The Molecular Mechanisms of Zinc Neurotoxicity and the Pathogenesis of Vascular Type Senile Dementia

Abstract
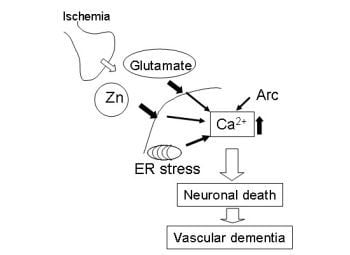
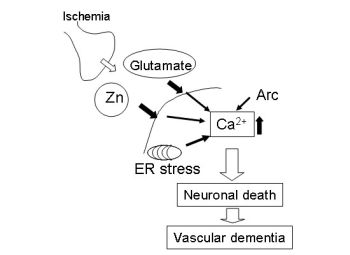
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Zinc and Vascular-Type Dementia
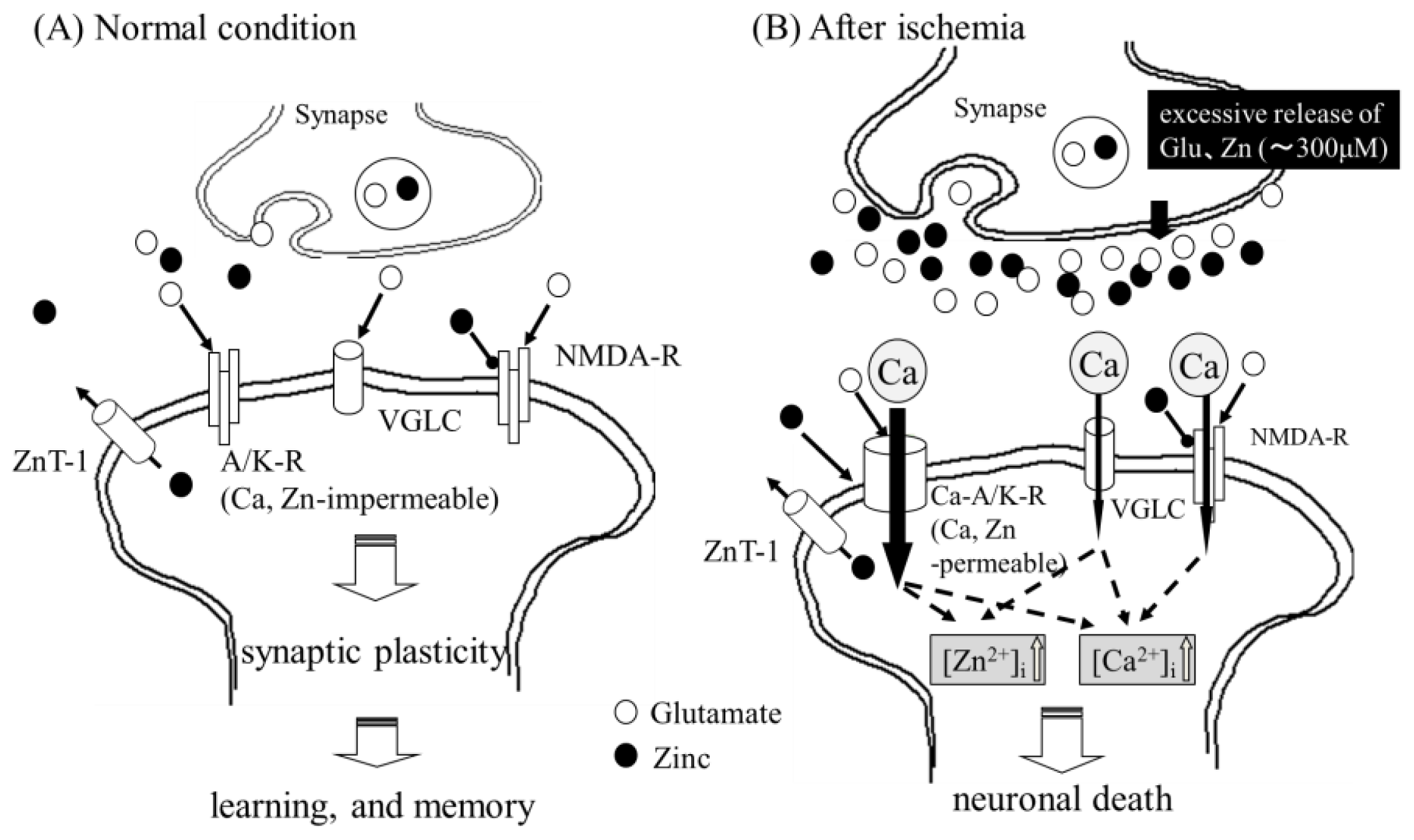
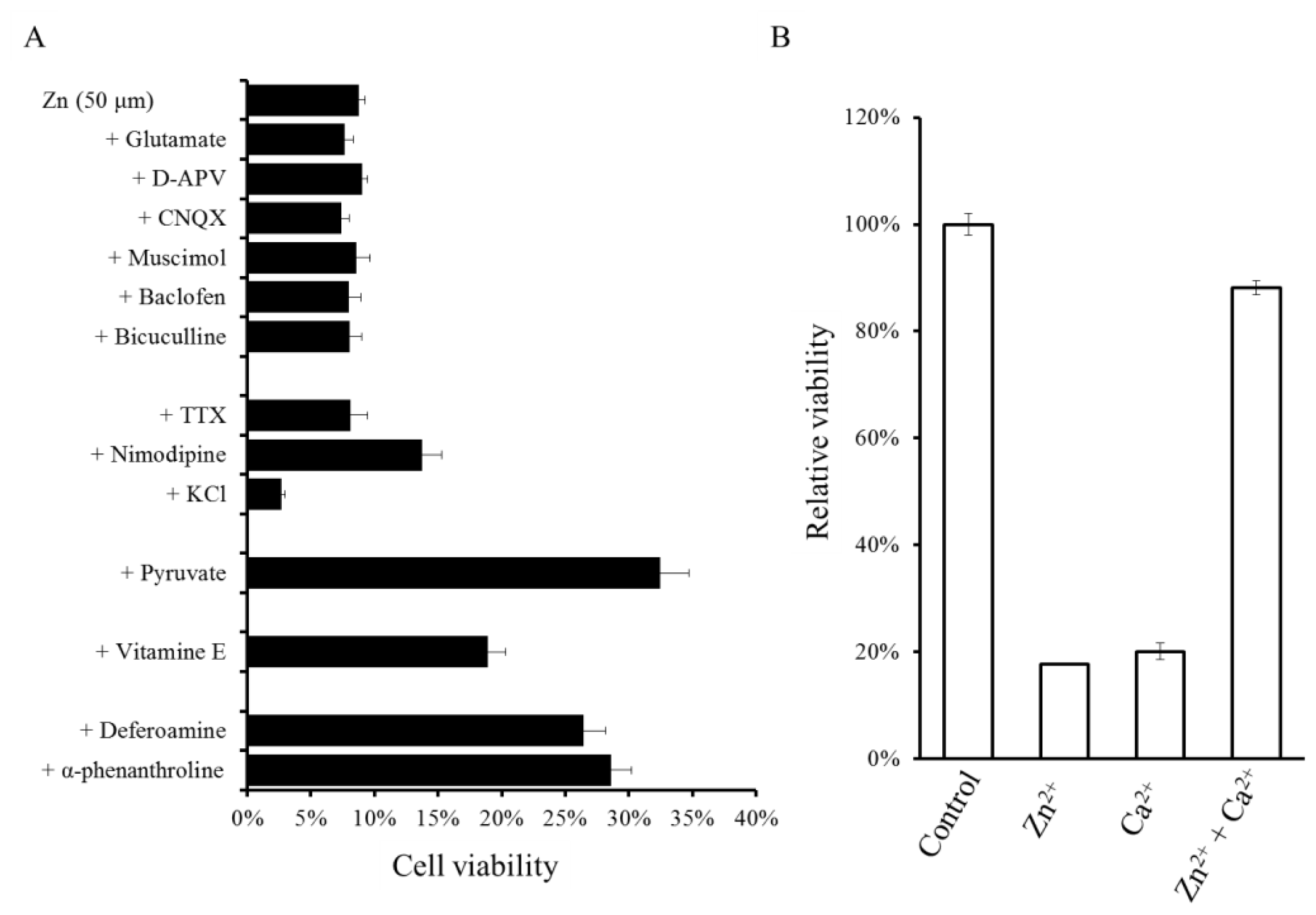
2.1. Zinc-Induced Neurodegeneration after Ischemia
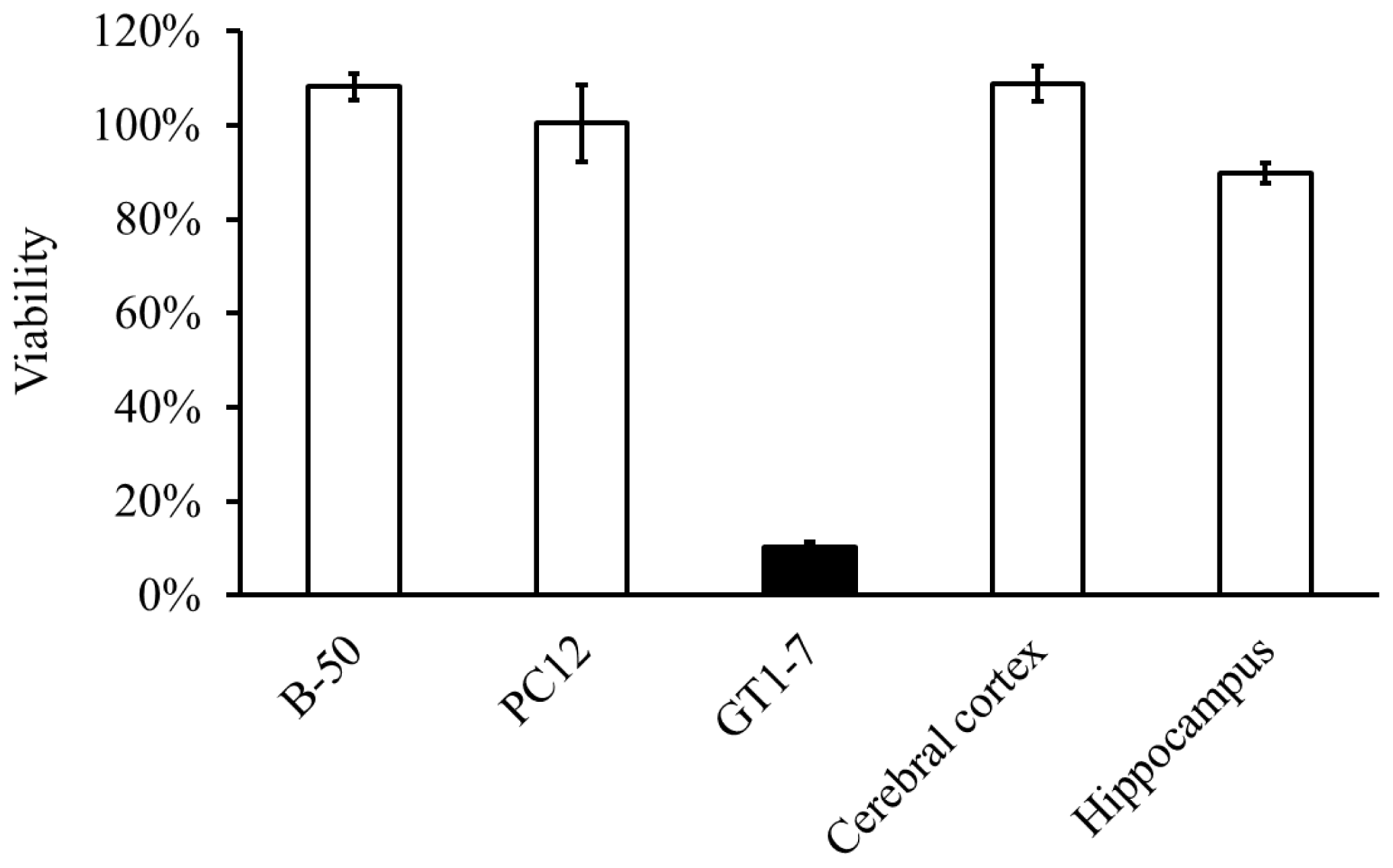
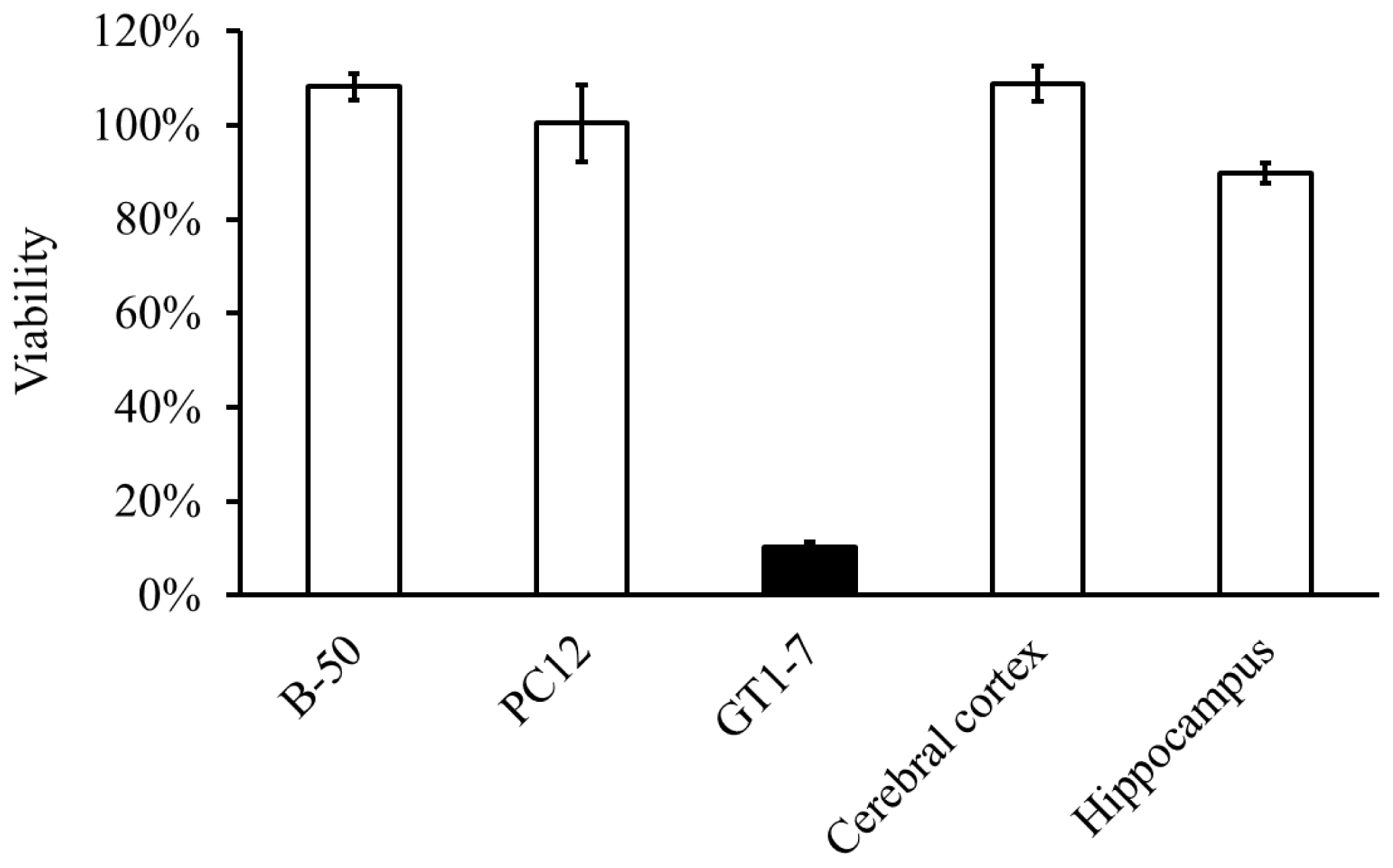
2.2. Molecular Mechanism of Zn-Induced Neuronal Neath: GT1–7 Cells as an In Vitro Model System
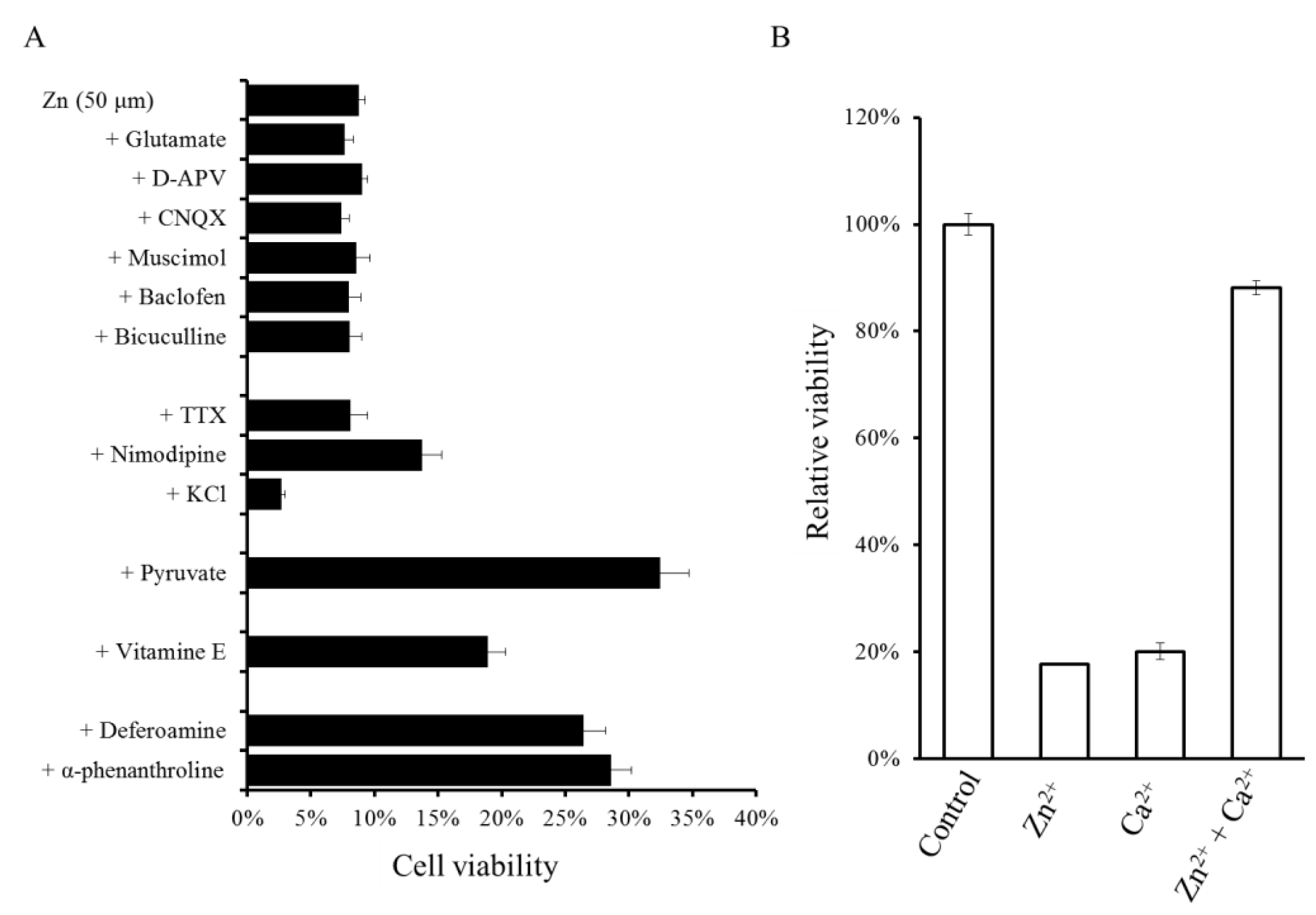
2.3. Implication of Ca Dyshomeostasis in Zn-Induced Neuron Death
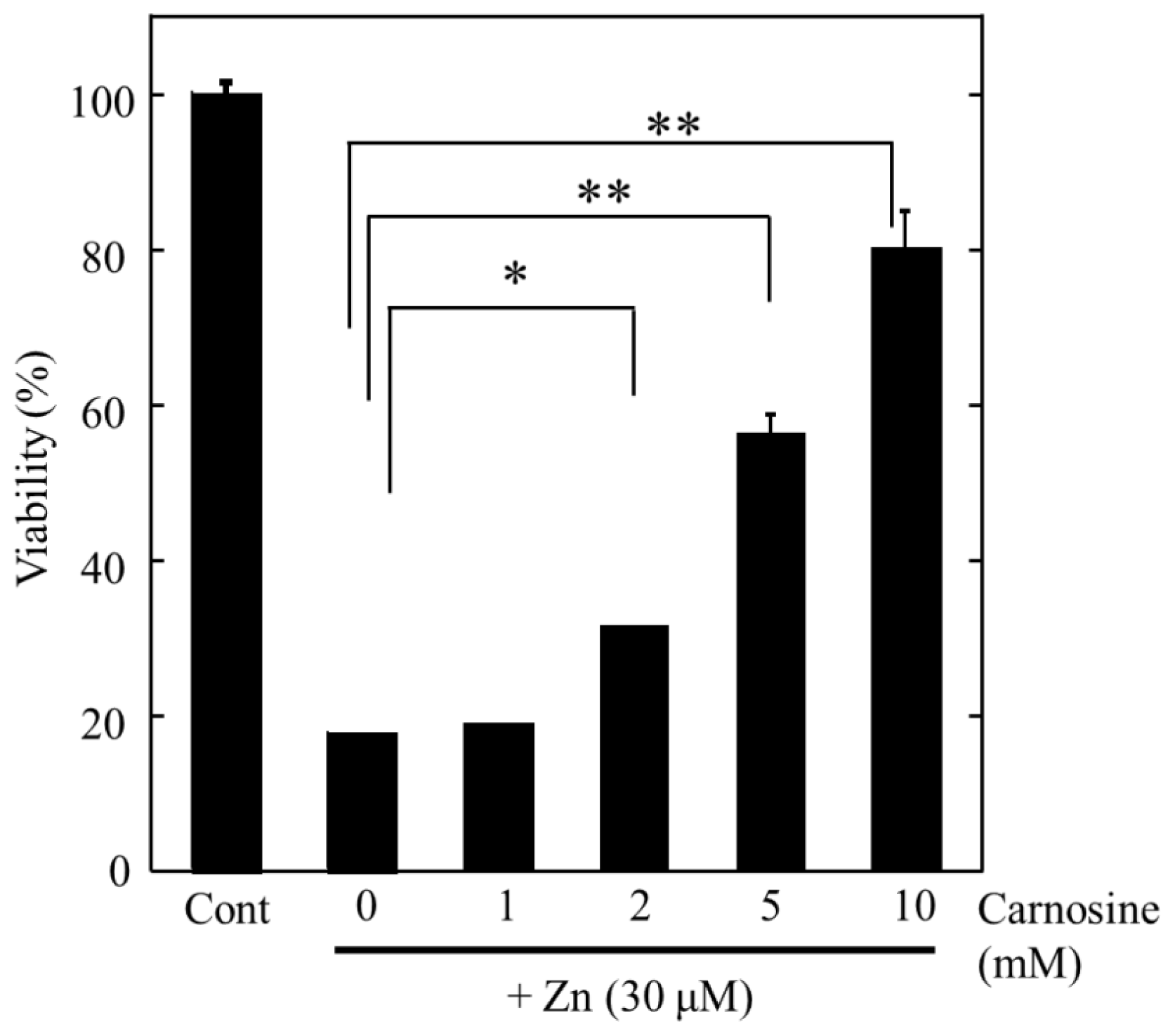
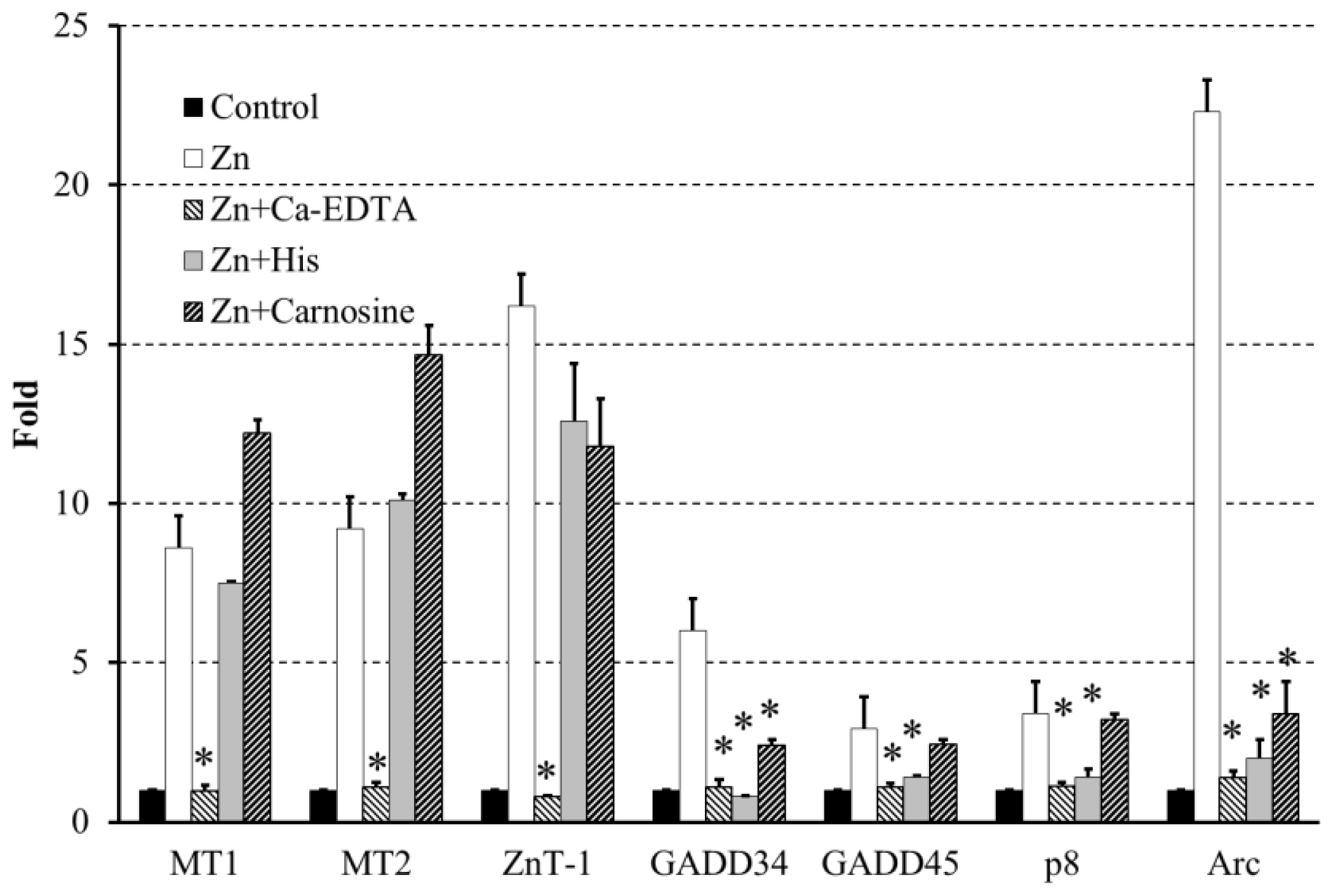
2.4. Altered Gene Expression during Zinc-Induced Neurotoxicity
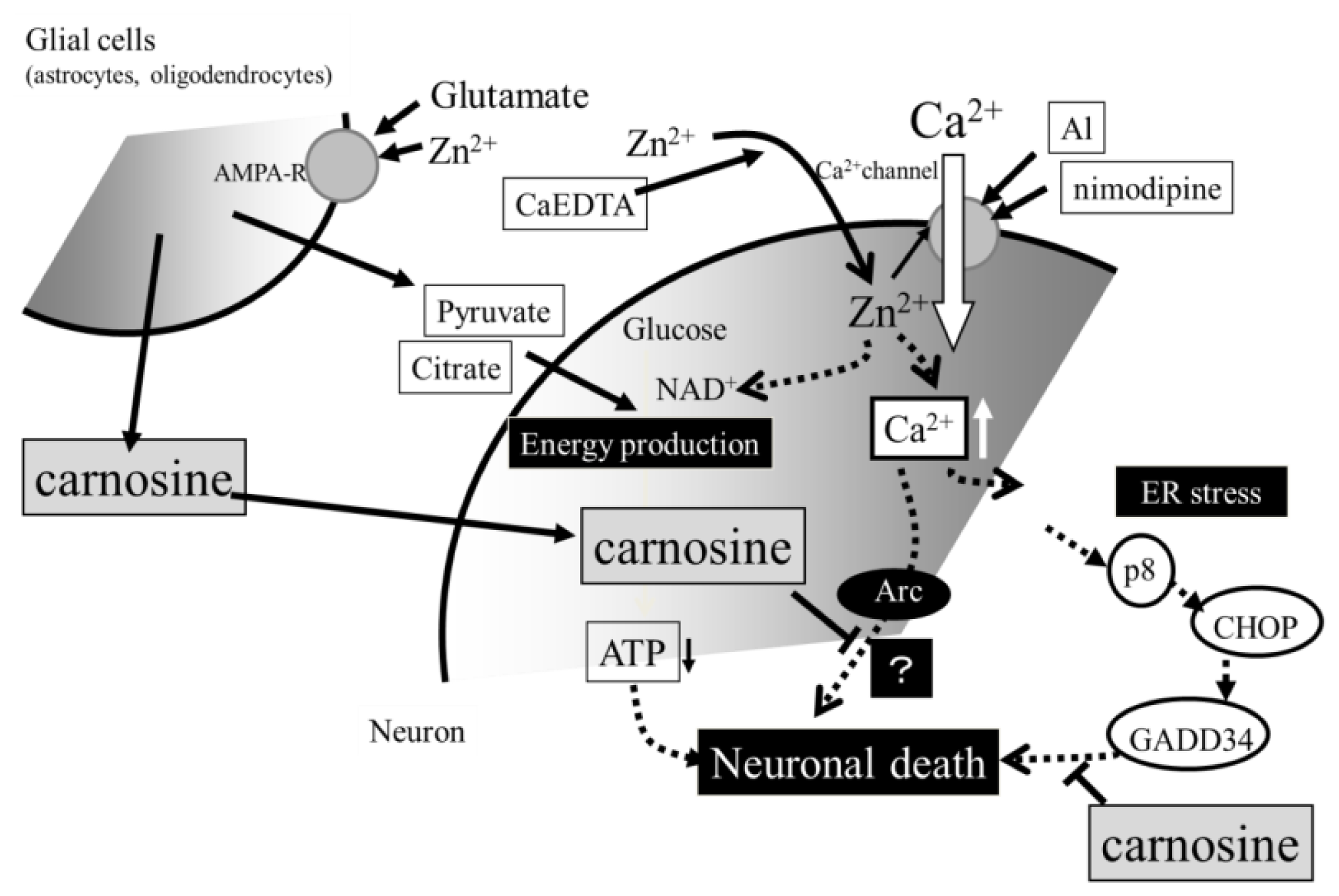
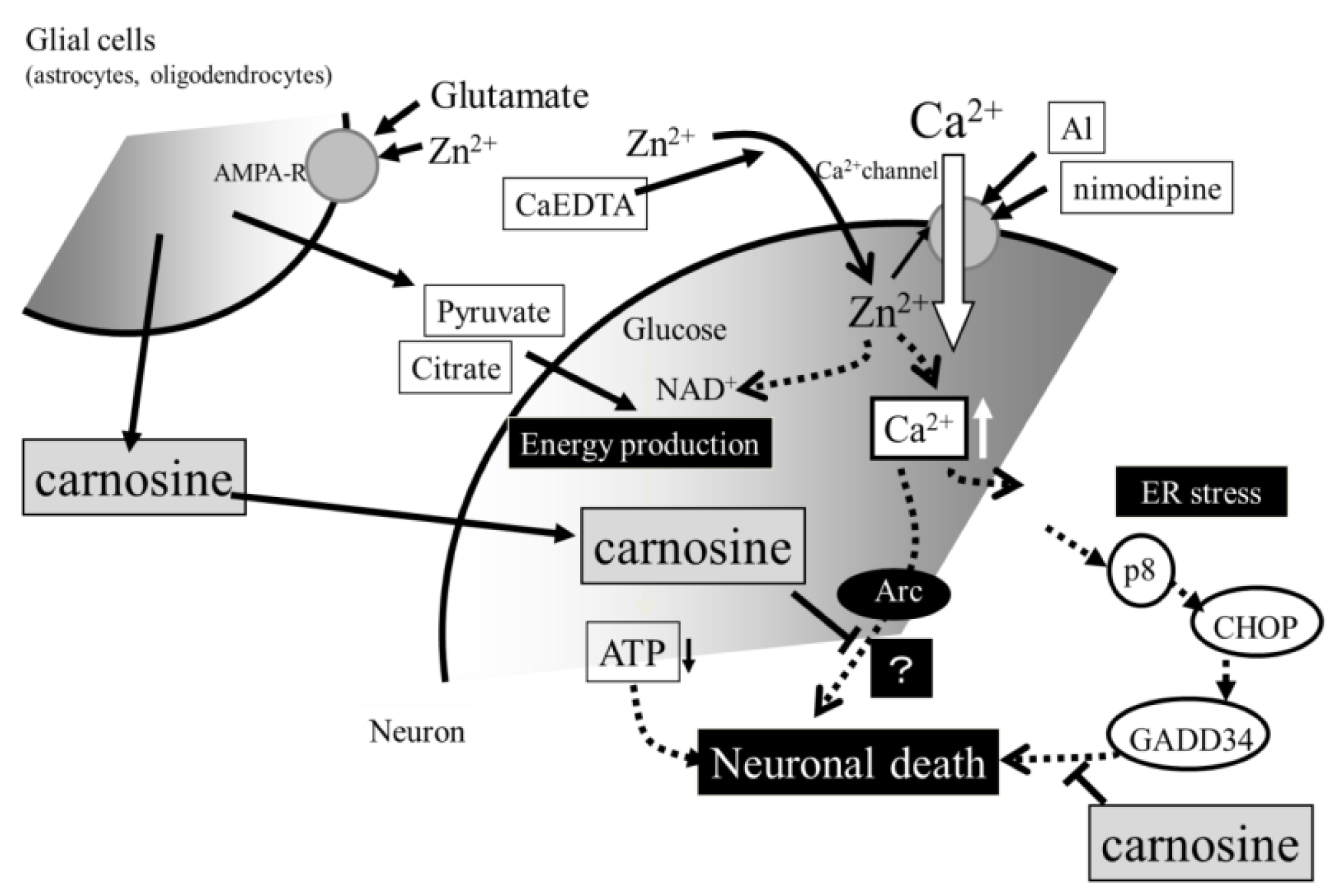
3. Hypothesis Regarding Zn-Induced Neurotoxicity
4. Conclusions



Acknowledgments
Conflicts of Interest
References
- Lee, J.M.; Grabb, M.C.; Zipfel, G.J.; Choi, D.W. Brain tissue responses to ischemia. J. Clin. Invest 2000, 106, 723–731. [Google Scholar]
- De Haan, E.H.; Nys, G.M.; van Zandvoort, M.J. Cognitive function following stroke and vascular cognitive impairment. Curr. Opin. Neurol 2006, 19, 559–564. [Google Scholar]
- Weiss, J.H.; Sensi, S.L.; Koh, J.Y. Zn2+: A novel ionic mediator of neural injury in brain disease. Trends Pharmacol. Sci 2000, 21, 395–401. [Google Scholar]
- Kawahara, M.; Kato-Negishi, M.; Kuroda, Y. Pyruvate blocks zinc-induced neurotoxicity in immortalized hypothalamic neurons. Cell Mol. Neurobiol 2002, 22, 87–93. [Google Scholar]
- Koyama, H.; Konoha, K.; Sadakane, Y.; Ohkawara, S.; Kawahara, M. Zinc neurotoxicity and the pathogenesis of vascular-type dementia: Involvement of calcium dyshomeostasis and carnosine. J. Clin. Toxicol 2011, S3. [Google Scholar] [CrossRef]
- Mellon, P.L.; Windle, J.J.; Goldsmith, P.C.; Padula, C.A.; Roberts, J.L.; Weiner, R.I. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 1990, 5, 1–10. [Google Scholar]
- Mahesh, V.B.; Zamorano, P.; De Sevilla, L.; Lewis, D.; Brann, D.W. Characterization of ionotropic glutamate receptors in rat hypothalamus, pituitary and immortalized gonadotropin-releasing hormone (GnRH) neurons (GT1–7 cells). Neuroendocrinology 1999, 69, 397–407. [Google Scholar]
- Sadakane, Y.; Konoha, K.; Kawahara, M. Protective activity of mango (Mangifera indica L.) fruit against a zinc-induced neuronal cell death is independent of its antioxidant activity. Trace Nutr. Res 2005, 22, 73–79. [Google Scholar]
- Konoha, K.; Sadakane, Y.; Kawahara, M. Carnosine protects GT1–7 cells against zinc-induced neurotoxicity: A possible candidate for treatment for vascular type of dementia. Trace Nutr. Res 2006, 23, 56–62. [Google Scholar]
- Sadakane, Y.; Konoha, K.; Nagata, T.; Kawahara, M. Protective activity of the extracts from Japanese eel (Anguilla japonica) against zinc-induced neuronal cell death: Carnosine and an unknown substance. Trace Nutr. Res 2007, 24, 98–105. [Google Scholar]
- Sadakane, Y.; Konoha, K.; Nagata, T.; Kawahara, M. Improvement of screening for protective substances against zinc-induced neuronal cell death. Trace Nutr. Res 2008, 25, 41–45. [Google Scholar]
- Kawahara, M.; Konoha, K.; Nagata, T.; Sadakane, Y. Protective substances against zinc-induced neuronal death after ischemia: Carnosine a target for drug of vascular type of dementia. Recent Pat. CNS Drug Discov 2007, 2, 145–149. [Google Scholar]
- Corona, C.; Frazzini, V.; Silvestri, E.; Lattanzio, R.; La Sorda, R.; Piantelli, M.; Canzoniero, L.M.; Ciavardelli, D.; Rizzarelli, E.; Sensi, S.L. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xTg-AD mice. PLoS One 2011, 6, e17971. [Google Scholar]
- Kawahara, M.; Koyama, H.; Nagata, T.; Sadakane, Y. Zinc, copper, and carnosine attenuate neurotoxicity of prion fragment PrP106–126. Metallomics 2011, 3, 726–734. [Google Scholar]
- Hipkiss, A.R. Carnosine and its possible roles in nutrition and health. Adv. Food Nutr. Res 2009, 57, 87–154. [Google Scholar]
- Hambidge, M. Human zinc deficiency. J. Nutr 2000, 130, 1344S–1349S. [Google Scholar]
- Hirano, T.; Murakami, M.; Fukada, T.; Nishida, K.; Yamasaki, S.; Suzuki, T. Roles of zinc and zinc signaling in immunity: Zinc as an intracellular signaling molecule. Adv. Immunol 2008, 97, 149–176. [Google Scholar]
- Prasad, A.S. Impact of the discovery of human zinc deficiency on health. J. Am. Coll. Nutr 2009, 28, 257–265. [Google Scholar]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr 2000, 130, 1471S–1483S. [Google Scholar]
- Tamano, H.; Takeda, A. Dynamic action of neurometals at the synapse. Metallomics 2011, 3, 656–661. [Google Scholar]
- Koh, J.Y.; Suh, S.W.; Gwag, B.J.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996, 272, 1013–1016. [Google Scholar]
- Calderone, A.; Jover, T.; Mashiko, T.; Noh, K.M.; Tanaka, H.; Bennett, M.V.; Zukin, R.S. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J. Neurosci 2004, 24, 9903–9913. [Google Scholar]
- Sensi, S.L.; Canzoniero, L.M.; Yu, S.P.; Ying, H.S.; Koh, J.Y.; Kerchner, G.A.; Choi, D.W. Measurement of intracellular free zinc in living cortical neurons: Routes of entry. J. Neurosci 1997, 17, 9554–9564. [Google Scholar]
- Pellegrini-Giampietro, D.E.; Gorter, J.A.; Bennett, M.V.; Zukin, R.S. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci 1997, 20, 464–470. [Google Scholar]
- Plum, L.M.; Rink, L.; Haase, H. The essential toxin: Impact of zinc on human health. Int. J. Environ. Res. Public Health 2010, 7, 1342–1365. [Google Scholar]
- Weiss, J.H.; Hartley, D.M.; Koh, J.Y.; Choi, D.W. AMPA receptor activation potentiates zinc neurotoxicity. Neuron 1993, 10, 43–49. [Google Scholar]
- Zhu, L.; Tang, Y.; Wang, H.D.; Zhang, Z.Y.; Pan, H. Immersion autometallographic demonstration of pathological zinc accumulation in human acute neural diseases. Neurol. Sci 2012, 33, 855–861. [Google Scholar]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease. J. Alzheimers. Dis 2006, 10, 145–163. [Google Scholar]
- Kawahara, M.; Arispe, N.; Kuroda, Y.; Rojas, E. Alzheimer’s disease amyloid ß-protein forms Zn2+-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys. J 1997, 73, 67–75. [Google Scholar]
- Lin, H.; Bhatia, R.; Lal, R. Amyloid beta protein forms ion channels: Implications for Alzheimer’s disease pathophysiology. FASEB J 2001, 15, 2433–2444. [Google Scholar]
- Leach, S.P.; Salman, M.D.; Hamar, D. Trace elements and prion diseases: A review of the interactions of copper, manganese and zinc with the prion protein. Anim. Health Res. Rev 2006, 7, 97–105. [Google Scholar]
- Valentine, J.S.; Hart, P.J. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 3617–3622. [Google Scholar]
- Brewer, G.J. Recognition, diagnosis, and management of Wilson’s disease. Proc. Soc. Exp. Biol. Med 2000, 223, 39–46. [Google Scholar]
- Koh, J.Y.; Choi, D.W. Zinc toxicity of cultured cortical neurons: Involvement of N-methyl-d-asparatate receptors. Neuroscience 1994, 4, 1049–1057. [Google Scholar]
- Kim, A.H.; Sheline, C.T.; Tian, M.; Higashi, T.; McMahon, R.J.; Cousins, R.J.; Choi, D.W. L-type Ca2+ channel-mediated Zn2+ toxicity and modulation by ZnT-1 in PC12 cells. Brain Res 2000, 886, 99–107. [Google Scholar]
- Lee, J.Y.; Kim, Y.H.; Koh, J.Y. Protection by pyruvate against transient forebrain ischemia in rats. J. Neurosci 2001, 21, RC171. [Google Scholar]
- Sheline, C.T.; Behrens, M.M.; Choi, D.W. Zinc-induced cortical neuronal death: Contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J. Neurosci 2000, 20, 3139–3146. [Google Scholar]
- Sensi, S.L.; Ton-That, D.; Sullivan, P.G.; Jonas, E.A.; Gee, K.R.; Kaczmarek, L.K.; Weiss, J.H. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc. Natl. Acad. Sci. USA 2003, 100, 6157–6162. [Google Scholar]
- Kawahara, M.; Kato-Negishi, M.; Hosoda, R.; Kuroda, Y. Characterization of zinc-induced apoptosis of GT1–7 cells. Biomed. Res. Trace Elements 2002, 13, 280–281. [Google Scholar]
- Konoha, K.; Sadakane, Y.; Kawahara, M. Effects of gadolinium and other metal on the neurotoxicity of immortalized hypothalamic neurons induced by zinc. Biomed. Res. Trace Elements 2004, 15, 275–277. [Google Scholar]
- Kim, E.Y.; Chang, S.Y.; Chung, J.M.; Ryu, B.R.; Joo, C.K.; Moon, H.S.; Kang, K.; Yoon, S.H.; Han, P.L.; Gwag, B.J. Attenuation of Zn2+ neurotoxicity by aspirin: Role of N-type Ca2+ channel and the carboxyl acid group. Neurobiol. Dis 2001, 8, 774–783. [Google Scholar]
- Kawahara, M.; Sadakane, Y.; Koyama, H.; Konoha, K.; Ohkawara, S. d-Histidine and l-histidine attenuate zinc-induced neuronal death in GT1–7 cells. Metallomics 2013, 5, 453–460. [Google Scholar]
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol 2012, 3, 263. [Google Scholar]
- Ferreiro, E.; Baldeiras, I.; Ferreira, I.L.; Costa, R.O.; Rego, A.C.; Pereira, C.F.; Oliveira, C.R. Mitochondrial- and endoplasmic reticulum-associated oxidative stress in Alzheimer’s disease: From pathogenesis to biomarkers. Int. J. Cell Biol 2012, 2012. [Google Scholar] [CrossRef]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol 2013, 12, 105–118. [Google Scholar]
- Moskalev, A.A.; Smit-McBride, Z.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Tacutu, R.; Fraifeld, V.E. Gadd45 proteins: Relevance to aging, longevity and age-related pathologies. Ageing Res. Rev 2012, 11, 51–66. [Google Scholar]
- Van Prooyen, N.; Andresen, V.; Gold, H.; Bialuk, I.; Pise-Masison, C.; Franchini, G. Hijacking the T-cell communication network by the human T-cell leukemia/lymphoma virus type 1 (HTLV-1) p12 and p8 proteins. Mol. Aspects Med 2010, 31, 333–343. [Google Scholar]
- Goruppi, S.; Iovanna, J.L. Stress-inducible protein p8 is involved in several physiological and pathological processes. J. Biol. Chem 2010, 285, 1577–1581. [Google Scholar]
- Onoue, S.; Kumon, Y.; Igase, K.; Ohnishi, T.; Sakanaka, M. Growth arrest and DNA damage-inducible gene 153 increases transiently in the thalamus following focal cerebral infarction. Brain Res. Mol. Brain Res 2005, 134, 189–197. [Google Scholar]
- Kunizuka, H.; Kinouchi, H.; Arai, S.; Izaki, K.; Mikawa, S.; Kamii, H.; Sugawara, T.; Suzuki, A.; Mizoi, K.; Yoshimoto, T. Activation of Arc gene, a dendritic immediate early gene, by middle cerebral artery occlusion in rat brain. Neuroreport 1999, 10, 1717–1722. [Google Scholar]
- Rickhag, M.; Teilum, M.; Wieloch, T. Rapid and long-term induction of effector immediate early genes (BDNF, Neuritin and Arc) in peri-infarct cortex and dentate gyrus after ischemic injury in rat brain. Brain Res 2007, 1151, 203–210. [Google Scholar]
- Bonfanti, L.; Peretto, P.; De Marchis, S.; Fasolo, A. Carnosine-related dipeptides in the mammalian brain. Prog. Neurobiol 1999, 59, 333–353. [Google Scholar]
- De Marchis, S.; Modena, C.; Peretto, P.; Giffard, C.; Fasolo, A. Carnosine-like immunoreactivity in the central nervous system of rats during postnatal development. J. Comp. Neurol 2000, 426, 378–390. [Google Scholar]
- Stuerenburg, H.J. The roles of carnosine in aging of skeletal muscle and in neuromuscular disease. Biochemistry (Mosc) 2000, 65, 862–865. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mizuno, D.; Kawahara, M. The Molecular Mechanisms of Zinc Neurotoxicity and the Pathogenesis of Vascular Type Senile Dementia. Int. J. Mol. Sci. 2013, 14, 22067-22081. https://doi.org/10.3390/ijms141122067
Mizuno D, Kawahara M. The Molecular Mechanisms of Zinc Neurotoxicity and the Pathogenesis of Vascular Type Senile Dementia. International Journal of Molecular Sciences. 2013; 14(11):22067-22081. https://doi.org/10.3390/ijms141122067
Chicago/Turabian StyleMizuno, Dai, and Masahiro Kawahara. 2013. "The Molecular Mechanisms of Zinc Neurotoxicity and the Pathogenesis of Vascular Type Senile Dementia" International Journal of Molecular Sciences 14, no. 11: 22067-22081. https://doi.org/10.3390/ijms141122067
APA StyleMizuno, D., & Kawahara, M. (2013). The Molecular Mechanisms of Zinc Neurotoxicity and the Pathogenesis of Vascular Type Senile Dementia. International Journal of Molecular Sciences, 14(11), 22067-22081. https://doi.org/10.3390/ijms141122067