Identification of Novel Small Molecules as Inhibitors of Hepatitis C Virus by Structure-Based Virtual Screening

Abstract
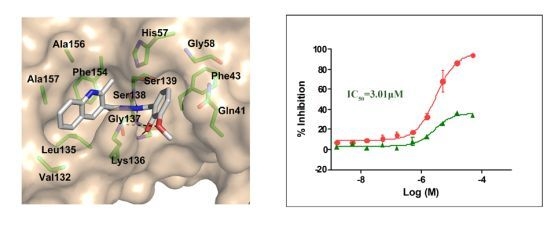
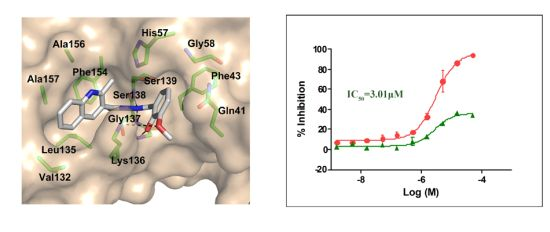
:
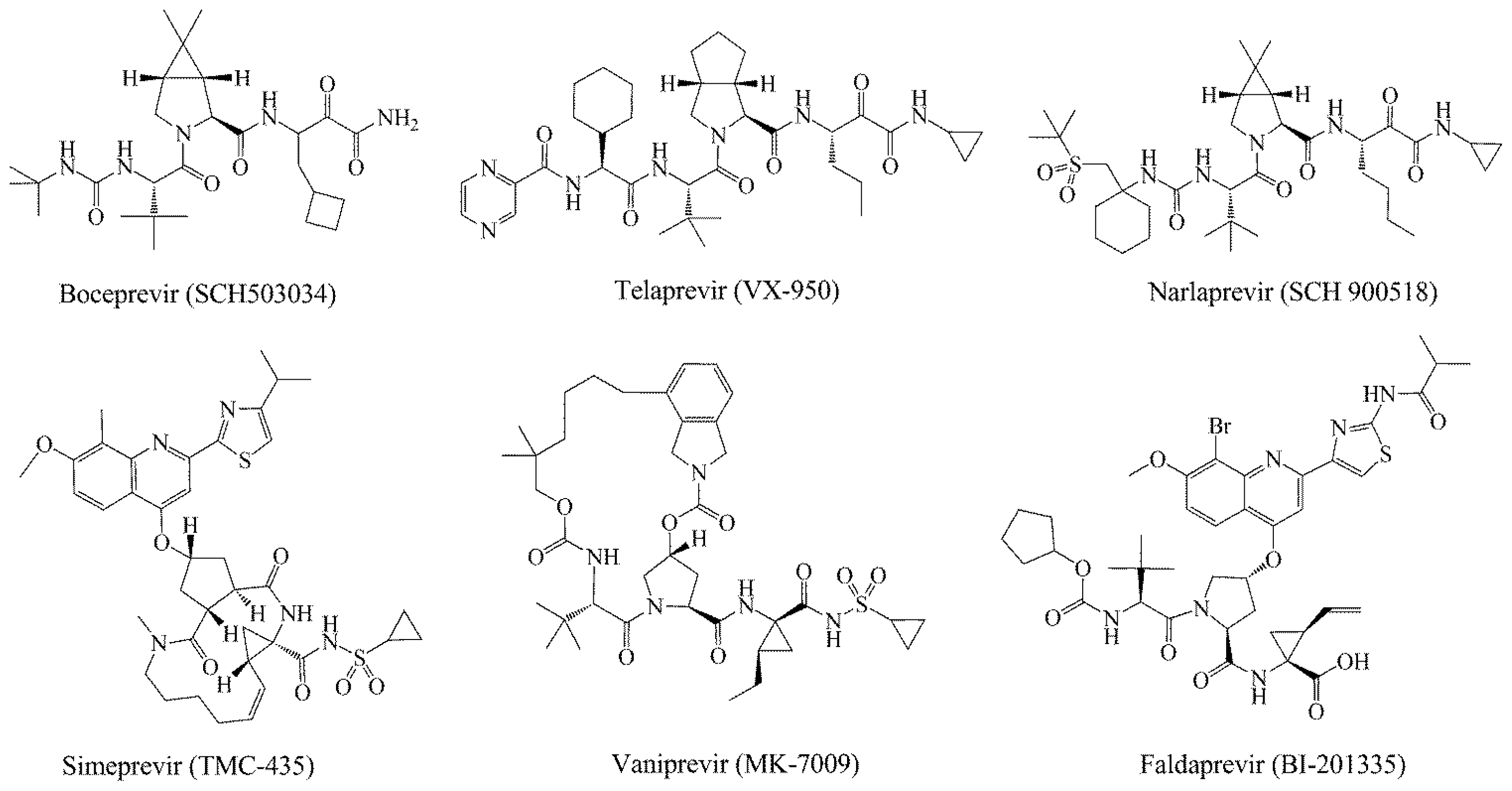
1. Introduction
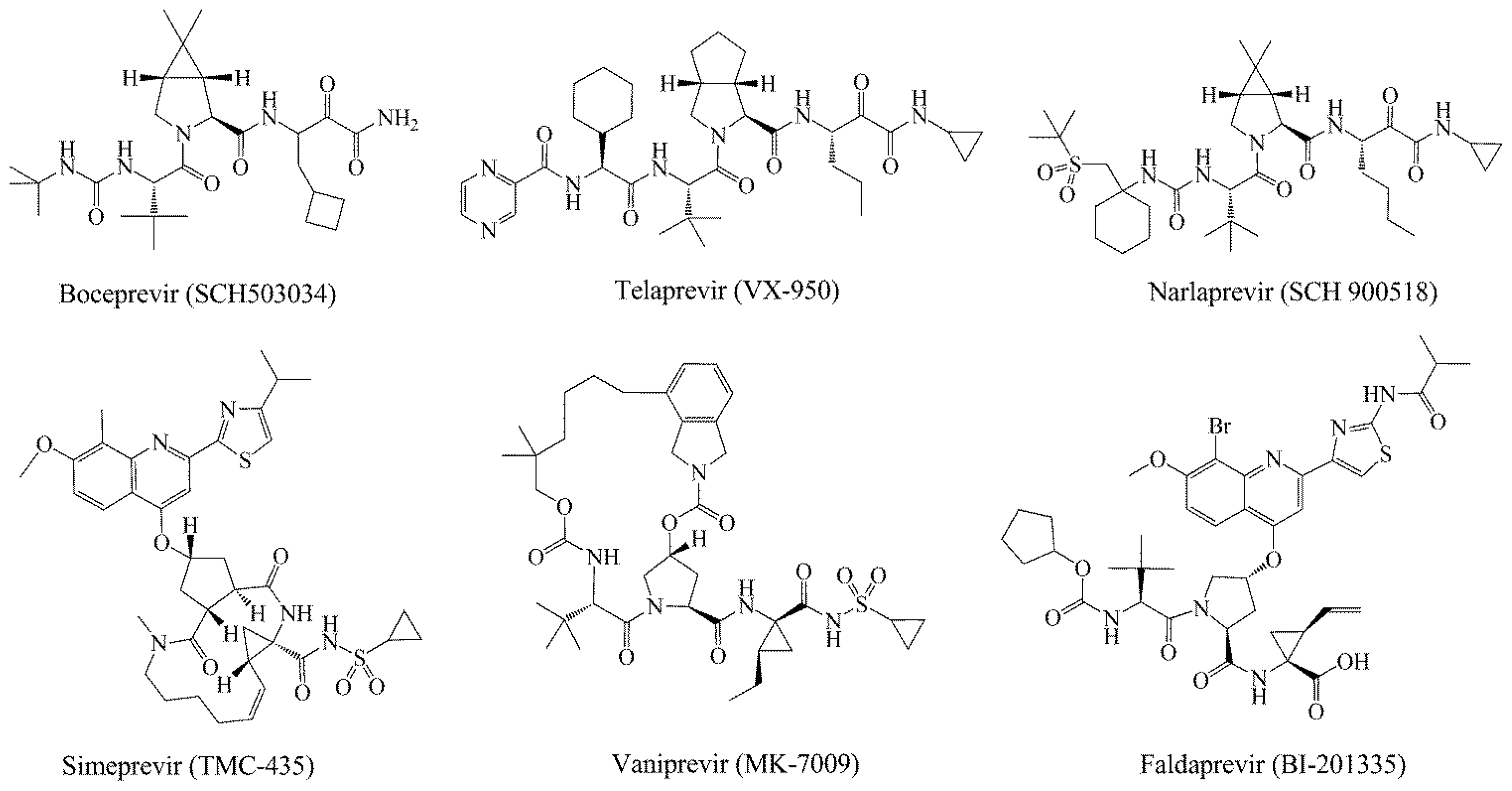
2. Results and Discussion
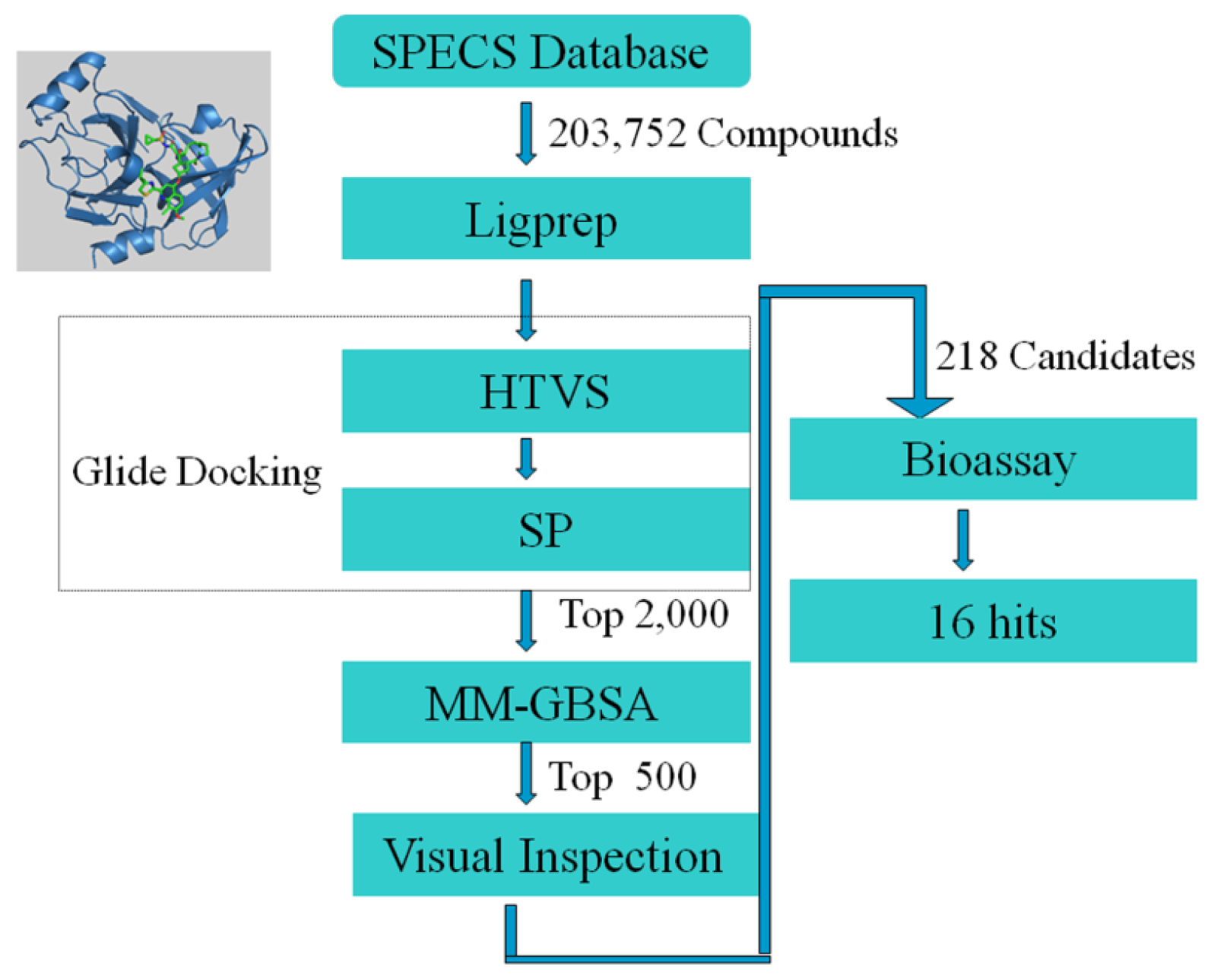
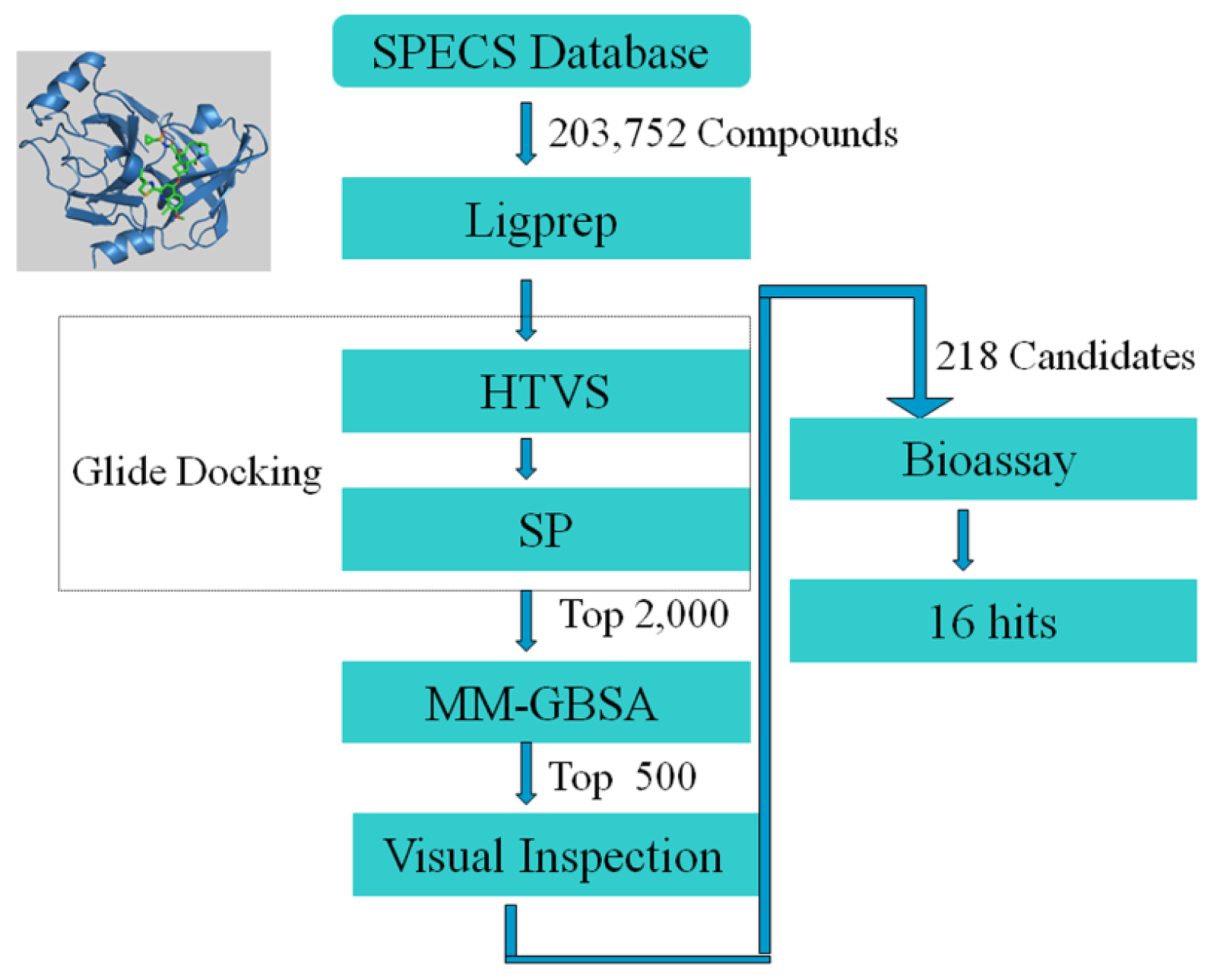
2.1. Structure-Based Virtual Screening of SPECS Database
2.2. In Vitro Evaluation of the Selected Compounds
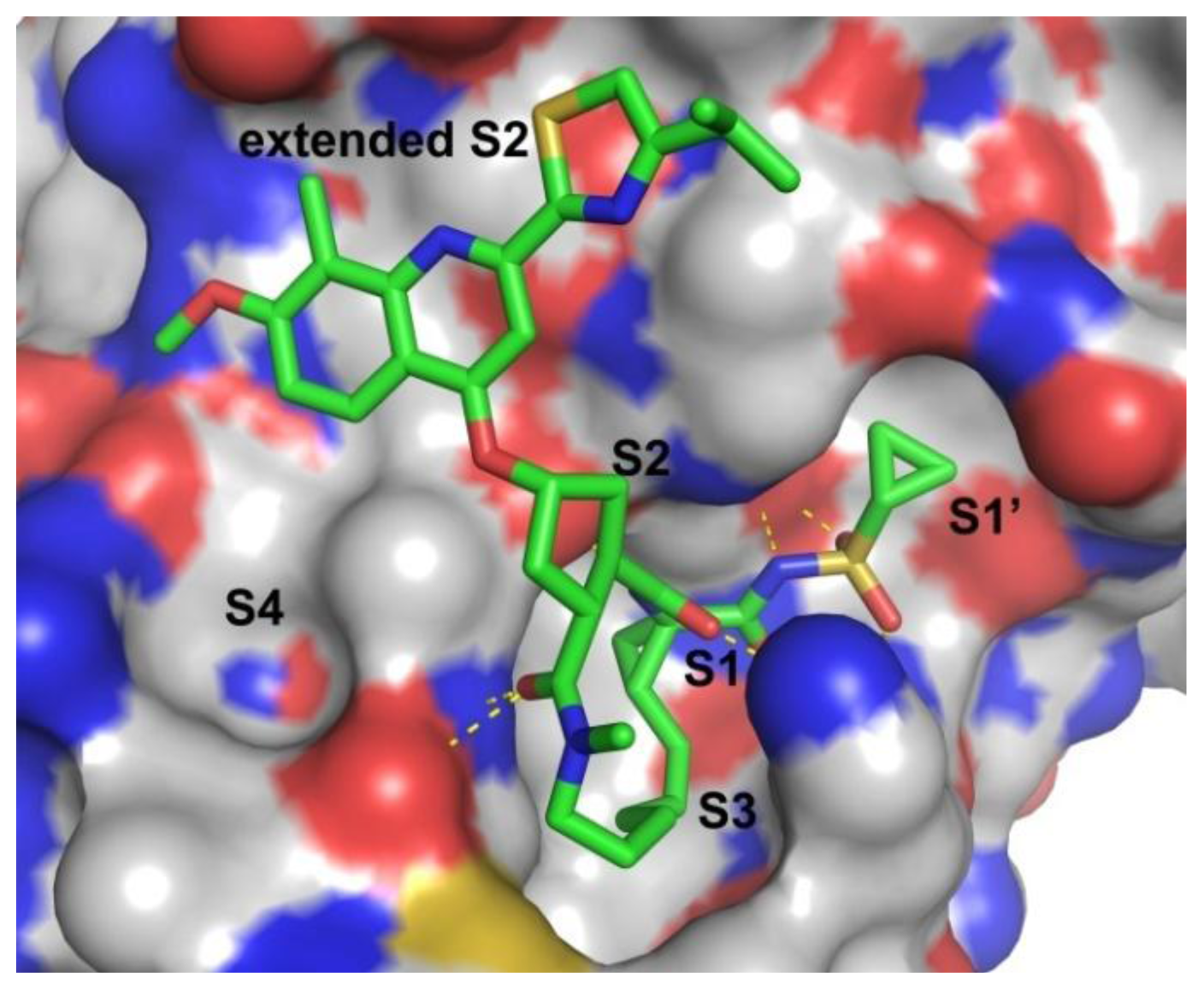
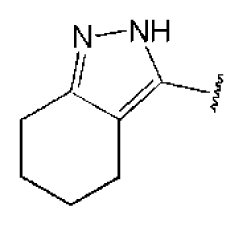
2.3. Molecular Modeling of the Hit Compounds
3. Computational and Experimental Methods
3.1. In Silico Experiment Schema
3.2. HCV Replicon Assay
3.3. Cell Cytotoxicity Assay
3.4. HCV NS3/4A Protease Assays
4. Conclusions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | R3 | IC50 (μM) | CC50 (MTT) | SI a | %Inh. b | MW | LogPc | LogSc |
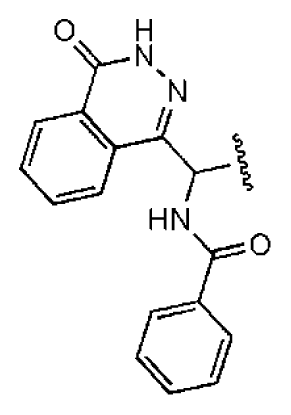
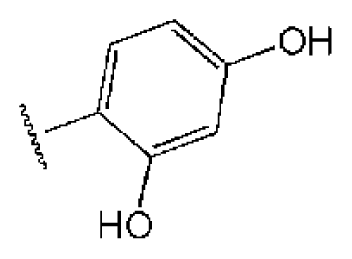
| 1 | 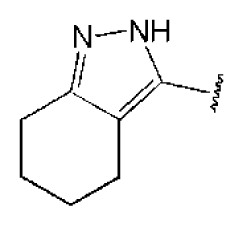 | 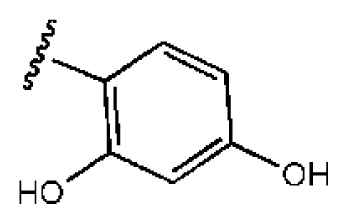 | O | 4.69 | 0.46 | 0.10 | 96.7 | 300 | 0.27 | −3.01 |
| 2 |  | 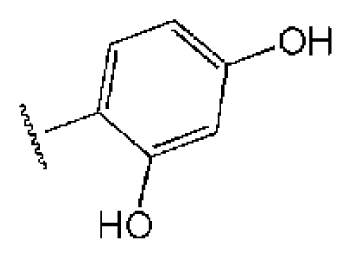 | O | 11.2 | >50 | >4.46 | 77.9 | 457 | 1.95 | −3.90 |
| 3 | 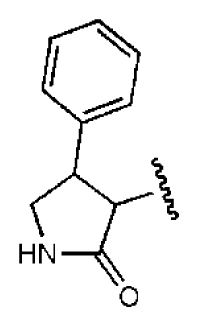 | 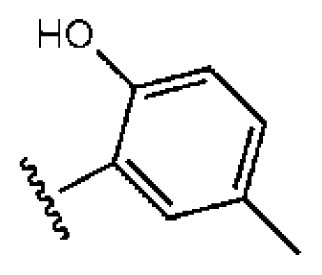 | O | 13.4 | >50 | >3.73 | 337 | 2.75 | −3.69 | |
| 4 | 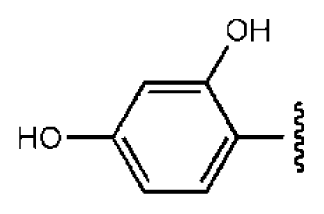 | 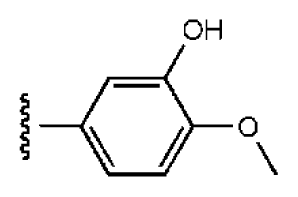 | O | 15.3 | >50 | >3.27 | 56.9 | 302 | 2.2 | −2.48 |
| 5 | 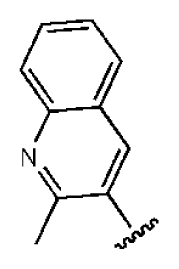 | 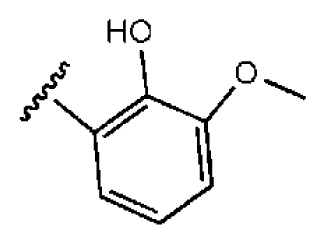 | O | 3.01 | >50 | >16.61 | 335 | 3.17 | −4.05 | |
| 6 | 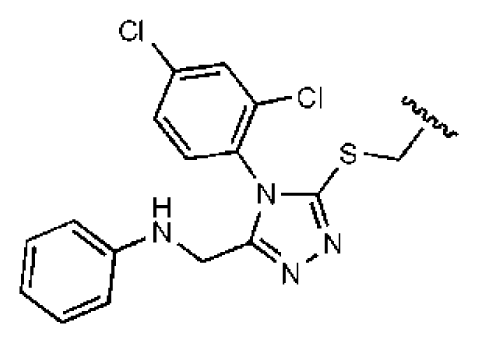 | 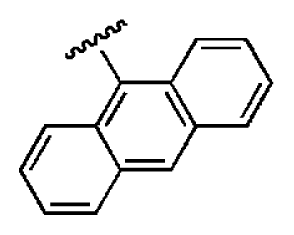 | O | 5.04 | 14.8 | 2.94 | 48.1 | 612 | 7.08 | −10.6 |
| 7 | 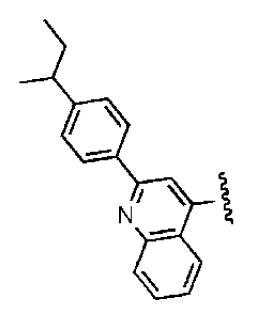 | 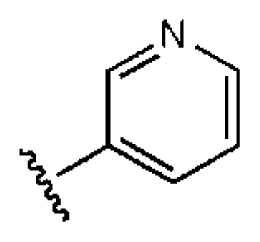 | O | 4.14 | 11.8 | 2.85 | 64.3 | 409 | 5.84 | −5.53 |
| 8 |  |  | S | 6.23 | 1.12 | 0.18 | 211 | 1.51 | −1.89 | |
| 9 | 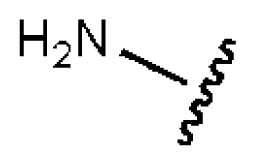 | 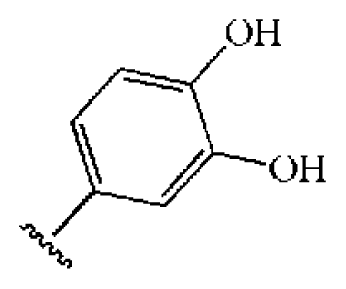 | S | 21.1 | >50 | >2.37 | 33.7 | 211 | 1.53 | −1.03 |
| 10 | 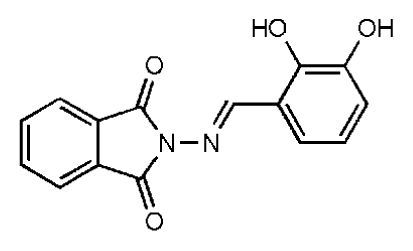 | 7.07 | >50 | >7.07 | 282 | 2.18 | −2.5 | |||
| CSA | 0.31 | >50 | >160 | |||||||
| No. | Structures | IC50 (μM) | CC50 (MTT) | SI a | %Inh. b | MW | LogPc | LogSc |
|---|---|---|---|---|---|---|---|---|
| 11 |  | 5.14 | >50 | >9.73 | 267 | 4.43 | −4.16 | |
| 12 | 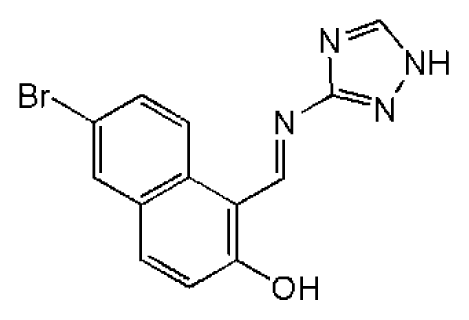 | 16.1 | >50 | >3.11 | 53.0 | 317 | 3.23 | −2.67 |
| 13 | 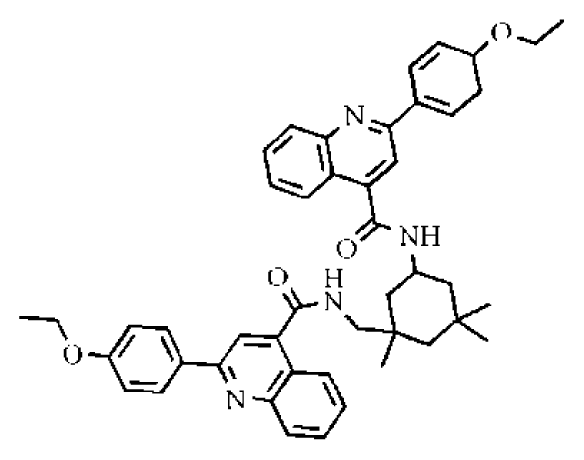 | 16.3 | >50 | >3.07 | 721 | 10.56 | −14.58 | |
| 14 | 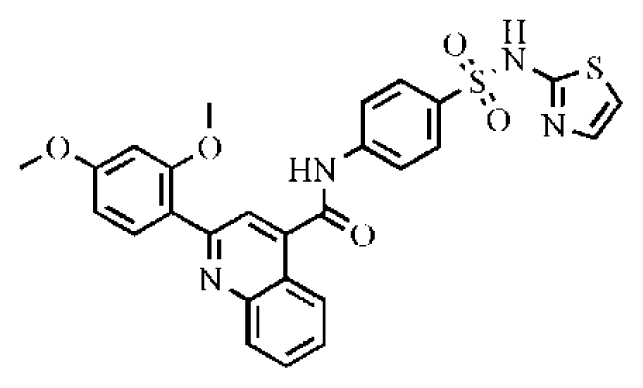 | 20.7 | >50 | >2.42 | 547 | 5.1 | −7.55 | |
| 15 | 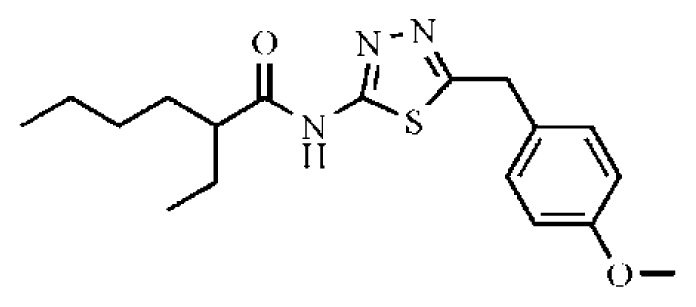 | 28.2 | >50 | >1.77 | 41.8 | 347 | 4.57 | −5.64 |
| 16 |  | 9.42 | >50 | >5.31 | 592 | 4.48 | −8.04 |
Acknowledgments
Conflicts of Interest
References
- Lamarre, D.; Anderson, P.C.; Bailey, M.; Beaulieu, P.; Bolger, G.; Bonneau, P.; Bos, M.; Cameron, D.R.; Cartier, M.; Cordingley, M.G.; et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003, 426, 186–189. [Google Scholar]
- Chen, S.H.; Tan, S.L. Discovery of small-molecule inhibitors of HCVNS3-4A protease as potential therapeutic agents against HCV infection. Curr. Med. Chem 2005, 12, 2317–2342. [Google Scholar]
- Qiu, P.; Cai, X.Y.; Ding, W.; Zhang, Q.; Norris, E.D.; Greene, J.R. HCV genotyping using statistical classification approach. J. Biomed. Sci 2009, 16, 62. [Google Scholar]
- Chen, Y.L.; Tang, J.; Kesler, M.J.; Sham, Y.Y.; Vince, R.; Geraghty, R.J.; Wang, Z.Q. The design, synthesis and biological evaluations of C-6 or C-7 substituted 2-hydroxyisoquinoline-1,3-diones as inhibitors of hepatitis C virus. Bioorg. Med. Chem 2012, 20, 467–479. [Google Scholar]
- Bacon, B.R.; Gordon, S.C.; Lawitz, E.; Marcellin, P.; Vierling, J.M.; Zeuzem, S.; Poordad, F.; Goodman, Z.D.; Sings, H.L.; Boparai, N.; et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med 2011, 364, 1207–1217. [Google Scholar]
- Elhefnawi, M.; ElGamacy, M.; Fares, M. Multiple virtual screening approaches for finding new hepatitis C virus RNA-dependent RNA polymerase inhibitors: Structure-based screens and molecular dynamics for the pursue of new poly pharmacological inhibitors. BMC Bioinf 2012, 13, S5. [Google Scholar]
- Patil, V.M.; Gupta, S.P.; Samanta, S.; Masand, N. Current perspective of HCV NS5B inhibitors: A review. Curr. Med. Chem 2011, 18, 5564–5597. [Google Scholar]
- Perni, R.B.; Kwong, A.D. Inhibitors of hepatitis C virus NS3-4A protease: An overdue line of therapy. Prog. Med. Chem 2002, 39, 215–255. [Google Scholar]
- Kim, J.L.; Morgenstern, K.A.; Lin, C.; Fox, T.; Dwyer, M.D.; Landro, J.A.; Chambers, S.P.; Markland, W.; Lepre, C.A.; OMalley, E.T.; et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 1996, 87, 343–355. [Google Scholar]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar]
- Steinkuhler, C.; Koch, U.; Narjes, F.; Matassa, V.G. Hepatitis C virus serine protease inhibitors: Current progress and future challenges. Curr. Med. Chem 2001, 8, 919–932. [Google Scholar]
- Lin, C.; Kwong, A.D.; Perni, R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect. Disord. Drug Targets 2006, 6, 3–16. [Google Scholar]
- Chen, B.N.; Mueller, C.; Willett, P. Combination rules for group fusion in similarity-based virtual screening. Mol. Inform 2010, 29, 533–541. [Google Scholar]
- Kwong, A.D.; Kauffman, R.S.; Hurter, P.; Mueller, P. Discovery and development of telaprevir: An NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol 2011, 29, 993–1003. [Google Scholar]
- Raney, K.D.; Sharma, S.D.; Moustafa, I.M.; Cameron, C.E. Hepatitis C virus non-structural protein 3 (HCV NS3): A multifunctional antiviral target. J. Biol. Chem 2010, 285, 22725–22731. [Google Scholar]
- Njoroge, F.G.; Chen, K.X.; Shih, N.Y.; Piwinski, J.J. Challenges in modern drug discovery: A case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection. Acc. Chem. Res 2008, 41, 50–59. [Google Scholar]
- Chen, S.H.; Lamar, J.; Yip, Y.; Victor, F.; Johnson, R.B.; Wang, Q.M.; Glass, J.I.; Heinz, B.; Colacino, J.; Guo, D.Q.; et al. P1 and P1′ optimization of [3,4]-bicycloproline P2 incorporated tetrapeptidyl alpha-ketoamide based HCV protease inhibitors. Lett. Drug Des. Discov 2005, 2, 118–123. [Google Scholar]
- Arasappan, A.; Bennett, F.; Bogen, S.L.; Venkatraman, S.; Blackman, M.; Chen, K.X.; Hendrata, S.; Huang, Y.H.; Huelgas, R.M.; Nair, L.; et al. Discovery of narlaprevir (SCH 900518): A potent, second generation HCV NS3 serine protease inhibitor. ACS Med. Chem. Lett 2010, 1, 64–69. [Google Scholar]
- Cummings, M.D.; Lindberg, J.; Lin, T.I.; de Kock, H.; Lenz, O.; Lilja, E.; Fellander, S.; Baraznenok, V.; Nystrom, S.; Nilsson, M.; et al. Induced-fit binding of the macrocyclic noncovalent inhibitor TMC435 to its HCV NS3/NS4A protease target. Angew. Chem. Int. Ed. Engl 2010, 49, 1652–1655. [Google Scholar]
- Liverton, N.J.; Carroll, S.S.; Dimuzio, J.; Fandozzi, C.; Graham, D.J.; Hazuda, D.; Holloway, M.K.; Ludmerer, S.W.; McCauley, J.A.; McIntyre, C.J.; et al. MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother 2010, 54, 305–311. [Google Scholar]
- Llinas-Brunet, M.; Bailey, M.D.; Goudreau, N.; Bhardwaj, P.K.; Bordeleau, J.; Bos, M.; Bousquet, Y.; Cordingley, M.G.; Duan, J.; Forgione, P.; et al. Discovery of a potent and selective noncovalent linear inhibitor of the hepatitis C virus NS3 protease (BI 201335). J. Med. Chem 2010, 53, 6466–6476. [Google Scholar]
- Perni, R.B.; Almquist, S.J.; Byrn, R.A.; Chandorkar, G.; Chaturvedi, P.R.; Courtney, L.F.; Decker, C.J.; Dinehart, K.; Gates, C.A.; Harbeson, S.L.; et al. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob. Agents Chemother 2006, 50, 899–909. [Google Scholar]
- Malcolm, B.A.; Liu, R.; Lahser, F.; Agrawal, S.; Belanger, B.; Butkiewicz, N.; Chase, R.; Gheyas, F.; Hart, A.; Hesk, D.; et al. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob. Agents Chemother 2006, 50, 1013–1020. [Google Scholar]
- Bachovchin, D.A.; Cravatt, B.F. The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discov 2012, 11, 52–68. [Google Scholar]
- Halfon, P.; Locarnini, S. Hepatitis C virus resistance to protease inhibitors. J. Hepatol 2011, 55, 192–206. [Google Scholar]
- Romano, K.P.; Ali, A.; Royer, W.E.; Schiffer, C.A. Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc. Natl. Acad. Sci. USA 2010, 107, 20986–20991. [Google Scholar]
- Doherty, K.M.; Nakka, P.; King, B.M.; Rhee, S.Y.; Holmes, S.P.; Shafer, R.W.; Radhakrishnan, M.L. A multifaceted analysis of HIV-1 protease multidrug resistance phenotypes. BMC Bioinf 2011, 12, 477. [Google Scholar]
- Venkatraman, S.; Velazquez, F.; Wu, W.; Blackman, M.; Chen, K.X.; Bogen, S.; Nair, L.; Tong, X.; Chase, R.; Hart, A.; et al. Discovery and structure-activity relationship of P1–P3 ketoamide derived macrocyclic inhibitors of hepatitis C virus NS3 protease. J. Med. Chem 2009, 52, 336–346. [Google Scholar]
- Karelson, M.; Dobchev, D.A.; Karelson, G.; Tamm, T.; Tamm, K.; Nikonov, A.; Mutso, M.; Merits, A. Fragment-based development of HCV protease inhibitors for the treatment of hepatitis C. Curr. Comput. Aided Drug Des 2012, 8, 55–61. [Google Scholar]
- Hagel, M.; Niu, D.; St Martin, T.; Sheets, M.P.; Qiao, L.; Bernard, H.; Karp, R.M.; Zhu, Z.; Labenski, M.T.; Chaturvedi, P.; et al. Selective irreversible inhibition of a protease by targeting a noncatalytic cysteine. Nat. Chem. Biol 2011, 7, 22–24. [Google Scholar]
- Takaya, D.; Yamashita, A.; Kamijo, K.; Gomi, J.; Ito, M.; Maekawa, S.; Enomoto, N.; Sakamoto, N.; Watanabe, Y.; Arai, R.; et al. A new method for induced fit docking (GENIUS) and its application to virtual screening of novel HCV NS3-4A protease inhibitors. Bioorg. Med. Chem 2011, 19, 6892–6905. [Google Scholar]
- Oliva, C.; Rodriguez, A.; Gonzalez, M.; Yang, W. A quantum mechanics/molecular mechanics study of the reaction mechanism of the hepatitis C virus NS3 protease with the NS5A/5B substrate. Proteins 2007, 66, 444–455. [Google Scholar]
- Rodriguez, A.; Oliva, C.; Gonzalez, M. A comparative QM/MM study of the reaction mechanism of the hepatitis C virus NS3/NS4A protease with the three main natural substrates NS5A/5B, NS4B/5A and NS4A/4B. Phys. Chem. Chem. Phys 2010, 12, 8001–8015. [Google Scholar]
- Lohmann, V.; Hoffmann, S.; Herian, U.; Penin, F.; Bartenschlager, R. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol 2003, 77, 3007–3019. [Google Scholar]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar]
- Romeiro, N.C.; Aguirre, G.; Hernandez, P.; Gonzalez, M.; Cerecetto, H.; Aldana, I.; Perez-Silanes, S.; Monge, A.; Barreiro, E.J.; Lima, L.M. Synthesis, trypanocidal activity and docking studies of novel quinoxaline-N-acylhydrazones, designed as cruzain inhibitors candidates. Bioorg. Med. Chem 2009, 17, 641–652. [Google Scholar]
- Schirmeister, T.; Kaeppler, U. Non-peptidic inhibitors of cysteine proteases. Mini Rev. Med. Chem 2003, 3, 361–373. [Google Scholar]
- Ifa, D.R.; Rodrigues, C.R.; de Alencastro, R.B.; Fraga, C.A.M.; Barreiro, E.J. A possible molecular mechanism for the inhibition of cysteine proteases by salicylaldehyde N-acylhydrazones and related compounds. J. Mol. Struct. Theochem 2000, 505, 11–17. [Google Scholar]
- Prongay, A.J.; Guo, Z.; Yao, N.; Pichardo, J.; Fischmann, T.; Strickland, C.; Myers, J., Jr.; Weber, P.C.; Beyer, B.M.; Ingram, R.; et al. Discovery of the HCV NS3/4A protease inhibitor (1R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino] carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxa mide (Sch 503034) II. Key steps in structure-based optimization. J. Med. Chem 2007, 50, 2310–2318. [Google Scholar]
- Chen, K.X.; Nair, L.; Vibulbhan, B.; Yang, W.; Arasappan, A.; Bogen, S.L.; Venkatraman, S.; Bennett, F.; Pan, W.; Blackman, M.L.; et al. Second-generation highly potent and selective inhibitors of the hepatitis C virus NS3 serine protease. J. Med. Chem 2009, 52, 1370–1379. [Google Scholar]
- LigPrep, version 2.0; Schrödinger Suite: Schrödinger, L.: New York, NY, USA, 2010.
- Glide, version 4.0; Schrödinger Suite: Schrödinger, L.: New York, NY, USA, 2010.
- Prime, version 1.5; Schrödinger Suite: Schrödinger, L.: New York, NY, USA, 2010.
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, J.; Liu, X.; Li, S.; Wang, Y.; Zhou, N.; Luo, C.; Luo, X.; Zheng, M.; Jiang, H.; Chen, K. Identification of Novel Small Molecules as Inhibitors of Hepatitis C Virus by Structure-Based Virtual Screening. Int. J. Mol. Sci. 2013, 14, 22845-22856. https://doi.org/10.3390/ijms141122845
Li J, Liu X, Li S, Wang Y, Zhou N, Luo C, Luo X, Zheng M, Jiang H, Chen K. Identification of Novel Small Molecules as Inhibitors of Hepatitis C Virus by Structure-Based Virtual Screening. International Journal of Molecular Sciences. 2013; 14(11):22845-22856. https://doi.org/10.3390/ijms141122845
Chicago/Turabian StyleLi, Jing, Xian Liu, Shanshan Li, Yulan Wang, Nannan Zhou, Cheng Luo, Xiaomin Luo, Mingyue Zheng, Hualiang Jiang, and Kaixian Chen. 2013. "Identification of Novel Small Molecules as Inhibitors of Hepatitis C Virus by Structure-Based Virtual Screening" International Journal of Molecular Sciences 14, no. 11: 22845-22856. https://doi.org/10.3390/ijms141122845