Oxidative Stress in Diabetes: Implications for Vascular and Other Complications


Abstract
:
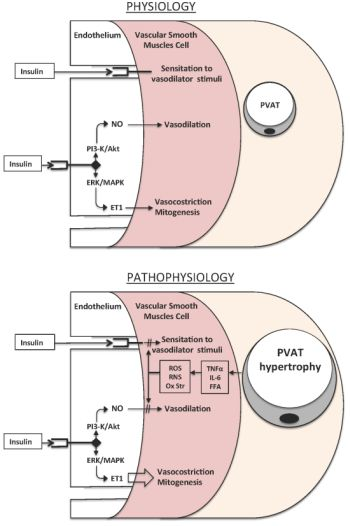{kind=link}
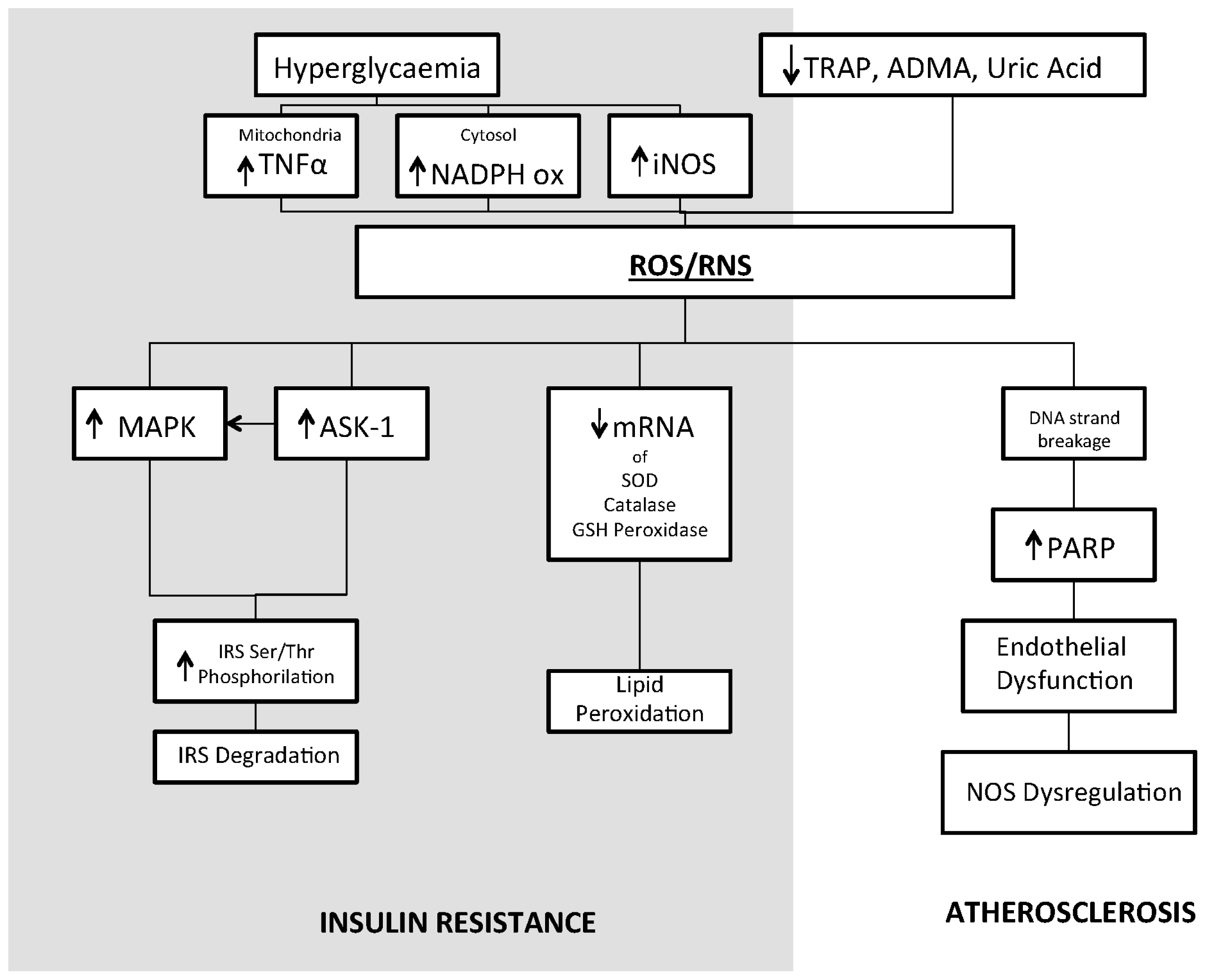{kind=link}
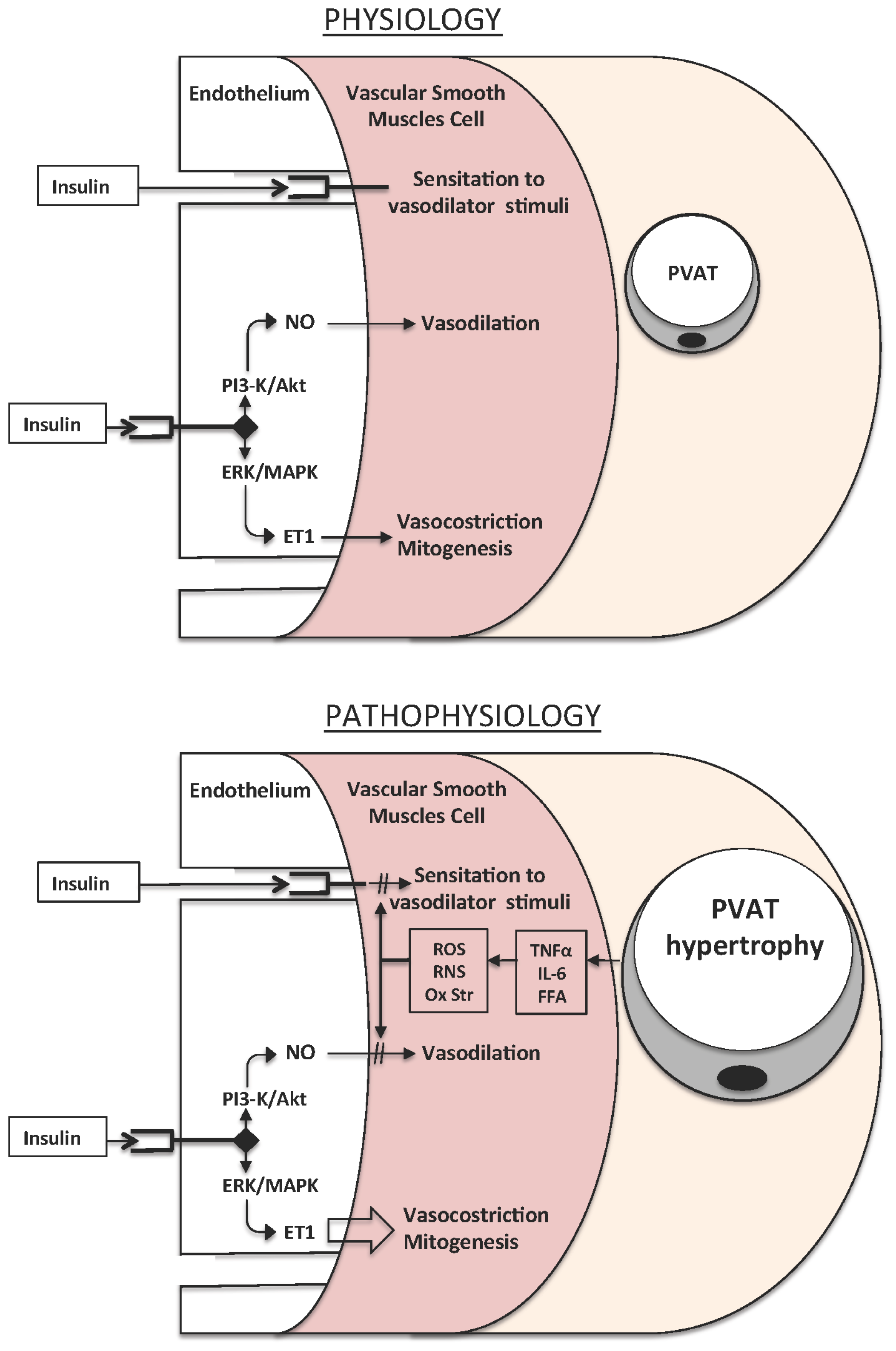{kind=link}
{kind=link}
1. Introduction
1.1. Methods for Detecting Oxidative Stress Metabolites
1.2. ROS and RNS in Physiology and Pathophysiology
2. Reactive Oxygen Species in Diabetes Mellitus
2.1. Oxidative Stress, β-Cells and Insulin Secretion
2.2. Oxidative Stress and Insulin Signal Transduction
2.3. Oxidative Stress and Insulin Resistance
2.4. Antioxidant Deficiency
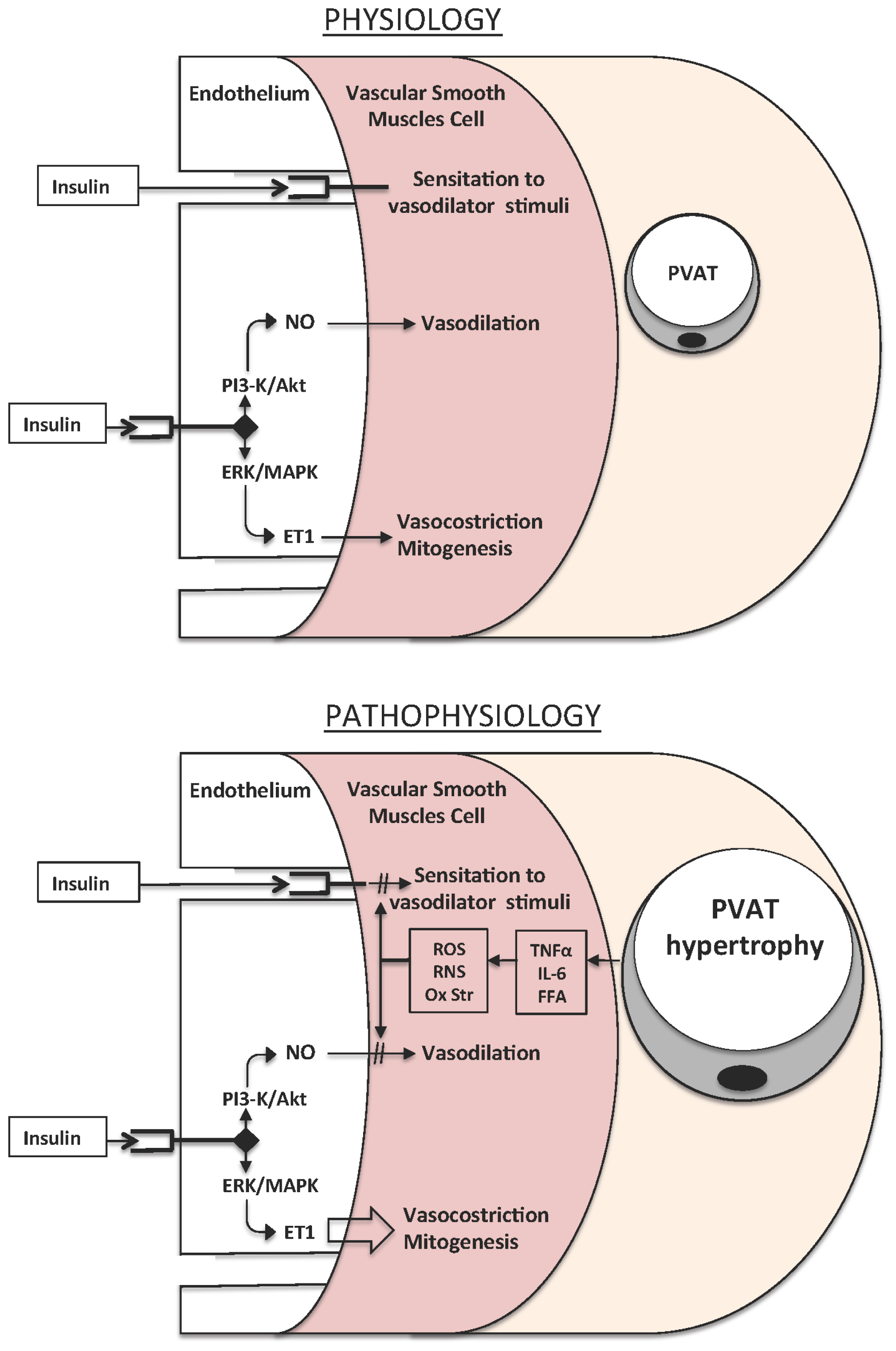
2.5. Oxidative Stress and Vascular Damage
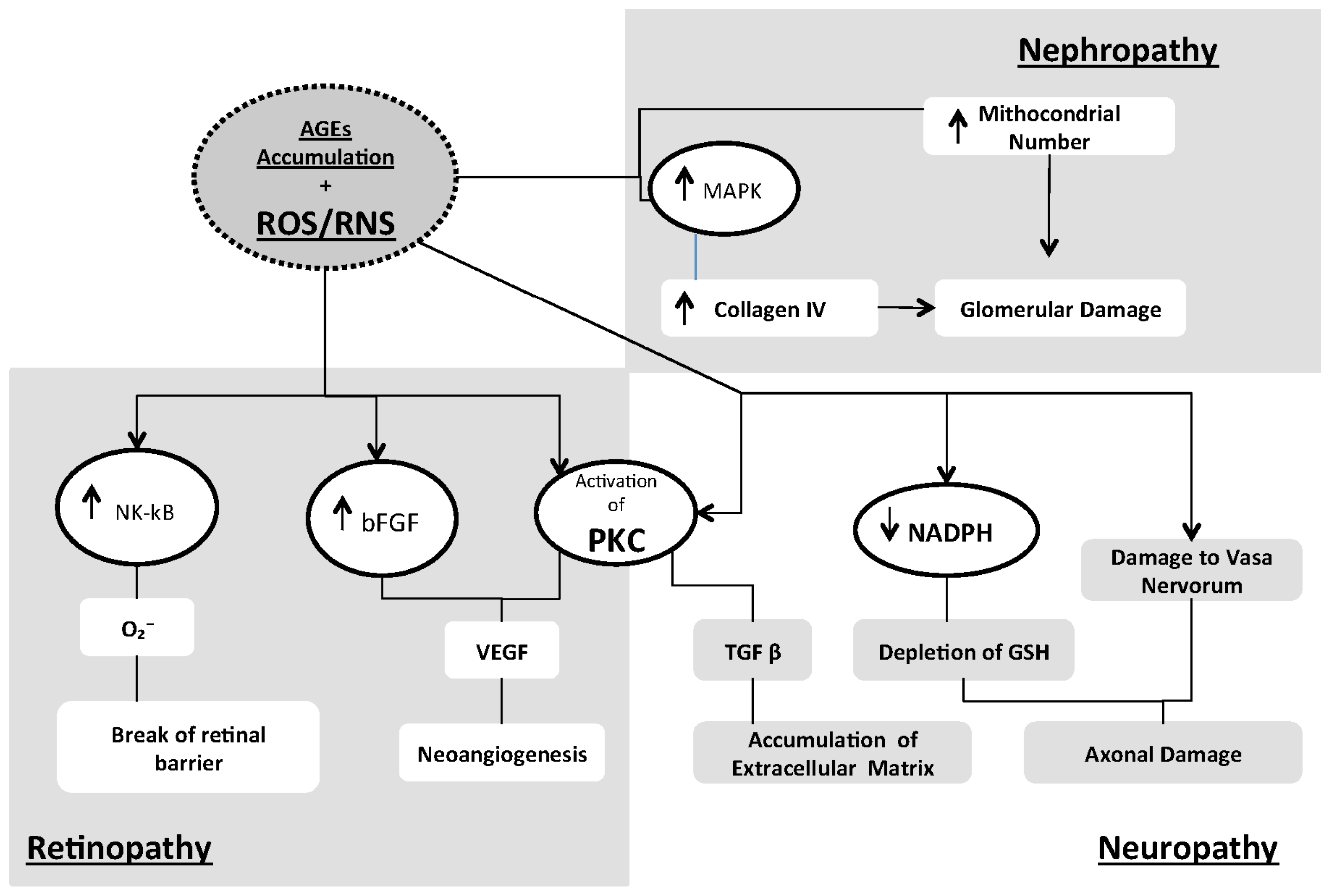
3. Oxidative Stress and Other Diabetic Complications
- Activation of the polyol pathway, probably by means of consumption of NADPH, an important scavenger of ROS [129].
- Increased PKC activation: PKCs are a family of at least 11 isoforms that can phosphorylate various target proteins [134]. Persistent and excessive activation of several PKC isoforms has been implicated in the decreased NO production in smooth muscle cells and has been shown to inhibit insulin-stimulated expression of eNOS in cultured endothelial cells. Activation of PKC by high glucose also induces expression VEGF, thereby enhancing permeability in vascular smooth muscle cells [135,136].
- Activation of the hexosamine pathway: hyperglycemia and insulin resistance-induced excess of fatty acid oxidation contributes to the pathogenesis of diabetic complications by increasing the flux of fructose-6-phosphate into the hexosamine pathway. Fructose 6-phosphate is the substrate for the rate-limiting enzyme of the glutamine:fructose 6-phosphate amidotransferase (GFAT) pathway, which converts fructose 6-phosphate into glucosamine 6-phosphate that is in turn converted into uridine-diphosphate (UDP)-N-acetylglucosamine. Inhibition of GFAT may block the hyperglycemia-induced increases in the transcription of both TGF-α and TGF-β1 [46]. In addition, specific inhibitors of aldose reductase activity, AGE formation, RAGE ligand binding, PKC activation and hexosamine pathway flux may ameliorate diabetes-induced abnormalities in cell culture or animal models [137].
3.1. Retinopathy
3.2. Nephropathy
3.3. Neuropathy
4. Antioxidant Therapy
5. Conclusions
Conflicts of Interest
References
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol 2007, 8, 813–824. [Google Scholar]
- Ibrahim, W.; Tatumi, V.; Yeh, C.C.; Hong, C.B.; Chow, C.K. Effects of dietary carnosine and vitamin E on antioxidant and oxidative status of rats. Int. J. Vitam. Nutr. Res 2008, 78, 230–237. [Google Scholar]
- Valdivia, P.A.; Zenteno-Savin, T.; Gardner, S.C.; Aguirre, A.A. Basic oxidative stress metabolites in eastern Pacific green turtles (Chelonia mydas agassizii). Comp. Biochem. Physiol. Toxicol. Pharmacol 2007, 146, 111–117. [Google Scholar]
- Camera, E.; Picardo, M. Analytical methods to investigate glutathione and related compounds in biological and pathological processes. J. Chromatogr. Anal. Technol. Biomed. Life Sci 2002, 781, 181–206. [Google Scholar]
- Tarpey, M.M.; Wink, D.A.; Grisham, M.B. Methods for detection of reactive metabolites of oxygen and nitrogen: In vitro and in vivo considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol 2004, 286, R431–R444. [Google Scholar]
- Palmieri, B.; Sblendorio, V. Oxidative stress tests: Overview on reliability and use. Eur. Rev. Med. Pharmacol. Sci 2007, 11, 309–342. [Google Scholar]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol 2002, 348, 93–112. [Google Scholar]
- Dambrova, M.; Baumane, L.; Kalvinsh, I.; Wikberg, J.E. Improved method for EPR detection of DEPMPO superoxide radicals by liquid nitrogen freezing. Biochem. Biophys. Res. Commun 2000, 275, 895–898. [Google Scholar]
- Laurindo, F.R.; Pedro, M.D.; Barbeiro, H.V.; Pileggi, F.; Carvalho, M.H.; Augusto, O.; da Luz, P.L. Vascular free radical release ex vivo and in vivo evidence for a flowdependent endothelial mechanism. Circ. Res 1994, 74, 700–709. [Google Scholar]
- Pryor, W.A.; Stanley, J.P.; Blair, E. Autoxidation of polyunsaturated fatty acids: II. A suggested mechanism for the formation of TBA-reactive materials from prostaglandin-like endoperoxides. Lipids 1976, 11, 370–379. [Google Scholar]
- Wallin, B.; Rosengren, B.; Shertzer, H.G.; Camejo, G. Lipoprotein oxidation and measurement of thiobarbituric acid reacting substances formation in a single microtiterplate: Its use for evaluation of antioxidants. Anal. Biochem 1993, 208, 10–15. [Google Scholar]
- Roberts, L.J.; Morrow, J.D. Measurement of F2-isoprostanes as an index of oxidative stress in vivo. Free Radic. Biol. Med. 2000, 28, 505–513. [Google Scholar]
- Aberti, A.; Bolognini, L.; Caratelli, M.; Della Bona, M.A.; Mavviantelli, D. Assessing Oxidative Stress with the DRoms Test. Some Mechanistic Consideration. Proceedings of the SFRR Summer Meeting; Europe Summer Meeting, Abano Terme, Padova, Italy, 26–28 June 1997; pp. 82–83.
- Ellis, G.; Adatia, I.; Yazdanpanah, M.; Makela, S.K. Nitrite and nitrate analyses: A clinical biochemistry perspective. Clin. Biochem 1998, 31, 195–220. [Google Scholar]
- Tsikas, D. Methods of quantitative analysis of the nitric oxide metabolites nitrite and nitrate in human biological fluids. Free Radic. Res 2005, 39, 797–815. [Google Scholar]
- Wennmalm, A.; Benthin, G.; Edlund, A.; Jungersten, L.; Kieler-Jensen, N.; Lundin, S.; Westfelt, U.N.; Petersson, A.S.; Waagstein, F. Metabolism and excretion of nitric oxide in humans. An experimental and clinical study. Circ. Res 1993, 73, 1121–1127. [Google Scholar]
- Leone, A.M.; Francis, P.L.; Rhodes, P.; Moncada, S. A rapid and simple method for the measurement of nitrite and nitrate in plasma by high performance capillary electrophoresis. Biochem. Biophys. Res. Commun 1994, 200, 951–957. [Google Scholar]
- Everett, S.A.; Dennis, M.F.; Tozer, G.M.; Prise, V.E.; Wardman, P.; Stratford, M.R. Nitric oxide in biological fluids: Analysis of nitrite and nitrate by high-performance ion chromatography. J. Chromatogr 1995, 706, 437–442. [Google Scholar]
- Tsikas, D.; Gutzki, F.M.; Rossa, S.; Bauer, H.; Neumann, C.; Dockendorff, K.; Sandmann, J.; Frolich, J.C. Measurement of nitrite and nitrate in biological fluids by gas chromatography-mass spectrometry and by the Griess assay: Problems with the Griess assay—Solutions by gas chromatography-mass spectrometry. Anal. Biochem 1997, 244, 208–220. [Google Scholar]
- Becker, A.J.; Uckert, S.; Tsikas, D.; Noack, H.; Stief, C.G.; Frolich, J.C.; Wolf, G.; Jonas, U. Determination of nitric oxide metabolites by means of the Griess assay and gas chromatography-mass spectrometry in the cavernous and systemic blood of healthy males and patients with erectile dysfunction during different functional conditions of the penis. Urol. Res 2000, 28, 364–369. [Google Scholar]
- Romitelli, F.; Santini, S.A.; Chierici, E.; Pitocco, D.; Tavazzi, B.; Amorini, A.M.; Lazzarino, G.; di Stasio, E. Comparison of nitrite/nitrate concentration in human plasma and serum samples measured by the enzymatic batch Griess assay, ion-pairing HPLC and ion-trap GC-MS: The importance of a correct removal of proteins in the Griess assay. J. Chromatogr. Analyt. Technol. Biomed. Life Sci 2007, 851, 257–267. [Google Scholar]
- Schwedhelm, E. Quantification of ADMA: Analytical approaches. Vasc. Med 2005, 10, S89–S95. [Google Scholar]
- Siroka, R.; Trefil, L.; Rajdl, D.; Racek, J.; Cibulka, R. Asymmetric dimethylarginine—Comparison of HPLC and ELISA methods. Chromatogr. Analyt. Technol. Biomed. Life Sci 2007, 850, 586–587. [Google Scholar]
- Borek, C. Dietary antioxidants and human cancer. Integr. Cancer Ther 2004, 3, 333–341. [Google Scholar]
- Said, T.M.; Kattal, N.; Sharma, R.K.; Sikka, S.C.; Thomas, A.J., Jr.; Mascha, E.; Agarwal, A. Enhanced chemiluminescence assay vs. colorimetric assay for measurement of the total antioxidant capacity of human seminal plasma. J. Androl 2003, 24, 676–680. [Google Scholar]
- Kanda, M.; Ihara, Y.; Murata, H.; Urata, Y.; Kono, T.; Yodoi, J.; Seto, S.; Yano, K.; Kondo, T. Glutaredoxin modulates platelet-derived growth factor-dependent cell signaling by regulating the redox status of low molecular weight protein-tyrosine phosphatase. J. Biol. Chem 2006, 281, 28518–28528. [Google Scholar]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar]
- Bedrad, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev 2007, 87, 245–313. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev 2007, 87, 315–424. [Google Scholar]
- Zhang, D.X.; Gutterman, D.D. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am. J. Physiol. Heart Circ. Physiol 2007, 292, H2023–H2031. [Google Scholar]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol 2004, 199, 316–331. [Google Scholar]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev 2002, 23, 599–622. [Google Scholar]
- Gutterman, D.D.; Miura, H.; Liu, Y. Redox modulation of vascular tone: Focus of potassium channel mechanisms of dilation. Arterioscler. Thromb. Vasc. Biol 2005, 25, 671–678. [Google Scholar]
- Chiarugi, P.; Pani, G.; Giannoni, E.; Taddei, L.; Colavitti, R.; Raugei, G.; Symons, M.; Borrello, S.; Galeotti, T.; Ramponi, G. Reactive oxygen species as essential mediators of cell adhesion: The oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell Biol 2003, 161, 933–944. [Google Scholar]
- Grisham, M.B. Reactive oxygen species in immune responses. Free Radic. Biol. Med 2004, 36, 1479–1480. [Google Scholar]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Lee, H.C.; Wie, Y.H. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp. Biol. Med 2007, 232, 592–606. [Google Scholar]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci 2005, 10, 1881–1896. [Google Scholar]
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative stress and vascular disease. Arterioscler. Thromb. Vasc. Biol 2005, 25, 29–38. [Google Scholar]
- Knight, J.A. Reactive oxygen species and the neurodegenerative disorders. Ann. Clin. Lab. Sci 1997, 27, 11–25. [Google Scholar]
- Bashan, N.; Kovsan, J.; Kachko, I.; Ovadia, H.; Rudich, A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol. Rev 2009, 89, 27–71. [Google Scholar]
- Du, X.; Stocklauser-Farber, K.; Rosen, P. Generation of reactive oxygen intermediates, activation of NF-κB and induction of apoptosis in human endothelial cells by glucose: Role of nitric oxide synthase? Free Radic. Biol. Med 1999, 27, 752–763. [Google Scholar]
- Gupta, S.; Chough, E.; Daley, J.; Oates, P.; Tornheim, K.; Ruderman, N.B.; Keaney, J.F., Jr. Hyperglycemia increases endothelial superoxide that impairs smooth muscle cell Na+-K+-ATPase activity. Am. J. Physiol. Cell Physiol 2002, 282, C560–C566. [Google Scholar]
- Mullarkey, C.J.; Edelstein, D.; Brownee, M. Free radical generation by early glycation products: A mechanism for accelerated atherogenesis in diabetes. Biochem. Biophys. Res. Commun 1990, 173, 932–939. [Google Scholar]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 1222–1226. [Google Scholar]
- Martens, G.A.; Cai, Y.; Hinke, S.; Stange, G.; van de Casteele, M.; Pipeleers, D. Glucose suppresses superoxide generation in metabolically responsive pancreatic β-cells. J. Biol. Chem 2005, 280, 20389–20396. [Google Scholar]
- Herlein, J.A.; Fink, B.D.; O’Malley, Y.; Sivitz, W.I. Superoxide and respiratory coupling in mitochondria of insulin-deficient diabetic rats. Endocrinology 2009, 150, 46–55. [Google Scholar]
- Hou, N.; Torii, S.; Saito, N.; Hosaka, M.; Takeuchi, T. Reactive oxygen species-mediated pancreatic β-cell death is regulated by interactions between stressactivated protein kinases, p38 and c-Jun N-terminal kinase and mitogen-activated protein kinase phosphatases. Endocrinology 2008, 149, 1654–1665. [Google Scholar]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and β-cell dysfunction. Endocr. Rev 2008, 29, 351–366. [Google Scholar]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic β-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar]
- Harmon, J.S.; Gleason, C.E.; Tanaka, Y.; Poitout, V.; Robertson, R.P. Antecedent hyperglycemia, not hyperllipidemia, is associated with increased islet triacylglycerol content and decreased insulin gene mRNA level in Zucker diabetic fatty rats. Diabetes 2001, 50, 2481–2486. [Google Scholar]
- Robertson, R.P.; Zhang, H.J.; Pyzdrowski, K.L.; Walseth, T.F. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J. Clin. Investig 1992, 90, 320–325. [Google Scholar]
- Kaneto, H.; Fujii, J.; Myint, T.; Miyazawa, N.; Islam, K.N.; Kawasaki, Y.; Suzuki, K.; Makamura, M.; Tatsumi, H.; Yamasaki, Y.; et al. Reducing sugars trigger oxidative modification and apoptosis in pancreatic β-cells by provoking oxidative stress through the glycation reaction. Biochem. J 1996, 320, 855–863. [Google Scholar]
- Tajiri, Y.; Moller, C.; Grill, V. Long term effects of aminoguanidine on insulin release and biosynthesis: Evidence that the formation of advanced glycosylation end products inhibits β-cell function. Endocrinology 1997, 138, 273–280. [Google Scholar]
- Tanaka, Y.; Gleason, C.E.; Tran, P.O.; Harmon, J.S.; Robertson, R.P. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc. Natl. Acad. Sci. USA 1999, 96, 10857–10862. [Google Scholar]
- Sakai, K.; Matsumoto, K.; Nishikawa, T.; Suefuji, M.; Nakamaru, K.; Hirashima, Y.; Kawashima, J.; Shirotani, T.; Ichinose, K.; Brownlee, M.; et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic β-cells. Biochem. Biophys. Res. Commun 2003, 300, 216–222. [Google Scholar]
- Krauss, S.; Zhang, C.Y.; Scorrano, L.; Dalgaard, L.T.; St-Pierre, J.; Grey, S.T.; Lowell, B.B. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic β-cell dysfunction. J. Clin. Investig 2003, 112, 1831–1842. [Google Scholar]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar]
- LeLoup, C.; Tourrel-Cuzin, C.; Magnan, C.; Karaca, M.; Castel, J.; Carneiro, L.; Colombani, A.L.; Ktorza, A.; Casteilla, L.; Pénicaud, L. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 2009, 58, 673–681. [Google Scholar]
- Li, N.; Brun, T.; Chop, M.; Cunha, D.A.; Eizirik, D.L.; Maechler, P. Transient exposure of β-cells to oxidative stress interrupts the transduction of signals normally coupling glucose metabolism to insulin secretion. J. Biol. Chem 2009, 284, 23602–23612. [Google Scholar]
- Fridlyand, L.E.; Philipson, L.H. Does the glucose-dependent insulin secretion mechanism itself cause oxidative stress in pancreatic β-cells? Diabetes 2004, 53, 1942–1948. [Google Scholar]
- Czech, M.P.; Lawrence, J.C., Jr.; Lynn, W.S. Evidence for the involvement of sulfhydryl oxidation in the regulation of fat cell hexose transport by insulin. Proc. Natl. Acad. Sci. USA 1974, 71, 4173–4177. [Google Scholar]
- Higaki, Y.; Mikami, T.; Fujii, N.; Hirshman, M.F.; Koyama, K.; Seino, T.; Tanaka, K.; Goodyear, L.J. Oxidative stress stimulates skeletal muscle glucose uptake through a phosphatidylinositol-3-kinase-dependent pathway. Am. J. Physiol. Endocrinol. Metab 2008, 294, E889–E897. [Google Scholar]
- May, J.M.; de Haen, C. The insulin-like effect of hydrogen peroxide on pathways of lipid synthesis in rat adipocytes. J. Biol. Chem 1979, 254, 9017–9021. [Google Scholar]
- Powell, D.J.; Hajduch, E.; Kular, G.; Hundal, H.S. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCζ-dependent mechanism. Mol. Cell Biol 2003, 23, 7794–7808. [Google Scholar]
- Imoto, K.; Kukidome, D.; Nishikawa, T.; Matsuhisa, T.; Sonoda, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Tsuruzoe, K.; et al. Impact of mitochondrial reactive oxygen species and apoptosis signal-regulating kinase 1 on insulin signaling. Diabetes 2006, 55, 1197–1204. [Google Scholar]
- Hotamisligil, G.S.; Budavari, A.; Murray, D.; Spiegelman, B.M. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-α. J. Clin. Investig 1994, 94, 1543–1549. [Google Scholar]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389, 610–614. [Google Scholar]
- Zhang, L.; Wheatley, C.M.; Richards, S.M.; Barrett, E.J.; Clark, M.G.; Rattigan, S. TNF-α acutely inhibits vascular effects of physiological but not high insulin or contraction. Am. J. Physiol. Endocrinol. Metab 2003, 285, E654–E660. [Google Scholar]
- Yoshizumi, M.; Perrella, M.A.; Burnett, J.C.; Lee, M.E. Tumor necrosis factor downregulates endothelial nitric oxide synthase mRNA by shortening its half-life. Circ. Res 1993, 73, 205–209. [Google Scholar]
- Mohamed, F.; Monge, J.C.; Gordon, A.; Cernacek, P.; Blais, D.; Stewart, D.J. Lack of role for nitric oxide (NO) in the selective destabilization of endothelial NO synthase mRNA by tumor necrosis factor-α. Arterioscler. Thromb. Vasc. Biol 1995, 15, 52–57. [Google Scholar]
- De Keulenaer, G.W.; Alexander, R.W.; Ushio-Fukai, M.; Ishizaka, N.; Griendling, K.K. Tumour necrosis factor α activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem. J 1998, 329, 653–657. [Google Scholar]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar]
- Rask-Madsen, C.; Dominguez, H.; Ihlemann, N.; Hermann, T.; Kober, L.; Torp-Pedersen, C. Tumor necrosis factor-alpha inhibits insulin’s stimulating effect on glucose uptake and endothelium-dependent vasodilation in humans. Circulation 2003, 108, 1815–1821. [Google Scholar]
- Ziccardi, P.; Nappo, F.; Giugliano, G.; Esposito, K.; Marfella, R.; Cioffi, M.; D’Andrea, F.; Molinari, A.M.; Giugliano, D. Reduction of inflammatory cytokine concentrations and improvement of endothelial functions in obese women after weight loss over one year. Circulation 2002, 105, 804–809. [Google Scholar]
- Tesauro, M.; Schinzari, F.; Rovella, V.; Melina, D.; Mores, N.; Barini, A.; Mettimano, M.; Lauro, D.; Iantorno, M.; Quon, M.J.; et al. Tumor necrosis factor-α antagonism improves vasodilation during hyperinsulinemia in metabolic syndrome. Diabetes Care 2008, 31, 1439–1441. [Google Scholar]
- Tesauro, M.; Canale, M.P.; Rodia, G.; di Daniele, N.; Lauro, D.; Scuteri, A.; Cardillo, C. Metabolic syndrome, chronic kidney, and cardiovascular diseases: Role of adipokines. Cardiol. Res. Pract 2011, 2011, 653182. [Google Scholar]
- Moriwaki, Y.; Inokuchi, T.; Yamamoto, A.; Ka, T.; Tsutsumi, Z.; Takahashi, S.; Yamamoto, T. Effect of TNF-α inhibition on urinary albumin excretion in experimental diabetic rats. Acta Diabetol 2007, 44, 215–218. [Google Scholar]
- Westermann, D.; van Linthout, S.; Dhayat, S.; Dhayat, N.; Schmidt, A.; Noutsias, M.; Song, X.Y.; Spillmann, F.; Riad, A.; Schultheiss, H.P.; et al. Tumor necrosis factor-α antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res. Cardiol 2007, 102, 500–507. [Google Scholar]
- Schaffer, S.W.; Jong, C.J.; Mozaffari, M. Role of oxidative stress in diabetes-mediated vascular dysfunction: Unifying hypothesis of diabetes revisited. Vascul. Pharmacol 2012, 57, 139–149. [Google Scholar]
- Campia, U.; Tesauro, M.; Cardillo, C. Human obesity and endothelium-dependent responsiveness. Br. J. Pharmacol 2012, 165, 561–573. [Google Scholar]
- Tesauro, M.; Cardillo, C. Obesity, blood vessels and metabolic syndrome. Acta Physiol. (Oxf.) 2011, 203, 279–286. [Google Scholar]
- Stadler, K. Oxidative stress in diabetes. Adv. Exp. Med. Biol 2012, 771, 272–287. [Google Scholar]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig 2004, 114, 1752–1761. [Google Scholar]
- Svegliati-Baroni, G.; Candelaresi, C.; Saccomanno, S.; Ferretti, G.; Bachetti, T.; Marzioni, M.; de Minicis, S.; Nobili, L.; Salzano, R.; Omenetti, A.; et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: Role of peroxisome proliferator-activated receptor-α and n-3 polyunsaturated fatty acid treatment on liver injury. Am. J. Pathol 2006, 169, 846–860. [Google Scholar]
- Davi, G.; Chiarelli, F.; Santilli, F.; Pomilio, M.; Vigneri, S.; Falco, A.; Basili, S.; Ciabattoni, G.; Patrono, C. Enhanced lipid peroxidation and platelet activation in the early phase of type 1 diabetes mellitus: Role of interleukin-6 and disease duration. Circulation 2003, 107, 3199–3203. [Google Scholar]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.P.; Colette, C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar]
- Davi, G.; Ciabattoni, G.; Consoli, A.; Mezzetti, A.; Falco, A.; Santarone, S.; Pennese, E.; Vitacolonna, E.; Bucciarelli, T.; Costantini, F.; et al. In vivo formation of 8-isoprostaglandin F2α and platelet activation in diabetes mellitus: Effects of improved metabolic control and vitamin E supplementation. Circulation 1999, 99, 224–229. [Google Scholar]
- Savu, O.; Ionescu-Tirgoviste, C.; Atanasiu, V.; Gaman, L.; Papacocea, R.; Stoian, I. Increase in total antioxidant capacity of plasma despite high levels of oxidative stress in uncomplicated type 2 diabetes mellitus. J. Int. Med. Res 2012, 40, 709–716. [Google Scholar]
- Marra, G.; Cotroneo, P.; Pitocco, D.; Manto, A.; di Leo, M.A.; Ruotolo, V.; Caputo, S.; Giardina, B.; Ghirlanda, G.; Santini, S.A. Early increase of oxidative stress and reduced antioxidant defenses in patients with uncomplicated type 1 diabetes: A case for gender difference. Diabetes Care 2002, 25, 370–375. [Google Scholar]
- Pitocco, D.; di Stasio, E.; Romitelli, F.; Zaccardi, F.; Tavazzi, B.; Manto, A.; Caputo, S.; Musella, T.; Zuppi, C.; Santini, S.A.; et al. Hypouricemia linked to an overproduction of nitric oxide is an early marker of oxidative stress in female subjects with type 1 diabetes. Diabetes Metab. Res. Rev 2008, 24, 318–323. [Google Scholar]
- Weber, C.; Zernecke, A.; Libby, P. The multifaceted contributions of leukocyte subsets to atherosclerosis: Lessons from mouse models. Nat. Rev. Immunol 2008, 8, 802–815. [Google Scholar]
- MacNaul, K.L.; Hutchinson, N.I. Differential expression of iNOS and cNOS mRNA in human vascular smooth muscle cells and endothelial cells under normal and inflammatory conditions. Biochem. Biophys. Res. Commun 1993, 196, 1330–1334. [Google Scholar]
- Ceriello, A.; Esposito, K.; Piconi, L.; Ihnat, M.A.; Thorpe, J.E.; Testa, R.; Boemi, M.; Giugliano, D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes 2008, 57, 1349–1354. [Google Scholar]
- Ceriello, A.; Mercuri, F.; Quagliaro, L.; Assaloni, R.; Motz, E.; Tonutti, L.; Taboga, C. Detection of nitrotyrosine in the diabetic plasma: Evidence of oxidative stress. Diabetologia 2001, 44, 834–838. [Google Scholar]
- Tannous, M.; Rabini, R.A.; Vignini, A.; Moretti, N.; Fumelli, P.; Zielinski, B.; Mazzanti, L.; Mutus, B. Evidence for iNOS-dependent peroxynitrite production in diabetic platelets. Diabetologia 1999, 42, 539–544. [Google Scholar]
- Frustaci, A.; Kajstura, J.; Chimenti, C.; Jakoniuk, I.; Leri, A.; Maseri, A.; Nadal-Ginard, B.; Anversa, P. Myocardial cell death in human diabetes. Circ. Res 2000, 87, 1123–1132. [Google Scholar]
- Pacher, P.; Obrosova, I.G.; Mabley, J.G.; Szabó, C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr. Med. Chem 2005, 12, 267–275. [Google Scholar]
- Pacher, P.; Szabó, C. Role of peroxynitrite in the pathogenesis of cardiovascular complications of diabetes. Curr. Opin. Pharmacol 2006, 6, 136–141. [Google Scholar]
- Pacher, P.; Schulz, R.; Liaudet, L.; Szabo, C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol. Sci 2005, 26, 302–310. [Google Scholar]
- Virág, L.; Szabó, C. The therapeutic potential of PARP inhibition. Pharmacol. Rev 2002, 54, 375–429. [Google Scholar]
- Szabó, C.; Zanchi, A.; Komjati, K.; Pacher, P.; Krolewski, A.S.; Quist, W.C.; LoGerfo, F.W.; Horton, E.S.; Veves, A. Poly(ADP-ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation 2002, 106, 2680–2686. [Google Scholar]
- Garcia Soriano, F.; Virág, L.; Jagtap, P.; Szabó, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly(ADP-ribose) polymerase activation. Nat. Med 2001, 7, 108–113. [Google Scholar]
- Pacher, P.; Liaudet, L.; Soriano, F.G.; Mabley, J.G.; Szabó, E.; Szabó, C. The role of poly(ADP-ribose) polymerase in the development of myocardial and endothelial dysfunction in diabetes mellitus. Diabetes 2002, 51, 514–521. [Google Scholar]
- Pacher, P.; Szabó, C. Role of poly(ADP-ribose) polymerase-1 (PARP) activation in the pathogenesis of diabetic complications: Endothelial dysfunction, as a common underlying theme. Antioxid. Redox Signal 2005, 7, 1568–1580. [Google Scholar]
- Kajstura, J.; Fiordaliso, F.; Andreoli, A.M.; Li, B.; Chimenti, S.; Medow, M.S.; Limana, F.; Nadal-Ginard, B.; Leri, A.; Anversa, P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes 2001, 50, 1414–1424. [Google Scholar]
- Szabó, C.; Mabley, J.G.; Moeller, S.M.; Shimanovich, R.; Pacher, P.; Virag, L.; Soriano, F.G.; van Duzer, J.H.; Williams, W.; Salzman, A.L.; et al. Pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: Studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol. Med 2002, 8, 571–580. [Google Scholar]
- Mihm, M.J.; Coyle, C.M.; Schanbacher, B.L.; Weinstein, D.M.; Bauer, J.A. Peroxynitrite induced nitration and inactivation of myofibrillar creatine kinase in experimental heart failure. Cardiovasc. Res 2001, 49, 798–807. [Google Scholar]
- Turko, I.V.; Marcondes, S.; Murad, F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase. Am. J. Physiol. Heart Circ. Physiol 2001, 281, 2289–2294. [Google Scholar]
- El-Remessy, A.B.; Behzadian, M.A.; Abou-Mohamed, G.; Franklin, T.; Caldwell, R.W.; Caldwell, R.B. Experimental diabetes causes breakdown of the blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am. J. Pathol 2003, 162, 1995–2004. [Google Scholar]
- El-Remessy, A.B.; Abou-Mohamed, G.; Caldwell, R.W.; Caldwell, R.B. High glucose-induced tyrosine nitration in endothelial cells: Role of eNOS uncoupling and aldose reductase activation. Investig. Ophthalmol. Vis. Sci 2003, 44, 3135–3143. [Google Scholar]
- Onozato, M.L.; Tojo, A.; Goto, A.; Fujita, T.; Wilcox, C.S. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: Effects of ACEI and ARB. Kidney Int 2002, 61, 186–194. [Google Scholar]
- Obrosova, I.G.; Mabley, J.G.; Zsengeller, Z.; Charniauskaya, T.; Abatan, O.I.; Groves, J.T.; Szabó, C. Role for nitrosative stress in diabetic neuropathy: Evidence from studies with a peroxynitrite decomposition catalyst. FASEB J 2005, 19, 401–403. [Google Scholar]
- Obrosova, I.G.; Drel, V.R.; Pacher, P.; Ilnytska, O.; Wang, Z.Q.; Stevens, M.J.; Yorek, M.A. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: The relation is revisited. Diabetes 2005, 54, 3435–3441. [Google Scholar]
- Johnson, E.L. Glycemic variability in type 2 diabetes mellitus: Oxidative stress and macrovascular complications. Adv. Exp. Med. Biol 2012, 771, 139–154. [Google Scholar]
- Piconi, L.; Quagliaro, L.; Da Ros, R.; Assaloni, R.; Giugliano, D.; Esposito, K.; Szabo, C.; Ceriello, A. Intermittent high glucose enhances ICAM-1, VCAM-1, E-selectin and interleukin-6 expression in human umbilical endothelial cells in culture: The role of poly(ADP-ribose) polymerase. J. Thromb. Haemost 2004, 2, 1453–1459. [Google Scholar]
- Durante, W.; Sen, A.K.; Sunahara, F.A. Impairment of endothelium-dependent relaxation in aortae from spontaneously diabetic rats. Br. J. Pharmacol 1988, 94, 463–468. [Google Scholar]
- Obrosova, I.G.; Minchenko, A.G.; Marinescu, V.; Fathallah, L.; Kennedy, A.; Stockert, C.M.; Frank, R.N.; Stevens, M.J. Antioxidants attenuate early up regulation of retinal vascular endothelial growth factor in streptozotocin-diabetic rats. Diabetologia 2001, 44, 1102–1110. [Google Scholar]
- Zhao, H.J.; Wang, S.; Cheng, H.; Zhang, M.Z.; Takahashi, T.; Fogo, A.B.; Breyer, M.D.; Harris, R.C. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J. Am. Soc. Nephrol 2006, 17, 2664–2669. [Google Scholar]
- Selvaraju, V.; Joshi, M.; Suresh, S.; Sanchez, J.A.; Maulik, N.; Maulik, G. Diabetes, oxidative stress, molecular mechanism, and cardiovascular disease—An overview. Toxicol. Mech. Methods 2012, 22, 330–335. [Google Scholar]
- Schinzari, F.; Tesauro, M.; Rovella, V.; Galli, A.; Mores, N.; Porzio, O.; Lauro, D.; Cardillo, C. Generalized impairment of vasodilator reactivity during hyperinsulinemia in patients with obesity-related metabolic syndrome. Am. J. Physiol. Endocrinol. Metab 2010, 299, E947–E952. [Google Scholar]
- Schinzari, F.; Tesauro, M.; Rovella, V.; di Daniele, N.; Gentileschi, P.; Mores, N.; Campia, U.; Cardillo, C. Rho-kinase inhibition improves vasodilator responsiveness during hyperinsulinemia in the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab 2012, 303, E806–E811. [Google Scholar]
- Tesauro, M.; Schinzari, F.; Adamo, A.; Rovella, V.; Martini, F.; Mores, N.; Barini, A.; Pitocco, D.; Ghirlanda, G.; Lauro, D.; et al. Effects of GLP-1 on forearm vasodilator function and glucose disposal during hyperinsulinemia in the metabolic syndrome. Diabetes Care 2013, 36, 683–689. [Google Scholar]
- Emerging, Risk; Factors, Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; di angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar]
- Centers for Disease Control and Prevention, National Diabetes Fact Sheet: National Estimates and General Information on Diabetes and Prediabetes in the United States, 2011; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2011.
- Huang, A.; Yang, Y.M.; Feher, A.; Bagi, Z.; Kaley, G.; Sun, D. Exacerbation of endothelial dysfunction during the progression of diabetes: Role of oxidative stress. Am. J. Physiol. Regul. Integr. Comp. Physiol 2012, 302, R674–R681. [Google Scholar]
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol 2003, 14, S233–S236. [Google Scholar]
- Wautier, J.L.; Schmidt, A.M. Protein glycation: A firm link to endothelial cell dysfunction. Circ. Res 2004, 95, 233–238. [Google Scholar]
- Candido, R.; Forbes, J.M.; Thomas, M.C.; Thallas, V.; Dean, R.G.; Burns, W.C.; Tikellis, C.; Ritchie, R.H.; Twigg, S.M.; Cooper, M.E.; et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ. Res 2003, 92, 785–792. [Google Scholar]
- Stitt, A.W.; Li, Y.M.; Gardiner, T.A.; Bucala, R.; Archer, D.B.; Vlassar, H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am. J. Pathol 1997, 150, 523–531. [Google Scholar]
- Nishino, T.; Horii, Y.; Shiiki, H.; Yamamoto, H.; Makita, Z.; Bucala, R.; Dohi, K. Immunohistochemical detection of advanced glycosylation end products within the vascular lesions and glomeruli in diabetic nephropathy. Hum. Pathol 1995, 26, 308–313. [Google Scholar]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res 2010, 106, 1319–1331. [Google Scholar]
- Inoguchi, T.; Battan, R.; Handler, E.; Sportsman, J.R.; Heath, W.; King, G.L. Preferential elevation of protein kinase C isoform β II and diacylglycerol levels in the aorta and heart of diabetic rats: Differential reversibility to glycemic control by islet cell transplantation. Proc. Natl. Acad. Sci. USA 1992, 89, 11059–11063. [Google Scholar]
- Ganz, M.B.; Seftel, A. Glucose-induced changes in protein kinase C and nitric oxide are prevented by vitamin E. Am. J. Physiol. Endocrinol. Metab 2000, 278, E146–E152. [Google Scholar]
- Brownlee, M. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med 1995, 46, 223–234. [Google Scholar]
- Wallace, D.C. Disease of the mitochondrial DNA. Annu. Rev. Biochem 1992, 61, 1175–1212. [Google Scholar]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 1997, 416, 15–18. [Google Scholar]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar]
- Ola, M.S.; Nawaz, M.I.; Khan, H.A.; Alhomida, A.S. Neurodegeneration and neuroprotection in diabetic retinopathy. Int. J. Mol. Sci 2013, 14, 2559–2572. [Google Scholar]
- Rodríguez-Carrizalez, A.D.; Castellanos-González, J.A.; Martínez-Romero, E.C.; Miller-Arrevillaga, G.; Villa-Hernández, D.; Hernández-Godínez, P.P.; Ortiz, G.G.; Pacheco-Moisés, F.P.; Cardona-Muñoz, E.G.; Miranda-Díaz, A.G. Oxidants, antioxidants and mitochondrial function in non-proliferative diabetic retinopathy. J. Diabetes 2013. [Google Scholar] [CrossRef]
- Yamagishi, S.; Maeda, S.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim. Biophys. Acta 2012, 1820, 663–671. [Google Scholar]
- Choudhuri, S.; Dutta, D.; Sen, A.; Chowdhury, I.H.; Mitra, B.; Mondal, L.K.; Saha, A.; Bhadhuri, G.; Bhattacharya, B. Role of N-ɛ-carboxy methyl lysine, advanced glycation end products and reactive oxygen species for the development of nonproliferative and proliferative retinopathy in type 2 diabetes mellitus. Mol. Vis 2013, 19, 100–113. [Google Scholar]
- Yafai, Y.; Iandiev, I.; Lange, J.; Yang, X.M.; Wiedemann, P.; Bringmann, A.; Eichler, W. Basic fibroblast growth factor contributes to a shift in the angioregulatory activity of retinal glial (Müller) cells. PLoS One 2013, 8, e68773. [Google Scholar]
- Taniguchi, K.; Xia, L.; Goldberg, H.J.; Lee, K.W.; Shah, A.; Stavar, L.; Masson, E.A.Y.; Momen, A.; Shikatani, E.A.; John, R.; et al. Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes 2013. [Google Scholar] [CrossRef]
- Al-Kafaji, G.; Golbahar, J. High glucose-induced oxidative stress increases the copy number of mitochondrial DNA in human mesangial cells. Biomed. Res. Int 2013, 2013, 754946. [Google Scholar]
- Van Dam, P.S.; Cotter, M.A.; Bravenboer, B.; Cameron, N.E. Pathogenesis of diabetic neuropathy: Focus on neurovascular mechanisms. Eur. J. Pharmacol 2013. [Google Scholar] [CrossRef]
- Hosseini, A.; Abdollahi, M. Diabetic neuropathy and oxidative stress: Therapeutic perspectives. Oxid. Med. Cell. Longev 2013, 2013, 168039:1–168039:15. [Google Scholar]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The molecular basis for oxidative stress-induced insulin resistance. Antioxid. Redox Signal 2005, 7, 1040–1052. [Google Scholar]
- Jacob, S.; Henriksen, E.J.; Tritschler, H.J.; Augustin, H.J.; Dietze, G.J. Improvement of insulin-stimulatedglucose-disposal in type 2 diabetes after repeated parenteral administration of thioctic acid. Exp. Clin. Endocrinol. Diabetes 1996, 104, 284–288. [Google Scholar]
- Jacob, S.; Ruus, P.; Hermann, R.; Tritschler, H.J.; Maerker, E.; Renn, W.; Augustin, H.J.; Dietze, G.J.; Rett, K. Oral administration of RAC-α-lipoic acid modulates insulin sensitivity in patients with type-2 diabetes mellitus: A placebo-controlled pilot trial. Free Radic. Biol. Med 1999, 27, 309–314. [Google Scholar]
- Konrad, T.; Vicini, P.; Kusterer, K.; Hoflich, A.; Assadkhani, A.; Bohles, H.J.; Sewell, A.; Tritschler, H.J.; Cobelli, C.; Usadel, K.H. α-Lipoic acid treatment decreases serum lactate and pyruvate concentrations and improves glucose effectiveness in lean and obese patients with type 2 diabetes. Diabetes Care 1999, 22, 280–287. [Google Scholar]
- Fulghesu, A.M.; Ciampelli, M.; Muzj, G.; Belosi, C.; Selvaggi, L.; Ayala, G.F.; Lanzone, A. N-acetyl-cysteine treatment improves insulin sensitivity in women with polycystic ovary syndrome. Fertil. Steril 2002, 77, 1128–1135. [Google Scholar]
- Caballero, B. Vitamin E improves the action of insulin. Nutr. Rev 1993, 51, 339–340. [Google Scholar]
- Paolisso, G.; D’Amore, A.; Galzerano, D.; Balbi, V.; Giugliano, D.; Varricchio, M.; D’Onofrio, F. Daily vitamin E supplements improve metabolic control but not insulin secretion in elderly type II diabetic patients. Diabetes Care 1993, 16, 1433–1437. [Google Scholar]
- Paolisso, G.; D’Amore, A.; Giugliano, D.; Ceriello, A.; Varricchio, M.; D’Onofrio, F. Pharmacologic doses of vitamin E improve insulin action in healthy subjects and non-insulin-dependent diabetic patients. Am. J. Clin. Nutr 1993, 57, 650–656. [Google Scholar]
- Paolisso, G.; di Maro, G.; Galzerano, D.; Cacciapuoti, F.; Varricchio, G.; Varricchio, M.; D’Onofrio, F. Pharmacological doses of vitamin E and insulin action in elderly subjects. Am. J. Clin. Nutr 1994, 59, 1291–1296. [Google Scholar]
- Hirashima, O.; Kawano, H.; Motoyama, T.; Hirai, N.; Ohgushi, M.; Kugiyama, K.; Ogawa, H.; Yasue, H. Improvement of endothelial function and insulin sensitivity with vitamin C in patients with coronary spastic angina: Possible role of reactive oxygen species. J. Am. Coll. Cardiol 2000, 35, 1860–1866. [Google Scholar]
- Chevion, M.; Berenshtein, E.; Stadtman, E.R. Human studies related to protein oxidation: Protein carbonyl content as a marker of damage. Free Radic. Res 2000, 33, S99–S108. [Google Scholar]
- Czernichow, S.; Couthouis, A.; Bertrais, S.; Vergnaud, A.C.; Dauchet, L.; Galan, P.; Hercberg, S. Antioxidant supplementation does not affect fasting plasma glucose in the Supplementation with Antioxidant Vitamins and Minerals (SU.VI.MAX) study in France: Association with dietary intake and plasma concentrations. Am. J. Clin. Nutr 2006, 84, 395–399. [Google Scholar]
- Gomez-Perez, F.J.; Valles-Sanchez, V.E.; Lopez-Alvarenga, J.C.; Choza-Romero, R.; Ibarra Pascuali, J.J.; Gonzalez Orellana, R.; Perez Ortiz, O.B.; Rodriguez Padilla, E.G.; Aguilar Salinas, C.A.; Rull, J.A. Vitamin E modifies neither fructosamine nor HbA1c levels in poorly controlled diabetes. Rev. Investig. Clin 1996, 48, 421–424. [Google Scholar]
- Liu, S.; Lee, I.M.; Song, Y.; van Denburgh, M.; Cook, N.R.; Manson, J.E.; Buring, J.E. Vitamin E and risk of type 2 diabetes in the women’s health study randomized controlled trial. Diabetes 2006, 55, 2856–2862. [Google Scholar]
- Scott, J.A.; King, G.L. Oxidative stress and antioxidant treatment in diabetes. Ann. N. Y. Acad. Sci 2004, 1031, 204–213. [Google Scholar]
- American diabetes association. Nutrition recommendations and interventions for diabetes: A position statement of the American Diabetes Association. Diabetes Care 2007, 30, S48–S65.
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar]
- Bleys, J.; Navas-Acien, A.; Guallar, E. Serum selenium and diabetes in US adults. Diabetes Care 2007, 30, 829–834. [Google Scholar]
- Miller, E.R., 3rd; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med 2005, 142, 37–46. [Google Scholar]
- Ceriello, A. Hypothesis: The “metabolic memory”, the new challenge of diabetes. Diabetes Res. Clin. Pract 2009, 86, S2–S6. [Google Scholar]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med 2008, 205, 2409–2417. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pitocco, D.; Tesauro, M.; Alessandro, R.; Ghirlanda, G.; Cardillo, C. Oxidative Stress in Diabetes: Implications for Vascular and Other Complications. Int. J. Mol. Sci. 2013, 14, 21525-21550. https://doi.org/10.3390/ijms141121525
Pitocco D, Tesauro M, Alessandro R, Ghirlanda G, Cardillo C. Oxidative Stress in Diabetes: Implications for Vascular and Other Complications. International Journal of Molecular Sciences. 2013; 14(11):21525-21550. https://doi.org/10.3390/ijms141121525
Chicago/Turabian StylePitocco, Dario, Manfredi Tesauro, Rizzi Alessandro, Giovanni Ghirlanda, and Carmine Cardillo. 2013. "Oxidative Stress in Diabetes: Implications for Vascular and Other Complications" International Journal of Molecular Sciences 14, no. 11: 21525-21550. https://doi.org/10.3390/ijms141121525
APA StylePitocco, D., Tesauro, M., Alessandro, R., Ghirlanda, G., & Cardillo, C. (2013). Oxidative Stress in Diabetes: Implications for Vascular and Other Complications. International Journal of Molecular Sciences, 14(11), 21525-21550. https://doi.org/10.3390/ijms141121525