DNA Damage Induced MutS Homologue hMSH4 Acetylation

Abstract
:1. Introduction
2. Results
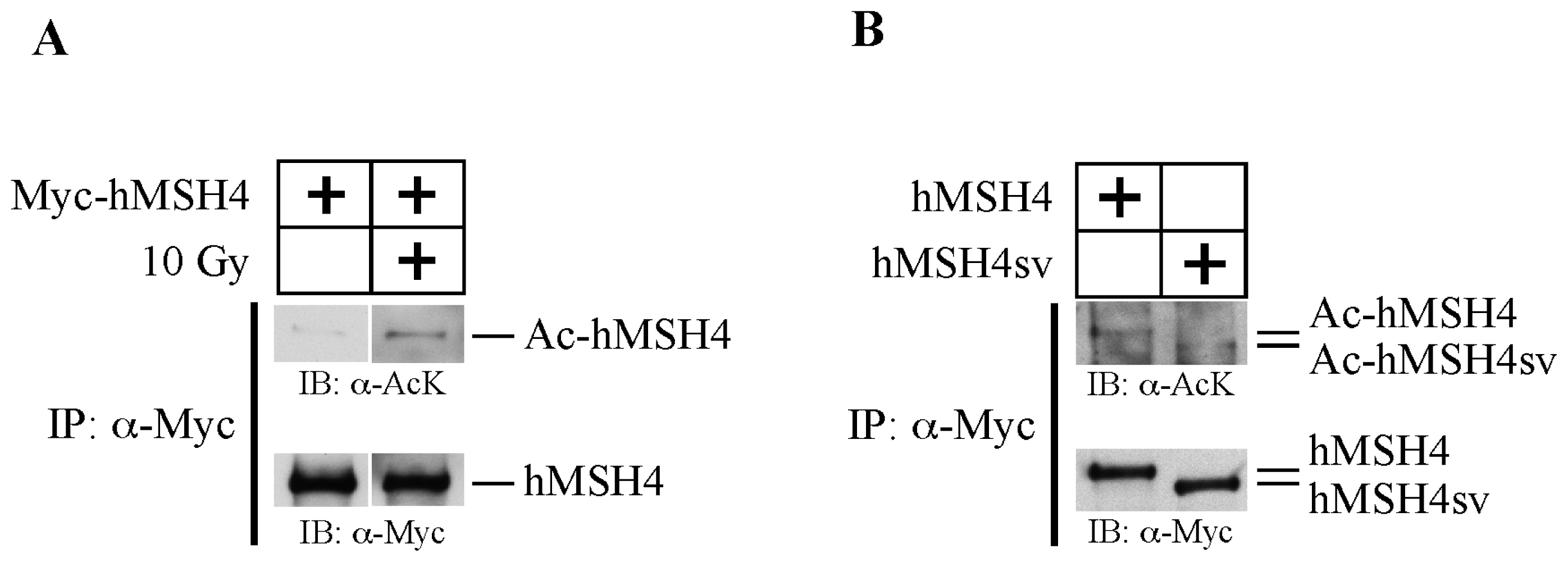
2.1. hMSH4 Is Acetylated in Response to DNA Damage
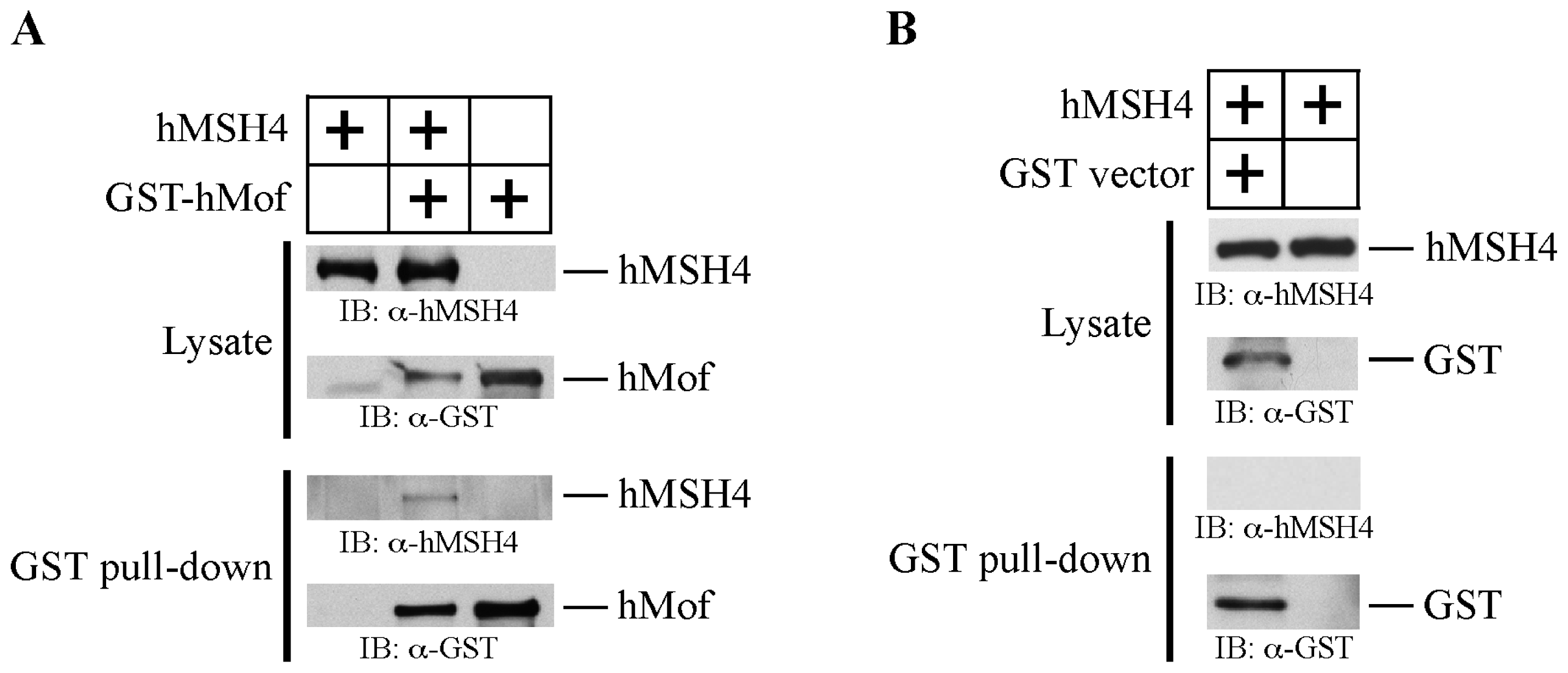
2.2. hMSH4 Physically Interacts with hMof
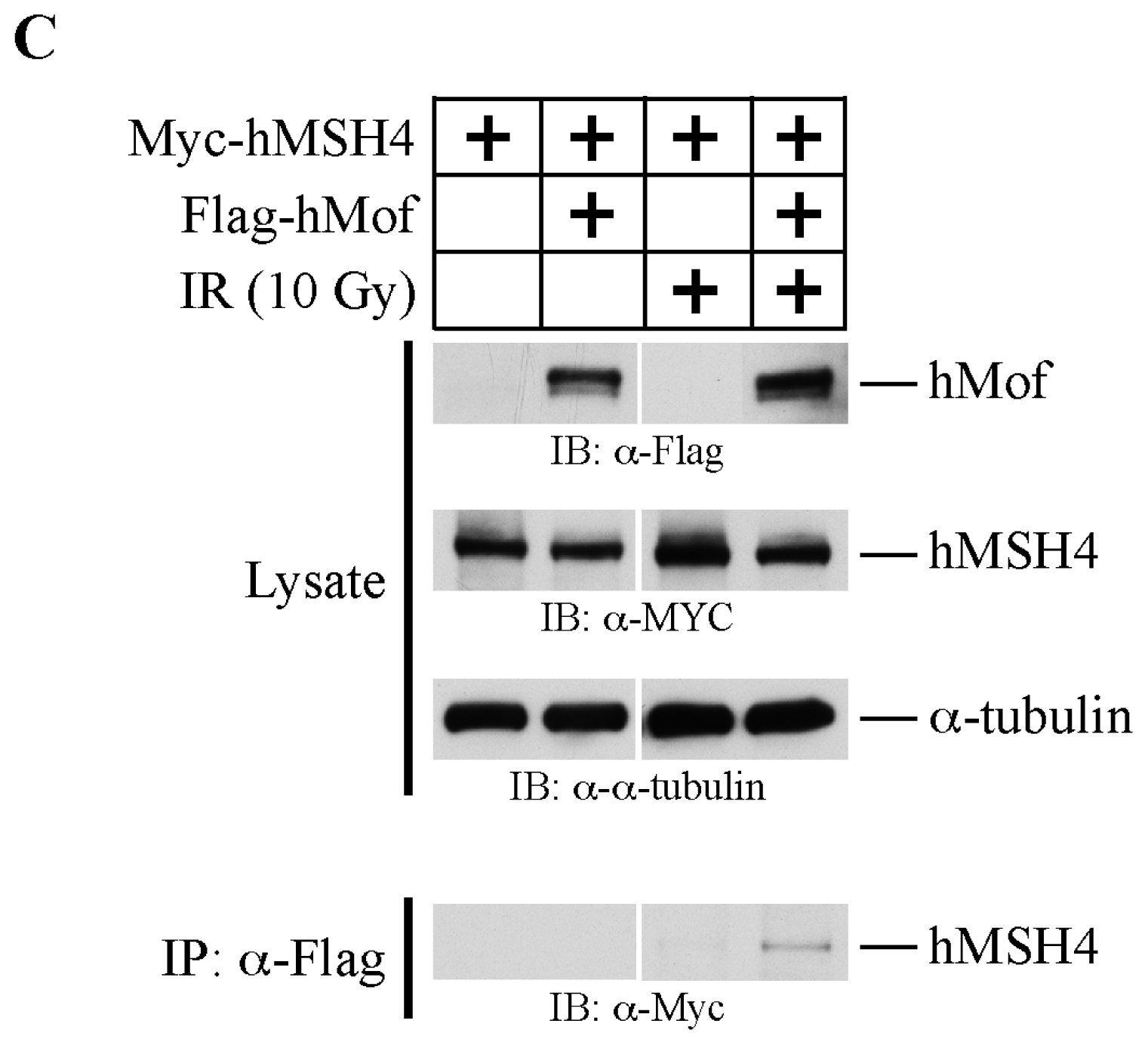
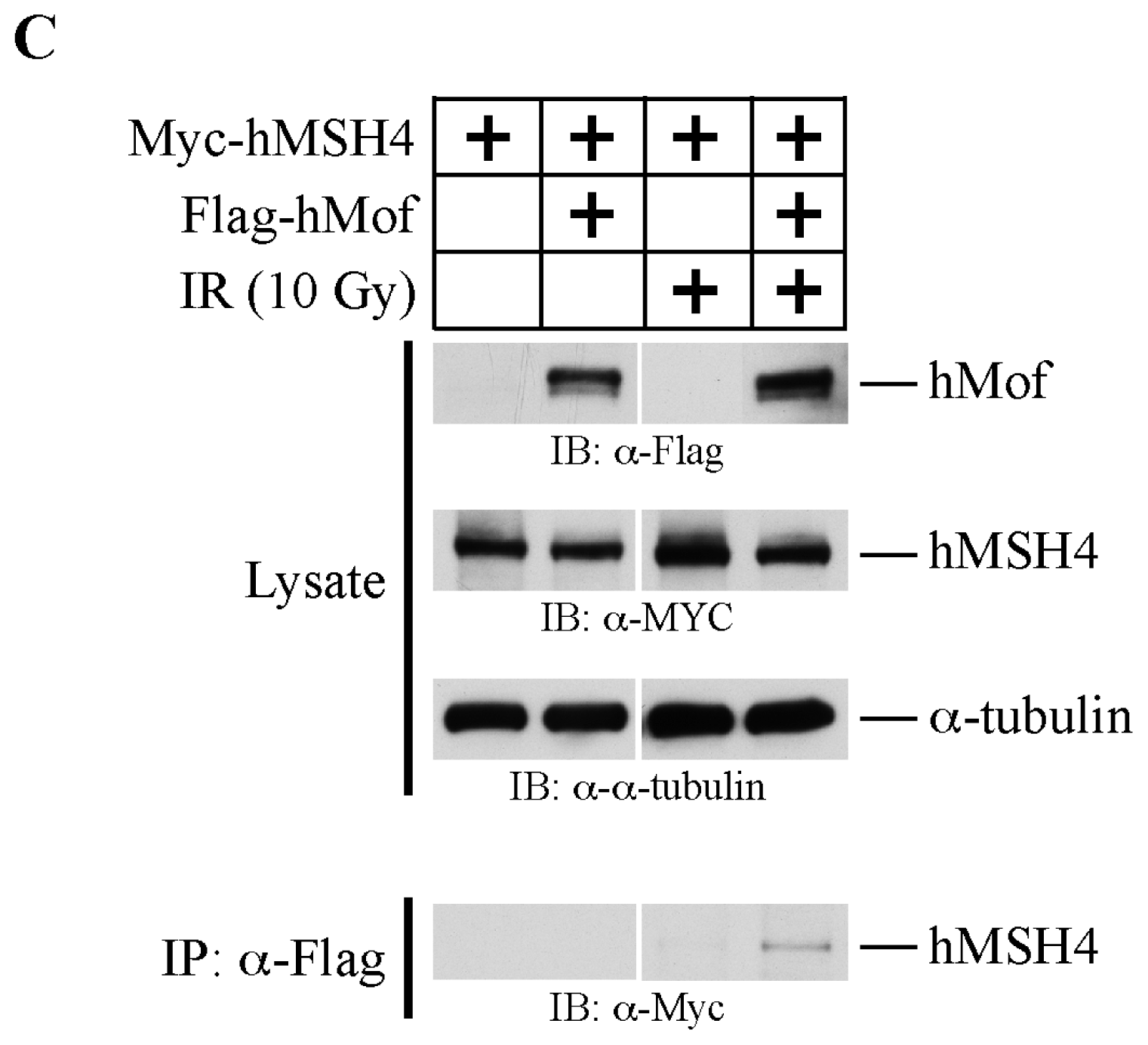
2.3. The hMSH4-hMof Interaction Is IR-Inducible in Human Cells
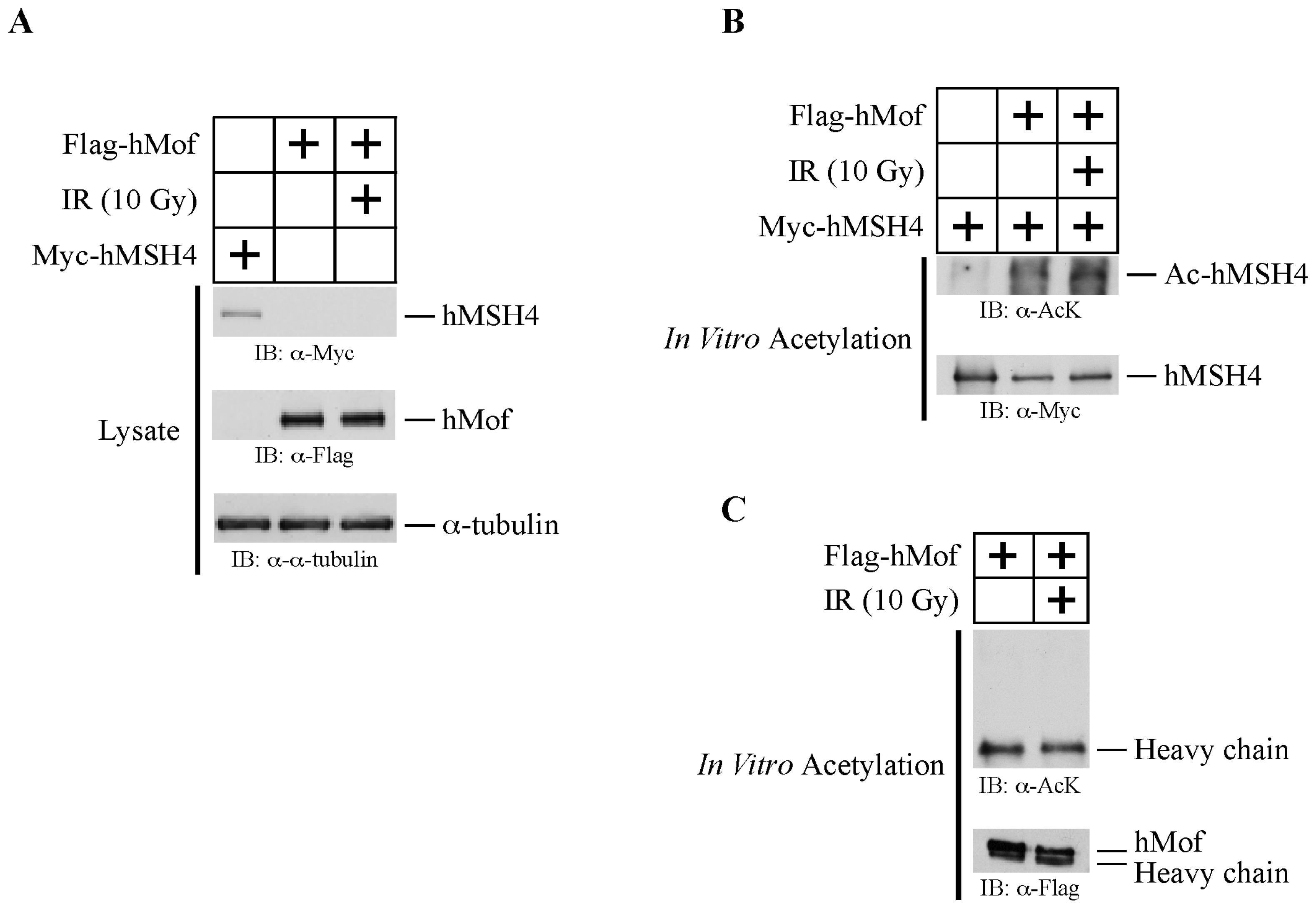
2.4. hMof Is Capable of Mediating hMSH4 Acetylation In Vitro
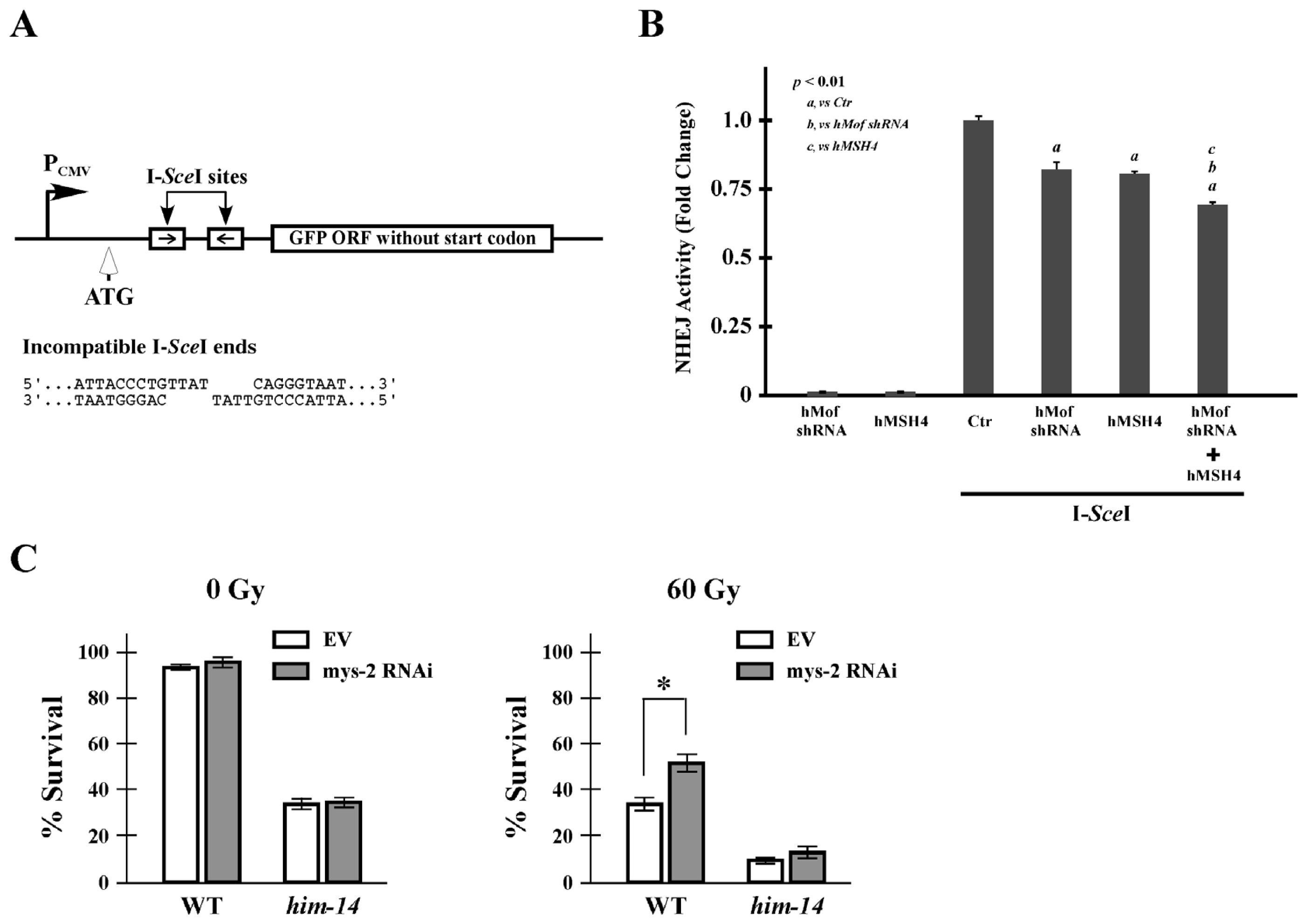
2.5. hMof Modulates the Effect of hMSH4 on NHEJ-Mediated DSB Repair and Cell Survival to IR
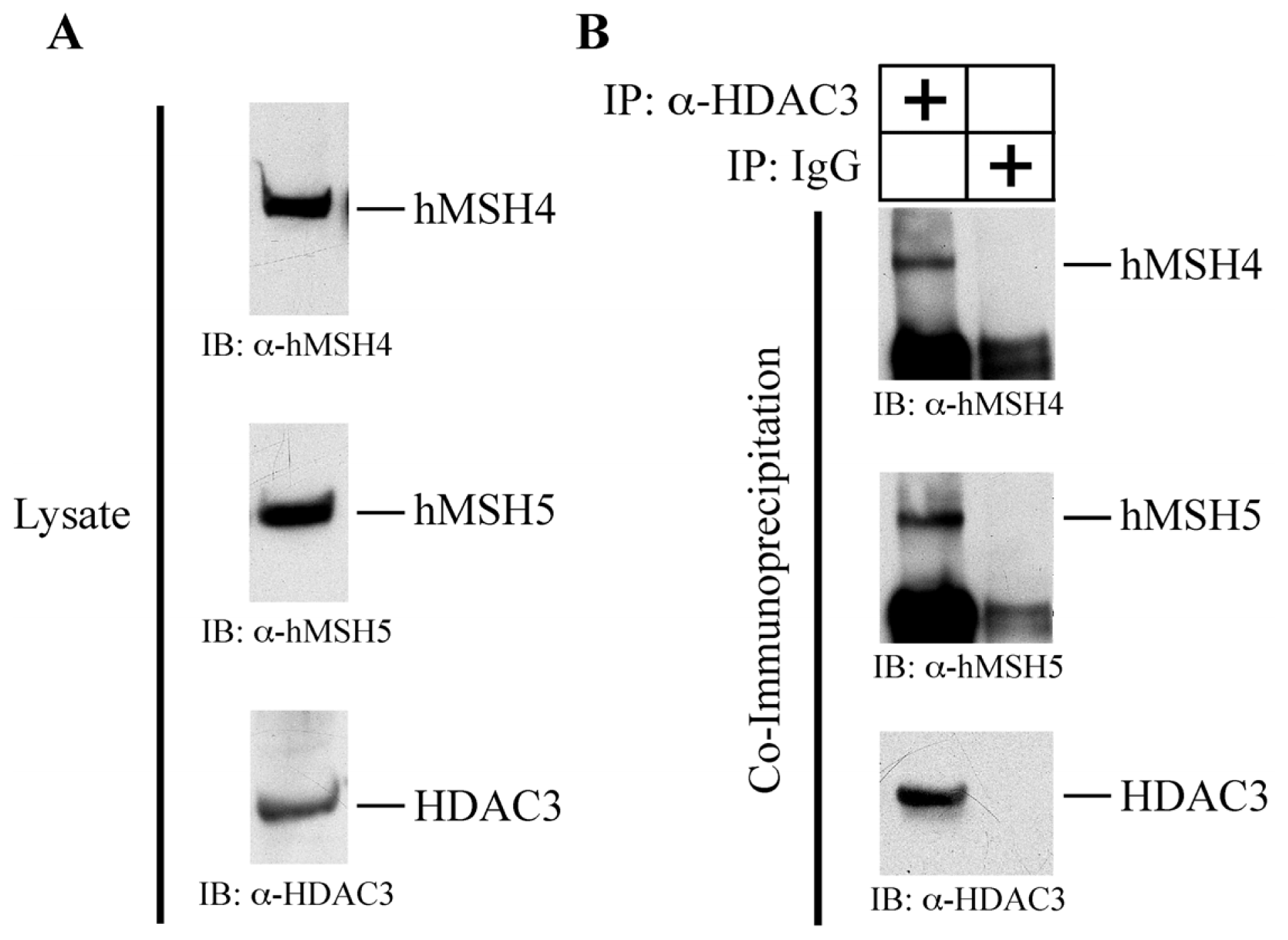
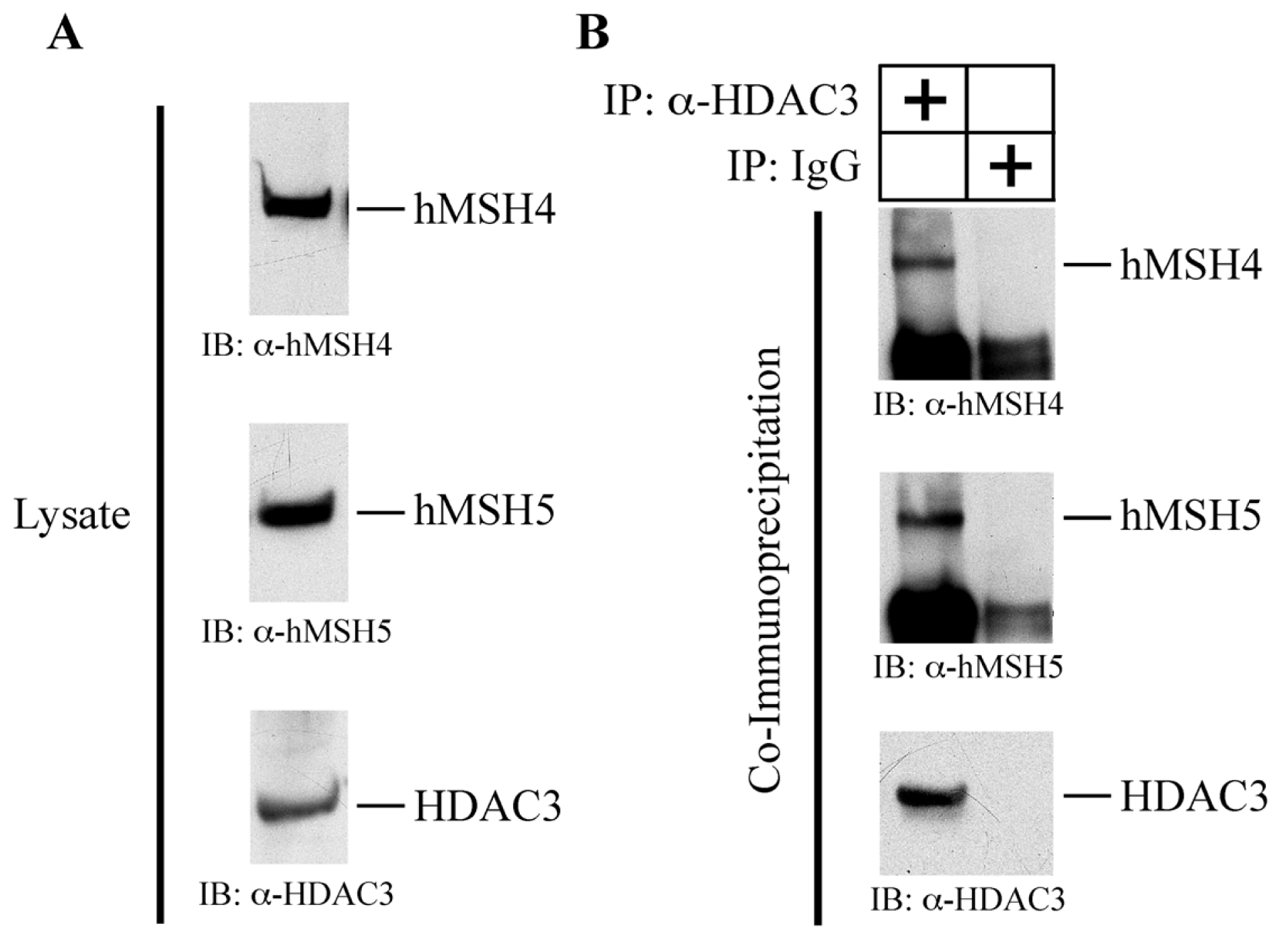
2.6. hMSH4 Interacts with Histone Deacetylase 3 (HDAC3)
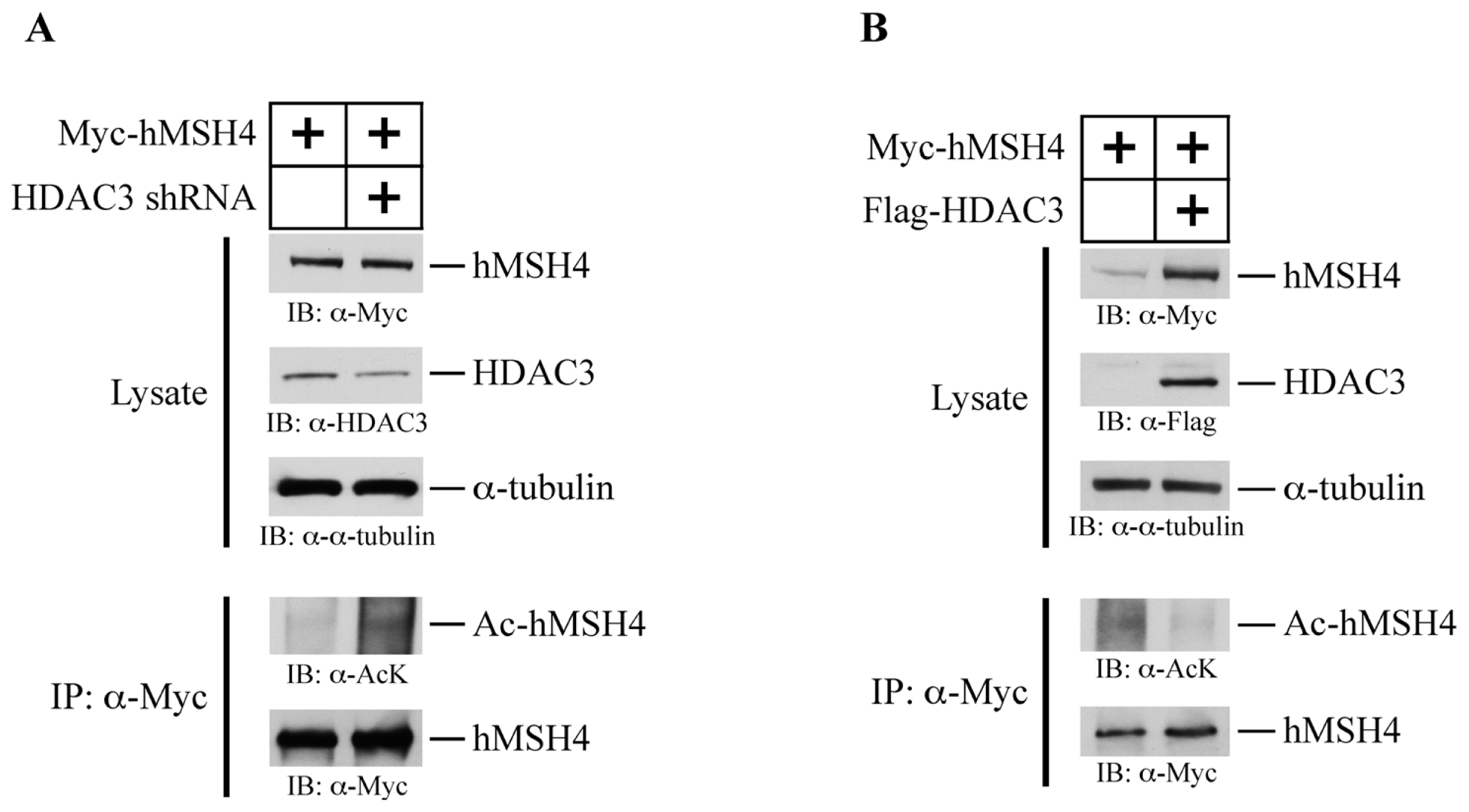
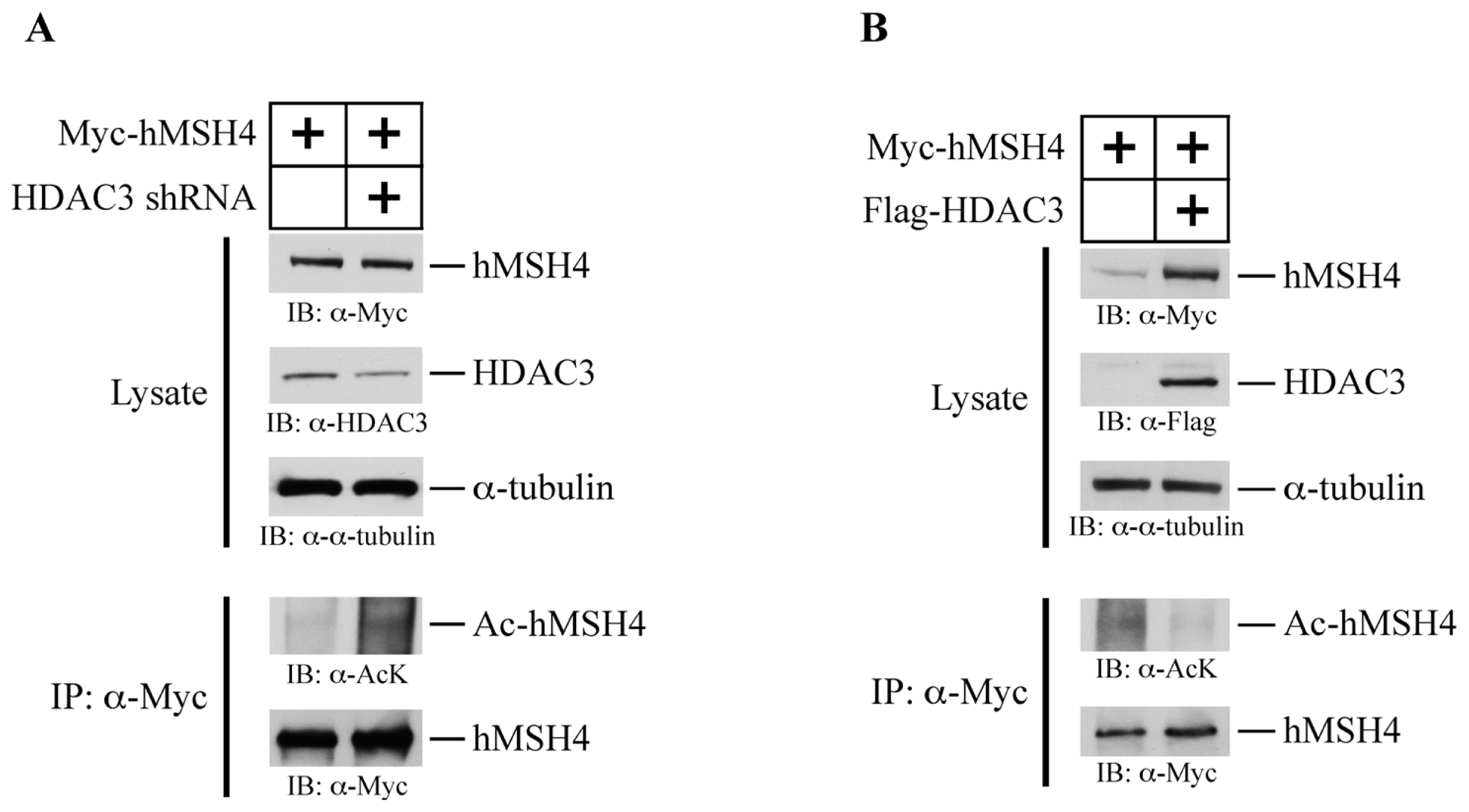
2.7. HDAC3 Facilitates hMSH4 Deacetylation
3. Discussion
4. Experimental Section
4.1. Cell Culture, Cell Extracts, and Induction of DNA Damage
4.2. SDS-PAGE, Western Blotting, Co-Immunoprecipitation (Co-IP) and Antibodies
4.3. Expression Constructs and Mammalian Transfection
4.4. GST Pull-Down Assay
4.5. Yeast Three-Hybrid Analysis
4.6. In Vitro Acetylation Assay
4.7. Survival Analysis
4.8. NHEJ Reporter Assay
5. Conclusions





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BD-fusion | “Native” HA-tagged | AD-fusion | His/Ade activation | |
|---|---|---|---|---|
| 1 | BD | HDAC3 | − | |
| 2 | hMSH4 | hMSH5 | AD | − |
| 3 | hMSH5 | hMSH4 | AD | − |
| 4 | hMSH4 | HDAC3 | − | |
| 5 | hMSH5 | HDAC3 | − | |
| 6 | hMSH4 | hMSH5 | HDAC3 | +++ |
| 7 | hMSH5 | hMSH4 | HDAC3 | − |
| 8 | hMSH4 | hMSH5 | GPS2 | +++ |
| 9 | hMSH5 | hMSH4 | GPS2 | +++ |
Acknowledgments
Conflicts of Interest
Abbreviations
| DSB | DNA double strand break |
| IR | ionizing radiation |
| NHEJ | nonhomologous end joining |
| MMR | DNA mismatch repair |
References
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol 2007, 8, 284–295. [Google Scholar]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar]
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem 2006, 281, 15741–15746. [Google Scholar]
- Yang, X.J. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res 2004, 32, 959–976. [Google Scholar]
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.M.; Sinclair, D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 2004, 13, 627–638. [Google Scholar]
- Yamaguchi, H.; Woods, N.T.; Piluso, L.G.; Lee, H.H.; Chen, J.; Bhalla, K.N.; Monteiro, A.; Liu, X.; Hung, M.C.; Wang, H.G. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J. Biol. Chem 2009, 284, 11171–11183. [Google Scholar]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.H.; Min, B.H.; Lee, K.M.; Cho, M.H.; Park, G.H.; Lee, K.H. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med 2007, 39, 8–13. [Google Scholar]
- Yuan, Z.; Zhang, X.; Sengupta, N.; Lane, W.S.; Seto, E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 2007, 27, 149–162. [Google Scholar]
- Terui, T.; Murakami, K.; Takimoto, R.; Takahashi, M.; Takada, K.; Murakami, T.; Minami, S.; Matsunaga, T.; Takayama, T.; Kato, J.; Niitsu, Y. Induction of PIG3 and NOXA through acetylation of p53 at 320 and 373 lysine residues as a mechanism for apoptotic cell death by histone deacetylase inhibitors. Cancer Res 2003, 63, 8948–8954. [Google Scholar]
- Gupta, A.; Sharma, G.G.; Young, C.S.; Agarwal, M.; Smith, E.R.; Paull, T.T.; Lucchesi, J.C.; Khanna, K.K.; Ludwig, T.; Pandita, T.K. Involvement of human MOF in ATM function. Mol. Cell Biol 2005, 25, 5292–5305. [Google Scholar]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar]
- Jiang, Z.; Kamath, R.; Jin, S.; Balasubramani, M.; Pandita, T.K.; Rajasekaran, B. Tip60-mediated acetylation activates transcription independent apoptotic activity of Abl. Mol. Cancer 2011, 10, 88. [Google Scholar]
- Sharma, G.G.; So, S.; Gupta, A.; Kumar, R.; Cayrou, C.; Avvakumov, N.; Bhadra, U.; Pandita, R.K.; Porteus, M.H.; Chen, D.J.; Cote, J.; Pandita, T.K. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell Biol 2010, 30, 3582–3595. [Google Scholar]
- Paquis-Flucklinger, V.; Santucci-Darmanin, S.; Paul, R.; Saunieres, A.; Turc-Carel, C.; Desnuelle, C. Cloning and expression analysis of a meiosis-specific MutS homolog: The human MSH4 gene. Genomics 1997, 44, 188–194. [Google Scholar]
- Her, C.; Zhao, N.; Wu, X.; Tompkins, J.D. MutS homologues hMSH4 and hMSH5: Diverse functional implications in humans. Front. Biosci 2007, 12, 905–911. [Google Scholar]
- Kneitz, B.; Cohen, P.E.; Avdievich, E.; Zhu, L.; Kane, M.F.; Hou, H., Jr.; Kolodner, R.D.; Kucherlapati, R.; Pollard, J.W.; Edelmann, W. MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev 2000, 14, 1085–1097. [Google Scholar]
- Snowden, T.; Acharya, S.; Butz, C.; Berardini, M.; Fishel, R. hMSH4-hMSH5 recognizes Holliday Junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol. Cell 2004, 15, 437–451. [Google Scholar]
- Lenzi, M.L.; Smith, J.; Snowden, T.; Kim, M.; Fishel, R.; Poulos, B.K.; Cohen, P.E. Extreme heterogeneity in the molecular events leading to the establishment of chiasmata during meiosis i in human oocytes. Am. J. Hum. Genet 2005, 76, 112–127. [Google Scholar]
- Adamo, A.; Montemauri, P.; Silva, N.; Ward, J.D.; Boulton, S.J.; La Volpe, A. BRC-1 acts in the inter-sister pathway of meiotic double-strand break repair. EMBO Rep 2008, 9, 287–292. [Google Scholar]
- Lipkin, S.M.; Wang, V.; Jacoby, R.; Banerjee-Basu, S.; Baxevanis, A.D.; Lynch, H.T.; Elliott, R.M.; Collins, F.S. MLH3: A DNA mismatch repair gene associated with mammalian microsatellite instability. Nat. Genet 2000, 24, 27–35. [Google Scholar]
- Santucci-Darmanin, S.; Neyton, S.; Lespinasse, F.; Saunieres, A.; Gaudray, P.; Paquis-Flucklinger, V. The DNA mismatch-repair MLH3 protein interacts with MSH4 in meiotic cells, supporting a role for this MutL homolog in mammalian meiotic recombination. Hum. Mol. Genet 2002, 11, 1697–1706. [Google Scholar]
- Santucci-Darmanin, S.; Walpita, D.; Lespinasse, F.; Desnuelle, C.; Ashley, T.; Paquis-Flucklinger, V. MSH4 acts in conjunction with MLH1 during mammalian meiosis. FASEB J 2000, 14, 1539–1547. [Google Scholar]
- Her, C.; Wu, X.; Bailey, S.M.; Doggett, N.A. Mouse MutS homolog 4 is predominantly expressed in testis and interacts with MutS homolog 5. Mamm. Genome 2001, 12, 73–76. [Google Scholar]
- Her, C.; Wu, X.; Griswold, M.D.; Zhou, F. Human MutS homologue MSH4 physically interacts with von Hippel-Lindau tumor suppressor-binding protein 1. Cancer Res 2003, 63, 865–872. [Google Scholar]
- Her, C.; Doggett, N.A. Cloning, structural characterization, and chromosomal localization of the human orthologue of Saccharomyces cerevisiae MSH5 gene. Genomics 1998, 52, 50–61. [Google Scholar]
- Lee, T.H.; Yi, W.; Griswold, M.D.; Zhu, F.; Her, C. Formation of hMSH4-hMSH5 heterocomplex is a prerequisite for subsequent GPS2 recruitment. DNA Repair 2006, 5, 32–42. [Google Scholar]
- Neyton, S.; Lespinasse, F.; Moens, P.B.; Paul, R.; Gaudray, P.; Paquis-Flucklinger, V.; Santucci-Darmanin, S. Association between MSH4 (MutS homologue 4) and the DNA strand-exchange RAD51 and DMC1 proteins during mammalian meiosis. Mol. Hum. Reprod 2004, 10, 917–924. [Google Scholar]
- Chu, Y.L.; Wu, X.; Xu, Y.; Her, C. MutS homologue hMSH4: Interaction with eIF3f and a role in NHEJ-mediated DSB repair. Mol. Cancer 2013, 12, 51. [Google Scholar]
- Wu, X.; Xu, Y.; Feng, K.; Tompkins, J.D.; Her, C. MutS Homologue hMSH5: Recombinational DSB Repair and Non-Synonymous Polymorphic Variants. PLoS One 2013, 8, e73284. [Google Scholar]
- MacQueen, A.J.; Colaiacovo, M.P.; McDonald, K.; Villeneuve, A.M. Synapsis-dependent and -independent mechanisms stabilize homolog pairing during meiotic prophase in C. elegans. Genes Dev 2002, 16, 2428–2442. [Google Scholar]
- Zalevsky, J.; MacQueen, A.J.; Duffy, J.B.; Kemphues, K.J.; Villeneuve, A.M. Crossing over during Caenorhabditis elegans meiosis requires a conserved MutS-based pathway that is partially dispensable in budding yeast. Genetics 1999, 153, 1271–1283. [Google Scholar]
- Zhang, J.; Kalkum, M.; Chait, B.T.; Roeder, R.G. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 2002, 9, 611–623. [Google Scholar]
- Hartman, H.B.; Yu, J.; Alenghat, T.; Ishizuka, T.; Lazar, M.A. The histone-binding code of nuclear receptor co-repressors matches the substrate specificity of histone deacetylase 3. EMBO Rep 2005, 6, 445–451. [Google Scholar]
- Rea, S.; Xouri, G.; Akhtar, A. Males absent on the first (MOF): From flies to humans. Oncogene 2007, 26, 5385–5394. [Google Scholar]
- Thomas, T.; Loveland, K.L.; Voss, A.K. The genes coding for the MYST family histone acetyltransferases, Tip60 and Mof, are expressed at high levels during sperm development. Gene Expr. Patterns 2007, 7, 657–665. [Google Scholar]
- Xu, Y.; Her, C. VBP1 facilitates proteasome and autophagy-mediated degradation of MutS homologue hMSH4. FASEB J 2013. [Google Scholar] [CrossRef]
- Singh, K.P.; Treas, J.; Tyagi, T.; Gao, W. DNA demethylation by 5-aza-2-deoxycytidine treatment abrogates 17 beta-estradiol-induced cell growth and restores expression of DNA repair genes in human breast cancer cells. Cancer Lett 2012, 316, 62–69. [Google Scholar]
- Conde, J.; Silva, S.N.; Azevedo, A.P.; Teixeira, V.; Pina, J.E.; Rueff, J.; Gaspar, J.F. Association of common variants in mismatch repair genes and breast cancer susceptibility: A multigene study. BMC Cancer 2009, 9, 344. [Google Scholar]
- Kim, J.; Lee, Y.G.; Kim, N. Bioinformatics interpretation of exome sequencing: Blood cancer. Genomics inform 2013, 11, 24–33. [Google Scholar]
- Chng, W.J.; Gertz, M.A.; Chung, T.H.; Van Wier, S.; Keats, J.J.; Baker, A.; Bergsagel, P.L.; Carpten, J.; Fonseca, R. Correlation between array-comparative genomic hybridization-defined genomic gains and losses and survival: Identification of 1p31–32 deletion as a prognostic factor in myeloma. Leukemia 2010, 24, 833–842. [Google Scholar]
- Tompkins, J.D.; Wu, X.; Chu, Y.L.; Her, C. Evidence for a direct involvement of hMSH5 in promoting ionizing radiation induced apoptosis. Exp. Cell Res 2009, 315, 2420–2432. [Google Scholar]
- Tompkins, J.D.; Wu, X.; Her, C. MutS homologue hMSH5: Role in cisplatin-induced DNA damage response. Mol. Cancer 2012, 11, 10. [Google Scholar]
- Meltser, V.; Ben-Yehoyada, M.; Reuven, N.; Shaul, Y. c-Abl downregulates the slow phase of double-strand break repair. Cell Death Dis 2010, 1, e20. [Google Scholar]
- Gonfloni, S.; Di Tella, L.; Caldarola, S.; Cannata, S.M.; Klinger, F.G.; Di Bartolomeo, C.; Mattei, M.; Candi, E.; De Felici, M.; Melino, G.; Cesareni, G. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat. Med 2009, 15, 1179–1185. [Google Scholar]
- Neyton, S.; Lespinasse, F.; Lahaye, F.; Staccini, P.; Paquis-Flucklinger, V.; Santucci-Darmanin, S. CRM1-dependent nuclear export and dimerization with hMSH5 contribute to the regulation of hMSH4 subcellular localization. Exp. Cell Res 2007, 313, 3680–3693. [Google Scholar]
- Cao, X.; Sudhof, T.C. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001, 293, 115–120. [Google Scholar]
- Emiliani, S.; Fischle, W.; Van Lint, C.; Al-Abed, Y.; Verdin, E. Characterization of a human RPD3 ortholog, HDAC3. Proc. Natl. Acad. Sci. USA 1998, 95, 2795–2800. [Google Scholar]
- Vo, A.T.; Zhu, F.; Wu, X.; Yuan, F.; Gao, Y.; Gu, L.; Li, G.M.; Lee, T.H.; Her, C. hMRE11 deficiency leads to microsatellite instability and defective DNA mismatch repair. EMBO Rep 2005, 6, 438–444. [Google Scholar]
- Ishizuka, T.; Lazar, M.A. The N-CoR/histone deacetylase 3 complex is required for repression by thyroid hormone receptor. Mol. Cell Biol 2003, 23, 5122–5131. [Google Scholar]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar]
- Fraser, A.G.; Kamath, R.S.; Zipperlen, P.; Martinez-Campos, M.; Sohrmann, M.; Ahringer, J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 2000, 408, 325–330. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chu, Y.-L.; Wu, X.; Xu, J.; Watts, J.L.; Her, C. DNA Damage Induced MutS Homologue hMSH4 Acetylation. Int. J. Mol. Sci. 2013, 14, 20966-20982. https://doi.org/10.3390/ijms141020966
Chu Y-L, Wu X, Xu J, Watts JL, Her C. DNA Damage Induced MutS Homologue hMSH4 Acetylation. International Journal of Molecular Sciences. 2013; 14(10):20966-20982. https://doi.org/10.3390/ijms141020966
Chicago/Turabian StyleChu, Yen-Lin, Xiling Wu, Jing Xu, Jennifer L. Watts, and Chengtao Her. 2013. "DNA Damage Induced MutS Homologue hMSH4 Acetylation" International Journal of Molecular Sciences 14, no. 10: 20966-20982. https://doi.org/10.3390/ijms141020966