usmar, V.M.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl. Acad. Sci. USA 2007, 104, 17977–17982. [Google Scholar]
- Kobayashi, K.; Urashima, K.; Shimada, N.; Chiba, K. Selectivities of human cytochrome p450 inhibitors toward rat p450 isoforms: Study with cdna-expressed systems of the rat. Drug Metab. Dispos 2003, 31, 833–836. [Google Scholar]
- Furge, L.L.; Guengerich, F.P. Cytochrome p450 enzymes in drug metabolism and chemical toxicology: An introduction. Biochem. Mol. Biol. Educ 2006, 34, 66–74. [Google Scholar]
- Oh, K.S.; Park, S.J.; Shinde, D.D.; Shin, J.G.; Kim, D.H. High-sensitivity liquid chromatography-tandem mass spectrometry for the simultaneous determination of five drugs and their cytochrome p450-specific probe metabolites in human plasma. J. Chromatogr. B 2012, 895–896, 56–64. [Google Scholar]
- Turpault, S.; Brian, W.; van Horn, R.; Santoni, A.; Poitiers, F.; Donazzolo, Y.; Boulenc, X. Pharmacokinetic assessment of a five-probe cocktail for CYPS 1A2, 2C9, 2C19, 2D6 and 3A. Br. J. Clin. Pharmacol 2009, 68, 928–935. [Google Scholar]
- Yin, O.Q.; Lam, S.S.; Lo, C.M.; Chow, M.S. Rapid determination of five probe drugs and their metabolites in human plasma and urine by liquid chromatography/tandem mass spectrometry: Application to cytochrome p450 phenotyping studies. Rapid Commun. Mass Spectrom 2004, 18, 2921–2933. [Google Scholar]
- Kozakai, K.; Yamada, Y.; Oshikata, M.; Kawase, T.; Suzuki, E.; Haramaki, Y.; Taniguchi, H. Reliable high-throughput method for inhibition assay of 8 cytochrome p450 isoforms using cocktail of probe substrates and stable isotope-labeled internal standards. Drug Metab. Pharmacokinet 2012, 27, 520–529. [Google Scholar]
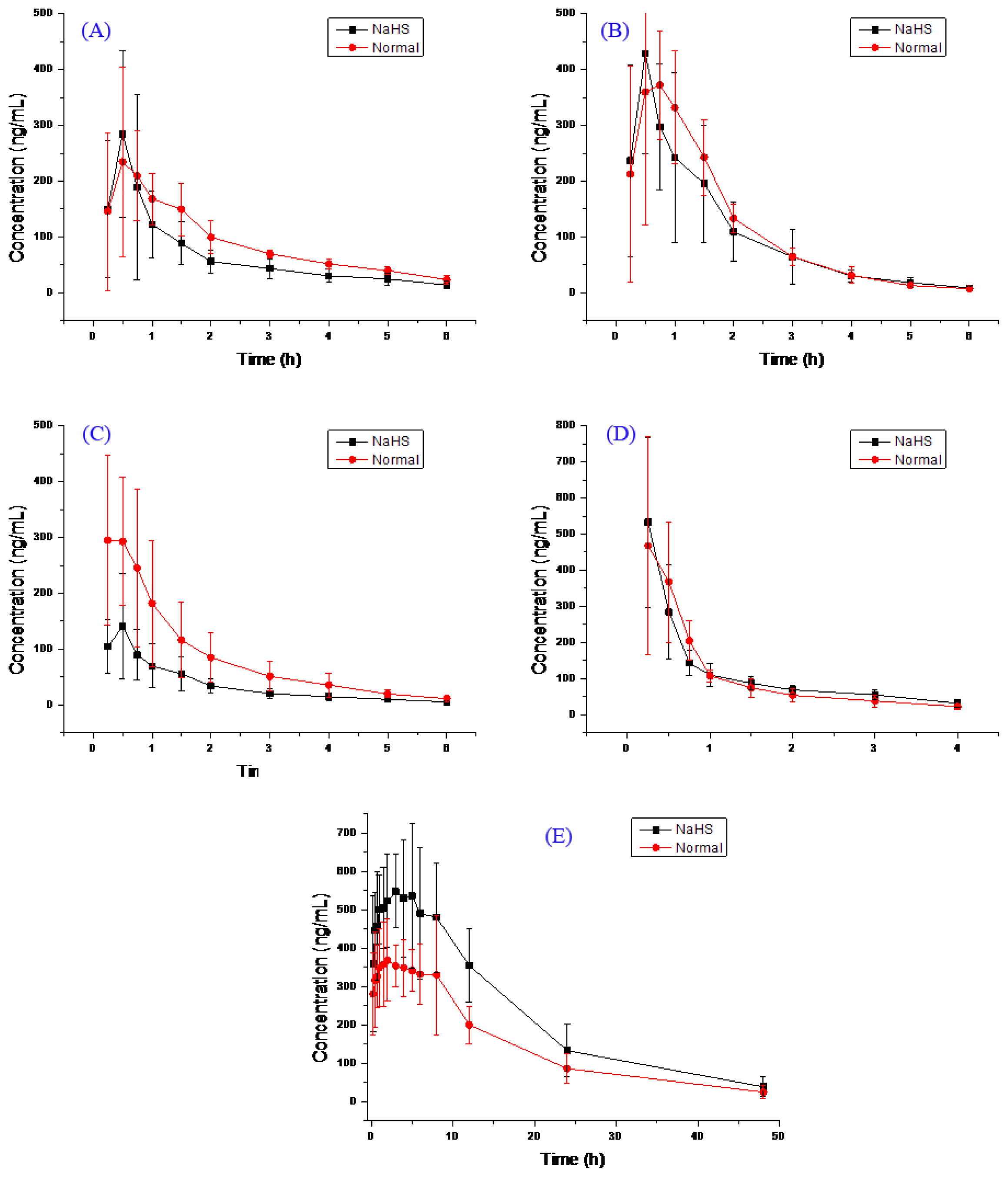

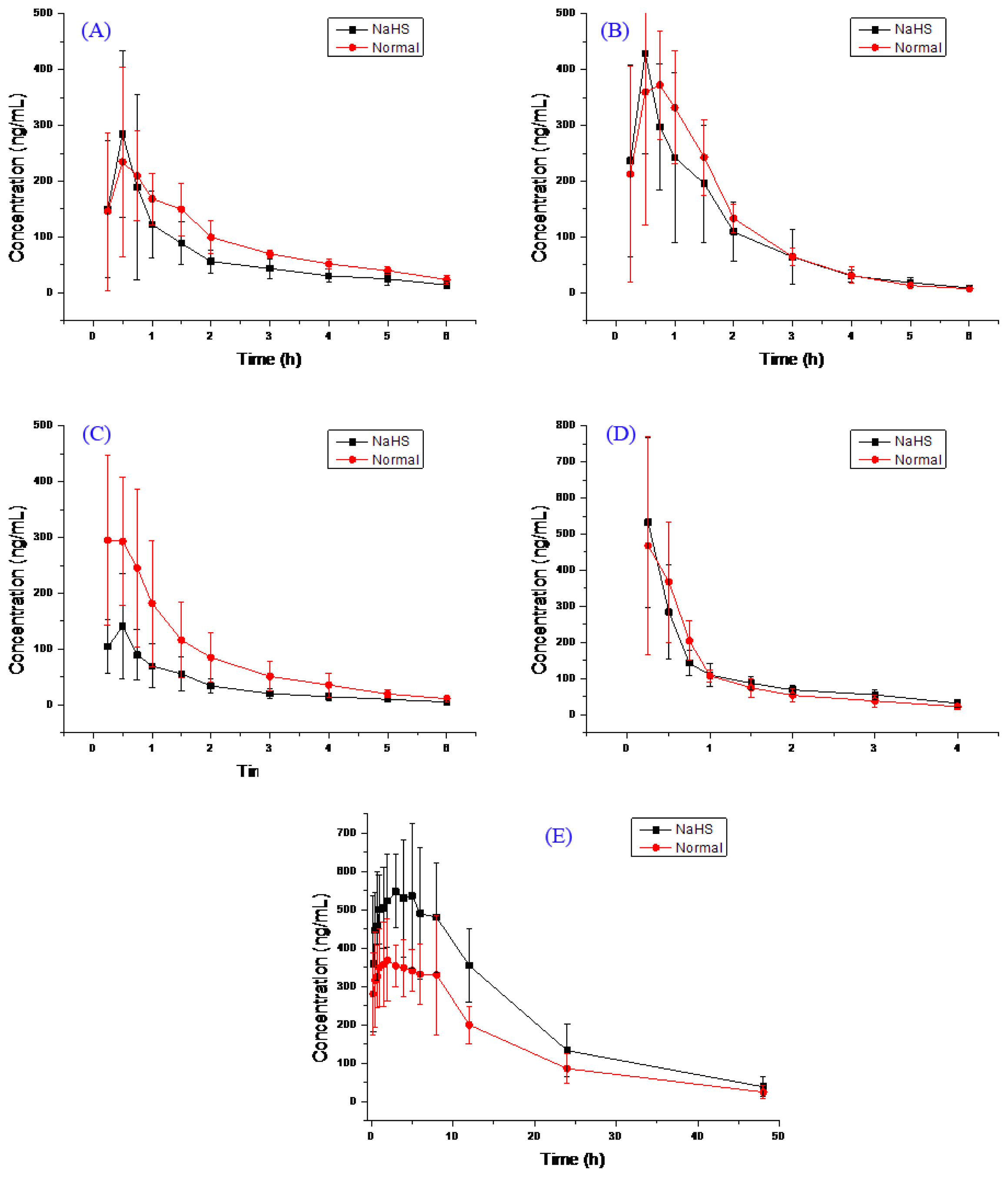{kind=link}
| Compound | Group | AUC (0–t) ng/mL h | AUC (0–∞) ng/mL h | MRT (0–t) h | MRT (0–∞) h | t1/2 h | Tmax h | CL L/h/kg | V L/kg | Cmax ng/mL |
|---|---|---|---|---|---|---|---|---|---|---|
| Bupropion | H2S | 393.0 ± 163.5 | 451.1 ± 176.3 | 1.8 ± 0.3 | 2.8 ± 1.3 | 2.3 ± 1.0 | 0.5 ± 0.1 | 21.7 ± 12.7 | 68.9 ± 40.3 | 308.4 ± 164.9 |
| Normal | 532.7 ± 141.4 | 603.6 ± 123.0 | 2.1 ± 0.3 | 3.0 ± 1.0 | 1.9 ± 0.7 | 0.8 ± 0.4 | 13.7 ± 2.8 | 39.3 ± 17.9 | 279.2 ± 119.3 | |
| Metoprolol | H2S | 627.7 ± 262.9 | 641.3 ± 265.9 | 1.5 ± 0.1 | 1.7 ± 0.1 | 1.1 ± 0.2 | 0.5 ± 0.1 | 14.3 ± 5.8 | 24.2 ± 12.1 | 447.1 ± 188.5 |
| Normal | 693.8 ± 172.4 | 704.6 ± 168.7 | 1.5 ± 0.3 | 1.6 ± 0.3 | 1.0 ± 0.3 | 0.7 ± 0.2 | 12.0 ± 3.0 | 18.2 ± 8.2 | 463.3 ± 139.1 | |
| Midazolam | H2S | 210.0 ± 95.5 * | 225.0 ± 100.7 * | 1.7 ± 0.3 | 2.2 ± 0.6 | 1.7 ± 0.3 | 0.4 ± 0.1 | 44.6 ± 25.8 * | 114.6 ± 92.6 | 152.9 ± 91.5 |
| Normal | 509.2 ± 244.8 | 535.1 ± 239.4 | 1.6 ± 0.1 | 2.2 ± 0.7 | 1.7 ± 1.1 | 0.3 ± 0.1 | 18.6 ± 10.5 | 58.1 ± 73.1 | 330.9 ± 117.1 | |
| Omeprazole | H2S | 446.7 ± 90.2 | 540.9 ± 90.5 | 1.2 ± 0.3 | 2.2 ± 0.7 | 1.9 ± 0.4 | 0.3 ± 0.2 | 15.2 ± 2.7 | 41.3 ± 11.8 | 536.7 ± 226.7 |
| Normal | 427.4 ± 155.2 | 479.9 ± 165.4 | 1.1 ± 0.2 | 1.7 ± 0.4 | 1.7 ± 0.6 | 0.4 ± 0.2 | 18.1 ± 5.0 | 43.9 ± 21.5 | 491.0 ± 276.0 | |
| Tolbutamide | H2S | 10,429.3 ± 2871.6 * | 11,099.1 ± 3218.4 * | 12.2 ± 2.7 | 14.8 ± 4.0 | 10.1 ± 2.7 | 2.9 ± 1.4 | 0.08 ± 0.02 * | 1.1 ± 0.3 | 640.4 ± 162.4 |
| Normal | 6799.3 ± 769.0 | 7261.5 ± 916.4 | 12.7 ± 1.9 | 16.0 ± 5.8 | 10.6 ± 4.7 | 3.6 ± 3.2 | 0.11 ± 0.02 | 1.6 ± 0.6 | 484.7 ± 98.3 | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, X.; Han, A.; Wen, C.; Chen, M.; Chen, X.; Yang, X.; Ma, J.; Lin, G. The Effects of H2S on the Activities of CYP2B6, CYP2D6, CYP3A4, CYP2C19 and CYP2C9 in Vivo in Rat. Int. J. Mol. Sci. 2013, 14, 24055-24063. https://doi.org/10.3390/ijms141224055
Wang X, Han A, Wen C, Chen M, Chen X, Yang X, Ma J, Lin G. The Effects of H2S on the Activities of CYP2B6, CYP2D6, CYP3A4, CYP2C19 and CYP2C9 in Vivo in Rat. International Journal of Molecular Sciences. 2013; 14(12):24055-24063. https://doi.org/10.3390/ijms141224055
Chicago/Turabian StyleWang, Xianqin, Anyue Han, Congcong Wen, Mengchun Chen, Xinxin Chen, Xuezhi Yang, Jianshe Ma, and Guanyang Lin. 2013. "The Effects of H2S on the Activities of CYP2B6, CYP2D6, CYP3A4, CYP2C19 and CYP2C9 in Vivo in Rat" International Journal of Molecular Sciences 14, no. 12: 24055-24063. https://doi.org/10.3390/ijms141224055
APA StyleWang, X., Han, A., Wen, C., Chen, M., Chen, X., Yang, X., Ma, J., & Lin, G. (2013). The Effects of H2S on the Activities of CYP2B6, CYP2D6, CYP3A4, CYP2C19 and CYP2C9 in Vivo in Rat. International Journal of Molecular Sciences, 14(12), 24055-24063. https://doi.org/10.3390/ijms141224055