Membrane Signaling Induced by High Doses of Ionizing Radiation in the Endothelial Compartment. Relevance in Radiation Toxicity

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Radiation-Induced Plasma Membrane Signaling in the Endothelial Compartment
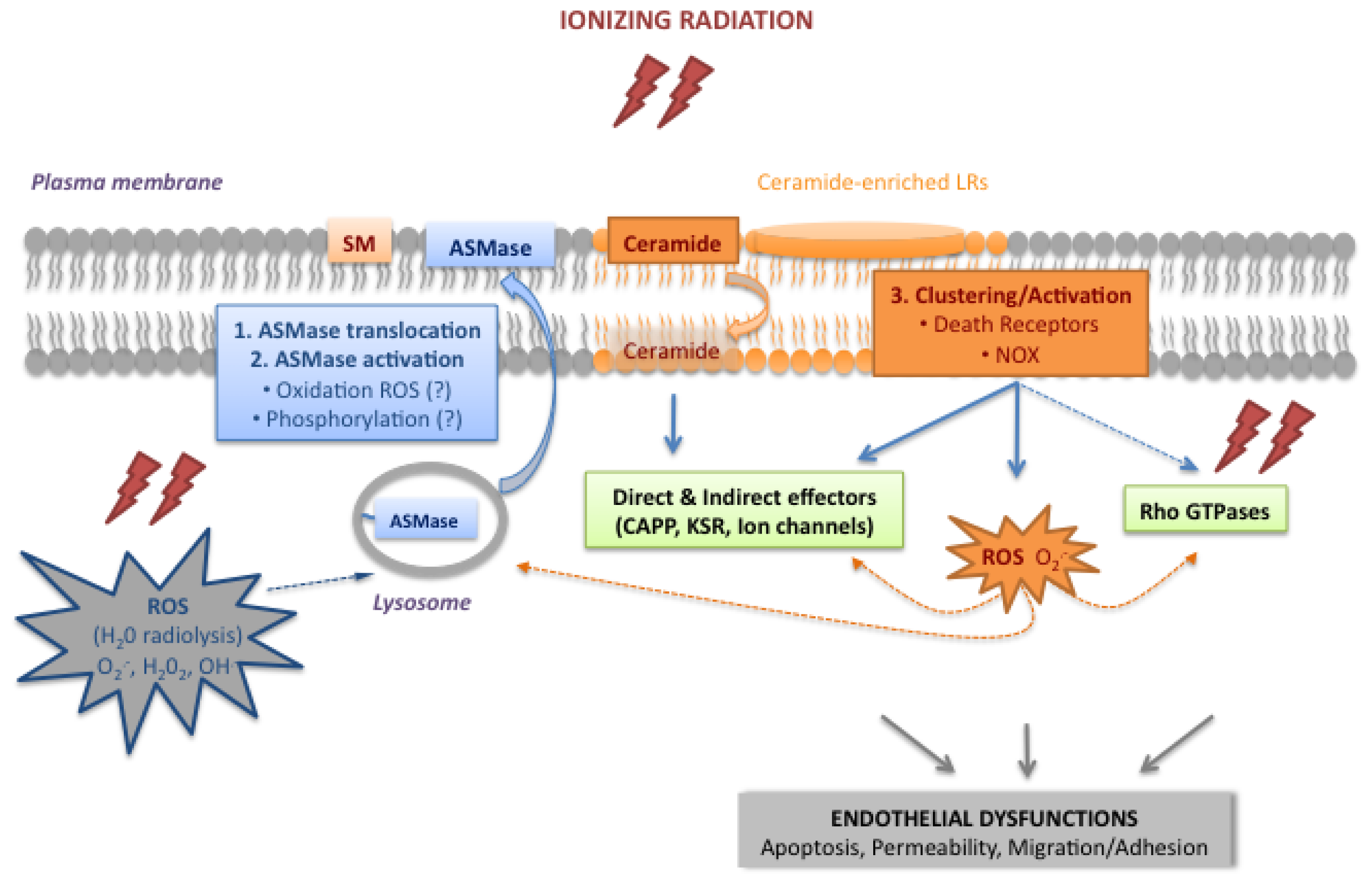
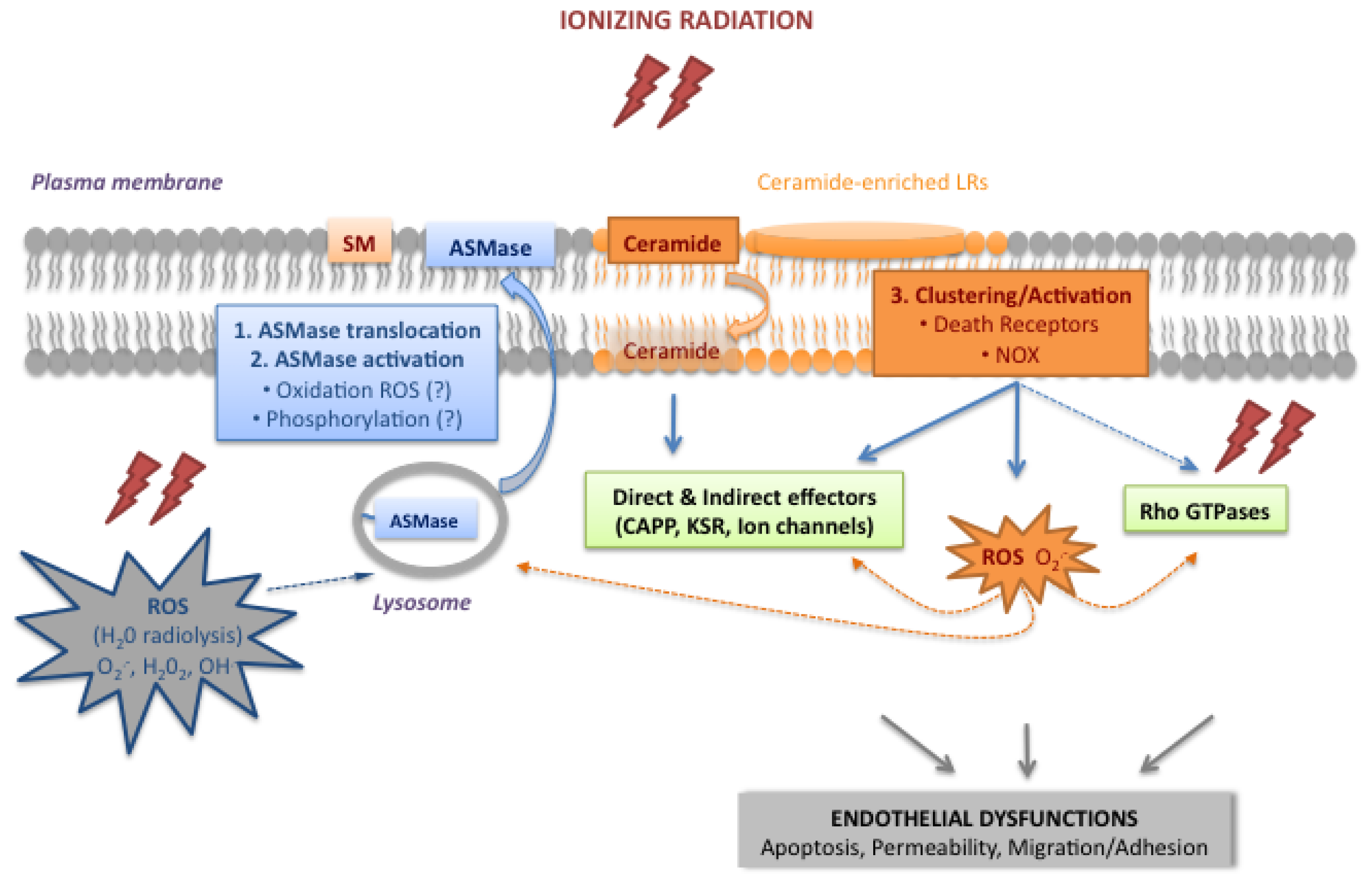
2.1. Activation of the Acid Sphingomyelinase Pathway
2.1.1. Translocation and Activation of ASMase
2.1.2. Ceramide, a Plasma Membrane Reorganizer
2.1.3. Ceramide, a Second Messenger
2.2. The Rho Family of Small GTPases
3. Protecting Tissues from Radiation Toxicity by Targeting the Endothelium
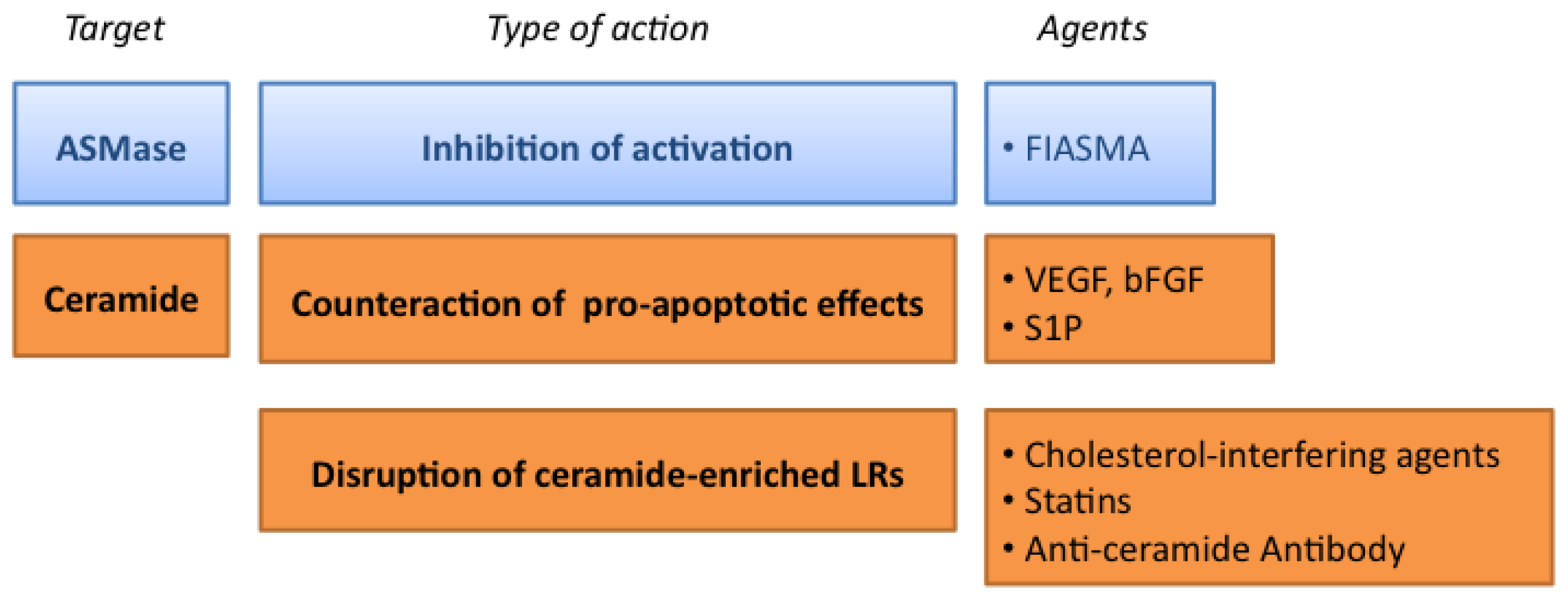
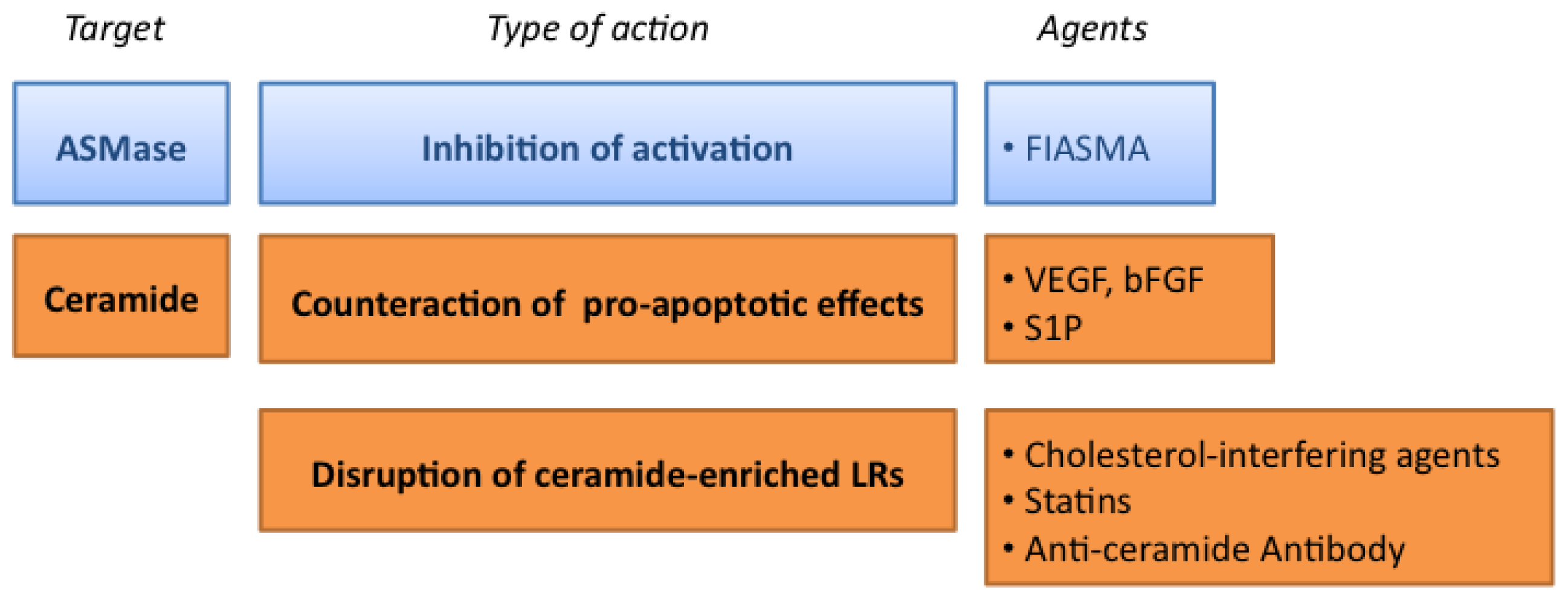
3.1. Targeting the ASMase Activity
3.2. Counteracting the Pro-Apoptotic Activity of Ceramide
3.3. Targeting the Formation of Ceramide-Enriched LRs
4. Conclusions

Acknowledgments
Abbreviations
| ASMase | acid sphingomyelinase |
| bFGF | basic fibroblast growth factor |
| CAPP | ceramide-activated protein phosphatase |
| EGF-R | epithelial growth factor-receptor |
| eNOS | endothelial nitric oxide synthase |
| FIASMA | functional inhibitors of acid sphingomyelinase |
| GIS | gastrointestinal syndrome |
| Gy | Gray |
| IGRT | image-guided radiotherapy |
| IMRT | intensity-modulated radiotherapy |
| KSR | kinase suppressor of Ras |
| ox-LDL | oxidized-low density lipoprotein |
| LRs | lipid rafts |
| LOX-1 | oxidized-low density lipoprotein receptor |
| MAPK | mitogen activated protein kinase |
| MβCD | methyl-β-cyclodextrin |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NAC | N-acetylcysteine |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| PI3K | phosphoinositide 3-kinase |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| SBRT | stereotactic body radiotherapy |
| S1P | sphingosine-1-phosphate |
| SM | sphingomyelin |
| TNF-α | tumor necrosis factor-α |
| TRAIL | TNF-related apoptosis-inducing ligand |
| UV | ultraviolet |
| VEGF | vascular endothelial growth factor. |
Conflicts of Interest
References
- Stone, H.B.; Coleman, C.N.; Anscher, M.S.; McBride, W.H. Effects of radiation on normal tissue: Consequences and mechanisms. Lancet Oncol 2003, 4, 529–536. [Google Scholar]
- Bhide, S.A.; Nutting, C.M. Recent advances in radiotherapy. BMC Med 2010, 8, 25. [Google Scholar]
- Almaghrabi, M.Y.; Supiot, S.; Paris, F.; Mahe, M.A.; Rio, E. Stereotactic body radiation therapy for abdominal oligometastases: A biological and clinical review. Radiat. Oncol 2012, 7. [Google Scholar] [CrossRef]
- Lo, S.S.; Fakiris, A.J.; Chang, E.L.; Mayr, N.A.; Wang, J.Z.; Papiez, L.; Teh, B.S.; McGarry, R.C.; Cardenes, H.R.; Timmerman, R.D. Stereotactic body radiation therapy: A novel treatment modality. Nat. Rev. Clin. Oncol 2010, 7, 44–54. [Google Scholar]
- Milano, M.T.; Constine, L.S.; Okunieff, P. Normal tissue toxicity after small field hypofractionated stereotactic body radiation. Radiat. Oncol 2008, 3, 36. [Google Scholar]
- Brown, J.M.; Koong, A.C. High-dose single-fraction radiotherapy: Exploiting a new biology? Int. J. Radiat. Oncol. Biol. Phys 2008, 71, 324–325. [Google Scholar]
- Pena, L.A.; Fuks, Z.; Kolesnick, R.N. Radiation-induced apoptosis of endothelial cells in the murine central nervous system: Protection by fibroblast growth factor and sphingomyelinase deficiency. Cancer Res 2000, 60, 321–327. [Google Scholar]
- Paris, F.; Fuks, Z.; Kang, A.; Capodieci, P.; Juan, G.; Ehleiter, D.; Haimovitz-Friedman, A.; Cordon-Cardo, C.; Kolesnick, R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 2001, 293, 293–297. [Google Scholar]
- Rodemann, H.P.; Blaese, M.A. Responses of normal cells to ionizing radiation. Semin. Radiat. Oncol 2007, 17, 81–88. [Google Scholar]
- Gaugler, M.H. A unifying system: Does the vascular endothelium have a role to play in multi-organ failure following radiation exposure? BJR Suppl 2005, 27, 100–105. [Google Scholar]
- Milliat, F.; Francois, A.; Tamarat, R.; Benderitter, M. Role of endothelium in radiation-induced normal tissue damages. Ann. Cardiol. Angeiol 2008, 57, 139–148. [Google Scholar]
- Oh, C.W.; Bump, E.A.; Kim, J.S.; Janigro, D.; Mayberg, M.R. Induction of a senescence-like phenotype in bovine aortic endothelial cells by ionizing radiation. Radiat. Res 2001, 156, 232–240. [Google Scholar]
- Supiot, S.; Paris, F. Radiobiology dedicated to endothelium. Cancer Radiother 2012, 16, 11–15. [Google Scholar]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. [Google Scholar]
- Fuks, Z.; Kolesnick, R. Engaging the vascular component of the tumor response. Cancer Cell 2005, 8, 89–91. [Google Scholar]
- Haimovitz-Friedman, A.; Kan, C.C.; Ehleiter, D.; Persaud, R.S.; McLoughlin, M.; Fuks, Z.; Kolesnick, R.N. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J. Exp. Med 1994, 180, 525–535. [Google Scholar]
- Valerie, K.; Yacoub, A.; Hagan, M.P.; Curiel, D.T.; Fisher, P.B.; Grant, S.; Dent, P. Radiation-induced cell signaling: Inside-out and outside-in. Mol. Cancer Ther 2007, 6, 789–801. [Google Scholar]
- Santana, P.; Pena, L.A.; Haimovitz-Friedman, A.; Martin, S.; Green, D.; McLoughlin, M.; Cordon-Cardo, C.; Schuchman, E.H.; Fuks, Z.; Kolesnick, R. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 1996, 86, 189–199. [Google Scholar]
- Milhas, D.; Clarke, C.J.; Hannun, Y.A. Sphingomyelin metabolism at the plasma membrane: implications for bioactive sphingolipids. FEBS Lett 2010, 584, 1887–1894. [Google Scholar]
- Schissel, S.L.; Jiang, X.; Tweedie-Hardman, J.; Jeong, T.; Camejo, E.H.; Najib, J.; Rapp, J.H.; Williams, K.J.; Tabas, I. Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH. Implications for atherosclerotic lesion development. J. Biol. Chem 1998, 273, 2738–2746. [Google Scholar]
- Xu, M.; Xia, M.; Li, X.X.; Han, W.Q.; Boini, K.M.; Zhang, F.; Zhang, Y.; Ritter, J.K.; Li, P.L. Requirement of translocated lysosomal V1 H(+)-ATPase for activation of membrane acid sphingomyelinase and raft clustering in coronary endothelial cells. Mol. Biol. Cell 2012, 23, 1546–1557. [Google Scholar]
- Charruyer, A.; Grazide, S.; Bezombes, C.; Muller, S.; Laurent, G.; Jaffrezou, J.P. UV-C light induces raft-associated acid sphingomyelinase and JNK activation and translocation independently on a nuclear signal. J. Biol. Chem 2005, 280, 19196–19204. [Google Scholar]
- Dumitru, C.A.; Gulbins, E. TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene 2006, 25, 5612–5625. [Google Scholar]
- Grassme, H.; Cremesti, A.; Kolesnick, R.; Gulbins, E. Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene 2003, 22, 5457–5470. [Google Scholar]
- Truman, J.P.; Garcia-Barros, M.; Kaag, M.; Hambardzumyan, D.; Stancevic, B.; Chan, M.; Fuks, Z.; Kolesnick, R.; Haimovitz-Friedman, A. Endothelial membrane remodeling is obligate for anti-angiogenic radiosensitization during tumor radiosurgery. PLoS One 2010, 5, e12310. [Google Scholar]
- Perrotta, C.; Bizzozero, L.; Cazzato, D.; Morlacchi, S.; Assi, E.; Simbari, F.; Zhang, Y.; Gulbins, E.; Bassi, M.T.; Rosa, P.; et al. Syntaxin 4 is required for acid sphingomyelinase activity and apoptotic function. J. Biol. Chem 2010, 285, 40240–40251. [Google Scholar]
- Han, W.Q.; Xia, M.; Xu, M.; Boini, K.M.; Ritter, J.K.; Li, N.J.; Li, P.L. Lysosome fusion to the cell membrane is mediated by the dysferlin C2A domain in coronary arterial endothelial cells. J. Cell Sci 2012, 125, 1225–1234. [Google Scholar]
- Edelmann, B.; Bertsch, U.; Tchikov, V.; Winoto-Morbach, S.; Perrotta, C.; Jakob, M.; Adam-Klages, S.; Kabelitz, D.; Schutze, S. Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. EMBO J 2011, 30, 379–394. [Google Scholar]
- Zeidan, Y.H.; Wu, B.X.; Jenkins, R.W.; Obeid, L.M.; Hannun, Y.A. A novel role for protein kinase Cdelta-mediated phosphorylation of acid sphingomyelinase in UV light-induced mitochondrial injury. FASEB J 2008, 22, 183–193. [Google Scholar]
- Qiu, H.; Edmunds, T.; Baker-Malcolm, J.; Karey, K.P.; Estes, S.; Schwarz, C.; Hughes, H.; van Patten, S.M. Activation of human acid sphingomyelinase through modification or deletion of C-terminal cysteine. J. Biol. Chem 2003, 278, 32744–32752. [Google Scholar]
- Lang, P.A.; Schenck, M.; Nicolay, J.P.; Becker, J.U.; Kempe, D.S.; Lupescu, A.; Koka, S.; Eisele, K.; Klarl, B.A.; Rubben, H.; et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat. Med 2007, 13, 164–170. [Google Scholar]
- Castillo, S.S.; Levy, M.; Thaikoottathil, J.V.; Goldkorn, T. Reactive nitrogen and oxygen species activate different sphingomyelinases to induce apoptosis in airway epithelial cells. Exp. Cell Res 2007, 313, 2680–2686. [Google Scholar]
- Zhang, A.Y.; Yi, F.; Jin, S.; Xia, M.; Chen, Q.Z.; Gulbins, E.; Li, P.L. Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid. Redox Signal 2007, 9, 817–828. [Google Scholar]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett 2012, 327, 48–60. [Google Scholar]
- Paris, F.; Nantes, F.R. CRCNA. Personal communication, 2013.
- Kolesnick, R.N.; Goni, F.M.; Alonso, A. Compartmentalization of ceramide signaling: Physical foundations and biological effects. J. Cell Physiol 2000, 184, 285–300. [Google Scholar]
- Holopainen, J.M.; Subramanian, M.; Kinnunen, P.K. Sphingomyelinase induces lipid microdomain formation in a fluid phosphatidylcholine/sphingomyelin membrane. Biochemistry 1998, 37, 17562–17570. [Google Scholar]
- Nurminen, T.A.; Holopainen, J.M.; Zhao, H.; Kinnunen, P.K. Observation of topical catalysis by sphingomyelinase coupled to microspheres. J. Am. Chem. Soc 2002, 124, 12129–12134. [Google Scholar]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar]
- Zhang, Y.; Li, X.; Becker, K.A.; Gulbins, E. Ceramide-enriched membrane domains—Structure and function. Biochim. Biophys. Acta 2009, 1788, 178–183. [Google Scholar]
- Stancevic, B.; Kolesnick, R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett 2010, 584, 1728–1740. [Google Scholar]
- Bionda, C.; Hadchity, E.; Alphonse, G.; Chapet, O.; Rousson, R.; Rodriguez-Lafrasse, C.; Ardail, D. Radioresistance of human carcinoma cells is correlated to a defect in raft membrane clustering. Free Radic. Biol. Med 2007, 43, 681–694. [Google Scholar]
- Rotolo, J.; Stancevic, B.; Zhang, J.; Hua, G.; Fuller, J.; Yin, X.; Haimovitz-Friedman, A.; Kim, K.; Qian, M.; Cardo-Vila, M.; et al. Anti-ceramide antibody prevents the radiation gastrointestinal syndrome in mice. J. Clin. Invest 2012, 122, 1786–1790. [Google Scholar]
- Gulbins, E.; Kolesnick, R. Raft ceramide in molecular medicine. Oncogene 2003, 22, 7070–7077. [Google Scholar]
- Kolesnick, R.; Fuks, Z. Radiation and ceramide-induced apoptosis. Oncogene 2003, 22, 5897–5906. [Google Scholar]
- Bao, J.X.; Jin, S.; Zhang, F.; Wang, Z.C.; Li, N.; Li, P.L. Activation of membrane NADPH oxidase associated with lysosome-targeted acid sphingomyelinase in coronary endothelial cells. Antioxid. Redox Signal 2010, 12, 703–712. [Google Scholar]
- Zhang, A.Y.; Yi, F.; Zhang, G.; Gulbins, E.; Li, P.L. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006, 47, 74–80. [Google Scholar]
- Viola, A. The amplification of TCR signaling by dynamic membrane microdomains. Trends Immunol 2001, 22, 322–327. [Google Scholar]
- Li, X.; Han, W.Q.; Boini, K.M.; Xia, M.; Zhang, Y.; Li, P.L. TRAIL death receptor 4 signaling via lysosome fusion and membrane raft clustering in coronary arterial endothelial cells: Evidence from ASM knockout mice. J. Mol. Med 2013, 91, 25–36. [Google Scholar]
- Jin, S.; Zhang, Y.; Yi, F.; Li, P.L. Critical role of lipid raft redox signaling platforms in endostatin-induced coronary endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol 2008, 28, 485–490. [Google Scholar]
- Collins-Underwood, J.R.; Zhao, W.; Sharpe, J.G.; Robbins, M.E. NADPH oxidase mediates radiation-induced oxidative stress in rat brain microvascular endothelial cells. Free Radic. Biol. Med 2008, 45, 929–938. [Google Scholar]
- Contreras, F.X.; Villar, A.V.; Alonso, A.; Kolesnick, R.N.; Goni, F.M. Sphingomyelinase activity causes transbilayer lipid translocation in model and cell membranes. J. Biol. Chem 2003, 278, 37169–37174. [Google Scholar]
- Chalfant, C.E.; Kishikawa, K.; Mumby, M.C.; Kamibayashi, C.; Bielawska, A.; Hannun, Y.A. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem 1999, 274, 20313–20317. [Google Scholar]
- Chalfant, C.E.; Szulc, Z.; Roddy, P.; Bielawska, A.; Hannun, Y.A. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J. Lipid Res 2004, 45, 496–506. [Google Scholar]
- Yan, F.; John, S.K.; Polk, D.B. Kinase suppressor of Ras determines survival of intestinal epithelial cells exposed to tumor necrosis factor. Cancer Res 2001, 61, 8668–8675. [Google Scholar]
- Zhang, Y.; Yao, B.; Delikat, S.; Bayoumy, S.; Lin, X.H.; Basu, S.; McGinley, M.; Chan-Hui, P.Y.; Lichenstein, H.; Kolesnick, R. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell 1997, 89, 63–72. [Google Scholar]
- Yin, X.; Zafrullah, M.; Lee, H.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. A ceramide-binding C1 domain mediates kinase suppressor of ras membrane translocation. Cell Physiol. Biochem 2009, 24, 219–230. [Google Scholar]
- Bourbon, N.A.; Yun, J.; Kester, M. Ceramide directly activates protein kinase C zeta to regulate a stress-activated protein kinase signaling complex. J. Biol. Chem 2000, 275, 35617–35623. [Google Scholar]
- Lozano, J.; Berra, E.; Municio, M.M.; Diaz-Meco, M.T.; Dominguez, I.; Sanz, L.; Moscat, J. Protein kinase C zeta isoform is critical for kappa B-dependent promoter activation by sphingomyelinase. J. Biol. Chem 1994, 269, 19200–19202. [Google Scholar]
- Muller, G.; Ayoub, M.; Storz, P.; Rennecke, J.; Fabbro, D.; Pfizenmaier, K. PKC zeta is a molecular switch in signal transduction of TNF-alpha, bifunctionally regulated by ceramide and arachidonic acid. EMBO J 1995, 14, 1961–1969. [Google Scholar]
- Heinrich, M.; Neumeyer, J.; Jakob, M.; Hallas, C.; Tchikov, V.; Winoto-Morbach, S.; Wickel, M.; Schneider-Brachert, W.; Trauzold, A.; Hethke, A.; et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and −3 activation. Cell Death Differ 2004, 11, 550–563. [Google Scholar]
- Church, L.D.; Hessler, G.; Goodall, J.E.; Rider, D.A.; Workman, C.J.; Vignali, D.A.; Bacon, P.A.; Gulbins, E.; Young, S.P. TNFR1-induced sphingomyelinase activation modulates TCR signaling by impairing store-operated Ca2+ influx. J. Leukoc. Biol 2005, 78, 266–278. [Google Scholar]
- Lepple-Wienhues, A.; Belka, C.; Laun, T.; Jekle, A.; Walter, B.; Wieland, U.; Welz, M.; Heil, L.; Kun, J.; Busch, G.; et al. Stimulation of CD95 (Fas) blocks T lymphocyte calcium channels through sphingomyelinase and sphingolipids. Proc. Natl. Acad. Sci. USA 1999, 96, 13795–13800. [Google Scholar]
- Szabo, I.; Gulbins, E.; Apfel, H.; Zhang, X.; Barth, P.; Busch, A.E.; Schlottmann, K.; Pongs, O.; Lang, F. Tyrosine phosphorylation-dependent suppression of a voltage-gated K+ channel in T lymphocytes upon Fas stimulation. J. Biol. Chem 1996, 271, 20465–20469. [Google Scholar]
- Wu, F.; Wilson, J.X. Peroxynitrite-dependent activation of protein phosphatase type 2A mediates microvascular endothelial barrier dysfunction. Cardiovasc. Res 2009, 81, 38–45. [Google Scholar]
- Haendeler, J.; Popp, R.; Goy, C.; Tischler, V.; Zeiher, A.M.; Dimmeler, S. Cathepsin D and H2O2 stimulate degradation of thioredoxin-1: Implication for endothelial cell apoptosis. J. Biol. Chem 2005, 280, 42945–42951. [Google Scholar]
- Oubaha, M.; Gratton, J.P. Phosphorylation of endothelial nitric oxide synthase by atypical PKC zeta contributes to angiopoietin-1-dependent inhibition of VEGF-induced endothelial permeability in vitro. Blood 2009, 114, 3343–3351. [Google Scholar]
- Friedman, M.; Ryan, U.S.; Davenport, W.C.; Chaney, E.L.; Strickland, D.L.; Kwock, L. Reversible alterations in cultured pulmonary artery endothelial cell monolayer morphology and albumin permeability induced by ionizing radiation. J. Cell Physiol 1986, 129, 237–249. [Google Scholar]
- Gabrys, D.; Greco, O.; Patel, G.; Prise, K.M.; Tozer, G.M.; Kanthou, C. Radiation effects on the cytoskeleton of endothelial cells and endothelial monolayer permeability. Int. J. Radiat. Oncol. Biol. Phys 2007, 69, 1553–1562. [Google Scholar]
- Rousseau, M.; Gaugler, M.H.; Rodallec, A.; Bonnaud, S.; Paris, F.; Corre, I. RhoA GTPase regulates radiation-induced alterations in endothelial cell adhesion and migration. Biochem. Biophys. Res. Commun 2011, 414, 750–755. [Google Scholar]
- Onoda, J.M.; Kantak, S.S.; Diglio, C.A. Radiation induced endothelial cell retraction in vitro: Correlation with acute pulmonary edema. Pathol. Oncol. Res 1999, 5, 49–55. [Google Scholar]
- Hall, A. Rho family GTPases. Biochem. Soc. Trans 2012, 40, 1378–1382. [Google Scholar]
- Blirando, K.; Hneino, M.; Martelly, I.; Benderitter, M.; Milliat, F.; Francois, A. Mast cells and ionizing radiation induce a synergistic expression of inflammatory genes in endothelial cells by a mechanism involving p38alpha MAP kinase and (p65) NF-kappaB activation. Radiat. Res 2012, 178, 556–567. [Google Scholar]
- Gupta, N.; Nodzenski, E.; Khodarev, N.N.; Yu, J.; Khorasani, L.; Beckett, M.A.; Kufe, D.W.; Weichselbaum, R.R. Angiostatin effects on endothelial cells mediated by ceramide and RhoA. EMBO Rep 2001, 2, 536–540. [Google Scholar]
- Brenner, B.; Koppenhoefer, U.; Weinstock, C.; Linderkamp, O.; Lang, F.; Gulbins, E. Fas- or ceramide-induced apoptosis is mediated by a Rac1-regulated activation of Jun N-terminal kinase/p38 kinases and GADD153. J. Biol. Chem 1997, 272, 22173–22181. [Google Scholar]
- Del Pozo, M.A.; Alderson, N.B.; Kiosses, W.B.; Chiang, H.H.; Anderson, R.G.; Schwartz, M.A. Integrins regulate Rac targeting by internalization of membrane domains. Science 2004, 303, 839–842. [Google Scholar]
- Plowman, S.J.; Muncke, C.; Parton, R.G.; Hancock, J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2005, 102, 15500–15505. [Google Scholar]
- Bentzen, S.M. Preventing or reducing late side effects of radiation therapy: Radiobiology meets molecular pathology. Nat. Rev. Cancer 2006, 6, 702–713. [Google Scholar]
- Adams, M.J.; Hardenbergh, P.H.; Constine, L.S.; Lipshultz, S.E. Radiation-associated cardiovascular disease. Crit. Rev. Oncol. Hematol 2003, 45, 55–75. [Google Scholar]
- Arenz, C. Small molecule inhibitors of acid sphingomyelinase. Cell Physiol. Biochem 2010, 26, 1–8. [Google Scholar]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Muhle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.W.; Gulbins, E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): A novel pharmacological group of drugs with broad clinical applications. Cell Physiol. Biochem 2010, 26, 9–20. [Google Scholar]
- Kornhuber, J.; Muehlbacher, M.; Trapp, S.; Pechmann, S.; Friedl, A.; Reichel, M.; Muhle, C.; Terfloth, L.; Groemer, T.W.; Spitzer, G.M.; et al. Identification of novel functional inhibitors of acid sphingomyelinase. PLoS One 2011, 6, e23852. [Google Scholar]
- Nahrlich, L.; Mainz, J.G.; Adams, C.; Engel, C.; Herrmann, G.; Icheva, V.; Lauer, J.; Deppisch, C.; Wirth, A.; Unger, K.; et al. Therapy of CF-patients with amitriptyline and placebo—A randomised, double-blind, placebo-controlled phase IIb multicenter, cohort-study. Cell Physiol. Biochem 2013, 31, 505–512. [Google Scholar]
- Xu, J.; Yan, X.; Gao, R.; Mao, L.; Cotrim, A.P.; Zheng, C.; Zhang, C.; Baum, B.J.; Wang, S. Effect of irradiation on microvascular endothelial cells of parotid glands in the miniature pig. Int. J. Radiat. Oncol. Biol. Phys 2010, 78, 897–903. [Google Scholar]
- Simons, M.; Annex, B.H.; Laham, R.J.; Kleiman, N.; Henry, T.; Dauerman, H.; Udelson, J.E.; Gervino, E.V.; Pike, M.; Whitehouse, M.J.; et al. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: Double-blind, randomized, controlled clinical trial. Circulation 2002, 105, 788–793. [Google Scholar]
- Ziobro, R.; Henry, B.; Edwards, M.J.; Lentsch, A.B.; Gulbins, E. Ceramide mediates lung fibrosis in cystic fibrosis. Biochem. Biophys. Res. Commun 2013, 434, 705–709. [Google Scholar]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar]
- Morita, Y.; Perez, G.I.; Paris, F.; Miranda, S.R.; Ehleiter, D.; Haimovitz-Friedman, A.; Fuks, Z.; Xie, Z.; Reed, J.C.; Schuchman, E.H.; et al. Oocyte apoptosis is suppressed by disruption of the acid sphingomyelinase gene or by sphingosine-1-phosphate therapy. Nat. Med 2000, 6, 1109–1114. [Google Scholar]
- Paris, F.; Perez, G.I.; Fuks, Z.; Haimovitz-Friedman, A.; Nguyen, H.; Bose, M.; Ilagan, A.; Hunt, P.A.; Morgan, W.F.; Tilly, J.L.; et al. Sphingosine 1-phosphate preserves fertility in irradiated female mice without propagating genomic damage in offspring. Nat. Med 2002, 8, 901–902. [Google Scholar]
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta 2002, 1585, 193–201. [Google Scholar]
- Bonnaud, S.; Niaudet, C.; Legoux, F.; Corre, I.; Delpon, G.; Saulquin, X.; Fuks, Z.; Gaugler, M.H.; Kolesnick, R.; Paris, F. Sphingosine-1-phosphate activates the AKT pathway to protect small intestines from radiation-induced endothelial apoptosis. Cancer Res 2010, 70, 9905–9915. [Google Scholar]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol 2012, 22, 50–60. [Google Scholar]
- Cho, C.H.; Kammerer, R.A.; Lee, H.J.; Yasunaga, K.; Kim, K.T.; Choi, H.H.; Kim, W.; Kim, S.H.; Park, S.K.; Lee, G.M.; et al. Designed angiopoietin-1 variant, COMP-Ang1, protects against radiation-induced endothelial cell apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 5553–5558. [Google Scholar]
- Bonnaud, S.; Niaudet, C.; Pottier, G.; Gaugler, M.H.; Millour, J.; Barbet, J.; Sabatier, L.; Paris, F. Sphingosine-1-phosphate protects proliferating endothelial cells from ceramide-induced apoptosis but not from DNA damage-induced mitotic death. Cancer Res 2007, 67, 1803–1811. [Google Scholar]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol 2000, 1, 31–39. [Google Scholar]
- Matarazzo, S.; Quitadamo, M.C.; Mango, R.; Ciccone, S.; Novelli, G.; Biocca, S. Cholesterol-lowering drugs inhibit lectin-like oxidized low-density lipoprotein-1 receptor function by membrane raft disruption. Mol. Pharmacol 2012, 82, 246–254. [Google Scholar]
- Wei, Y.M.; Li, X.; Xiong, J.; Abais, J.M.; Xia, M.; Boini, K.M.; Zhang, Y.; Li, P.L. Attenuation by statins of membrane raft-redox signaling in coronary arterial endothelium. J. Pharmacol. Exp. Ther 2013, 345, 170–179. [Google Scholar]
- Gaugler, M.H.; Vereycken-Holler, V.; Squiban, C.; Vandamme, M.; Vozenin-Brotons, M.C.; Benderitter, M. Pravastatin limits endothelial activation after irradiation and decreases the resulting inflammatory and thrombotic responses. Radiat. Res 2005, 163, 479–487. [Google Scholar]
- Holler, V.; Buard, V.; Gaugler, M.H.; Guipaud, O.; Baudelin, C.; Sache, A.; Perez Mdel, R.; Squiban, C.; Tamarat, R.; Milliat, F.; et al. Pravastatin limits radiation-induced vascular dysfunction in the skin. J. Invest. Dermatol 2009, 129, 1280–1291. [Google Scholar]
- Jain, M.K.; Ridker, P.M. Anti-inflammatory effects of statins: Clinical evidence and basic mechanisms. Nat. Rev. Drug Discov 2005, 4, 977–987. [Google Scholar]
- Bourgier, C.; Haydont, V.; Milliat, F.; Francois, A.; Holler, V.; Lasser, P.; Bourhis, J.; Mathe, D.; Vozenin-Brotons, M.C. Inhibition of Rho kinase modulates radiation induced fibrogenic phenotype in intestinal smooth muscle cells through alteration of the cytoskeleton and connective tissue growth factor expression. Gut 2005, 54, 336–343. [Google Scholar]
- Haydont, V.; Bourgier, C.; Pocard, M.; Lusinchi, A.; Aigueperse, J.; Mathe, D.; Bourhis, J.; Vozenin-Brotons, M.C. Pravastatin Inhibits the Rho/CCN2/extracellular matrix cascade in human fibrosis explants and improves radiation-induced intestinal fibrosis in rats. Clin. Cancer Res 2007, 13, 5331–5340. [Google Scholar]
- Moding, E.J.; Kastan, M.B.; Kirsch, D.G. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat. Rev. Drug Discov 2013, 12, 526–542. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Corre, I.; Guillonneau, M.; Paris, F. Membrane Signaling Induced by High Doses of Ionizing Radiation in the Endothelial Compartment. Relevance in Radiation Toxicity. Int. J. Mol. Sci. 2013, 14, 22678-22696. https://doi.org/10.3390/ijms141122678
Corre I, Guillonneau M, Paris F. Membrane Signaling Induced by High Doses of Ionizing Radiation in the Endothelial Compartment. Relevance in Radiation Toxicity. International Journal of Molecular Sciences. 2013; 14(11):22678-22696. https://doi.org/10.3390/ijms141122678
Chicago/Turabian StyleCorre, Isabelle, Maëva Guillonneau, and François Paris. 2013. "Membrane Signaling Induced by High Doses of Ionizing Radiation in the Endothelial Compartment. Relevance in Radiation Toxicity" International Journal of Molecular Sciences 14, no. 11: 22678-22696. https://doi.org/10.3390/ijms141122678