The Epidermal Growth Factor Receptor and Its Ligands in Cardiovascular Disease

Abstract
:
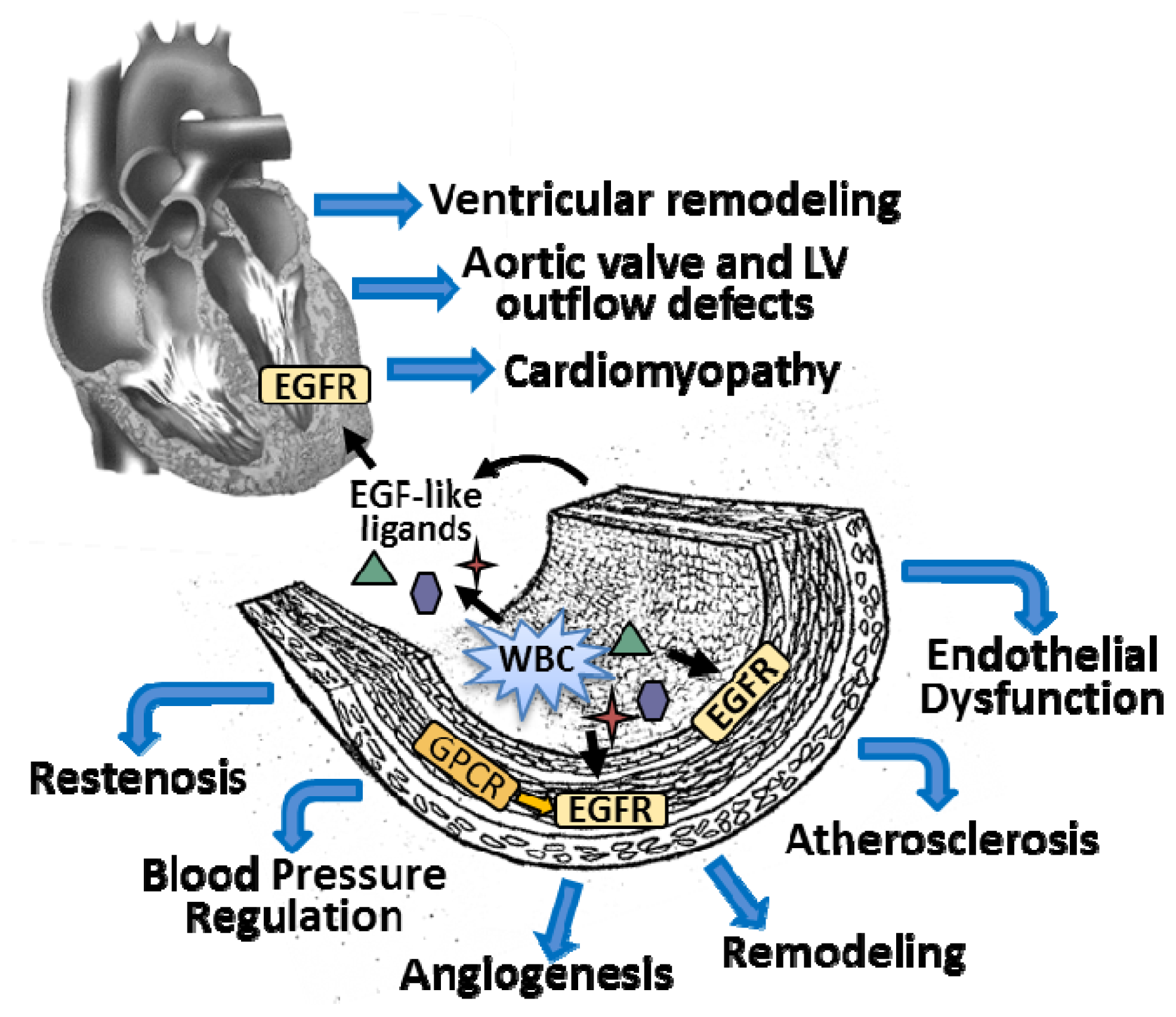
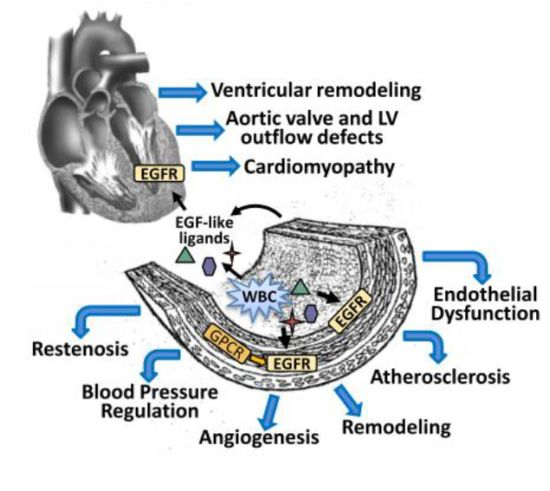{kind=link}
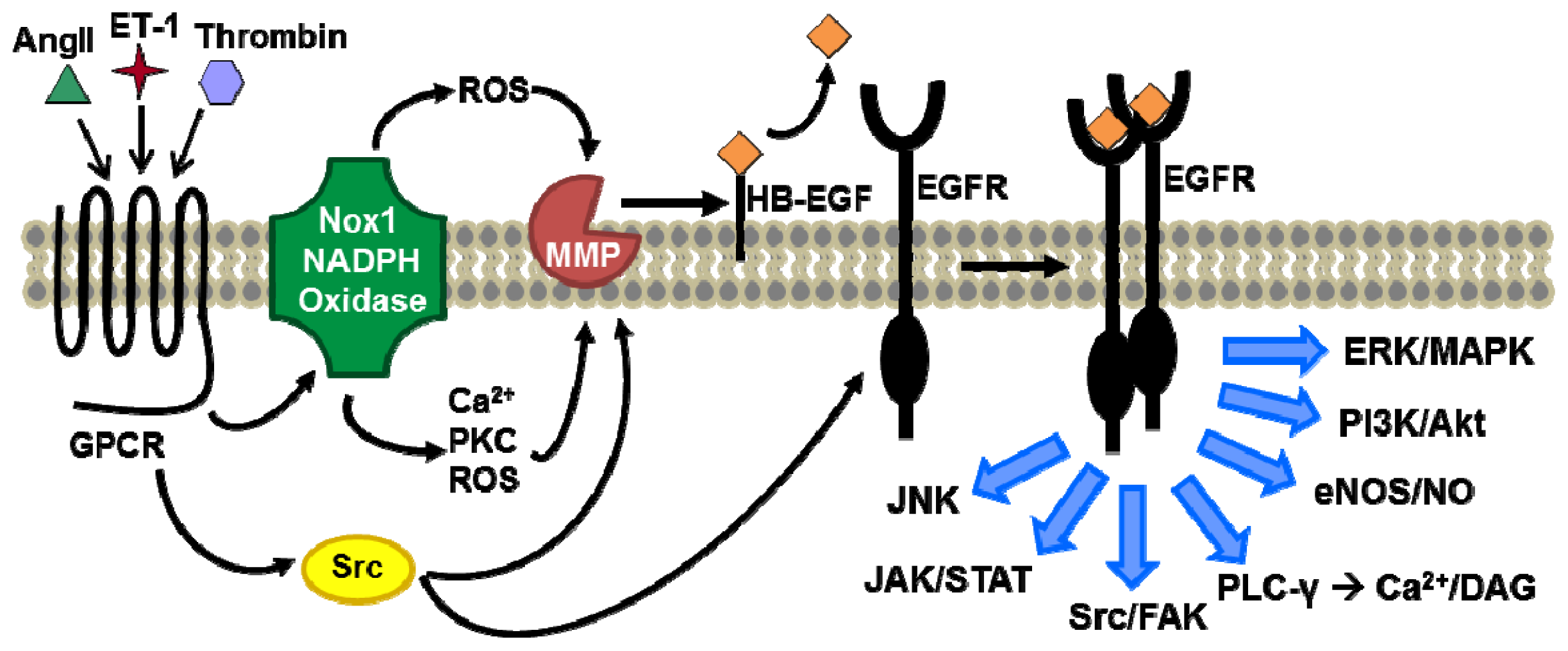
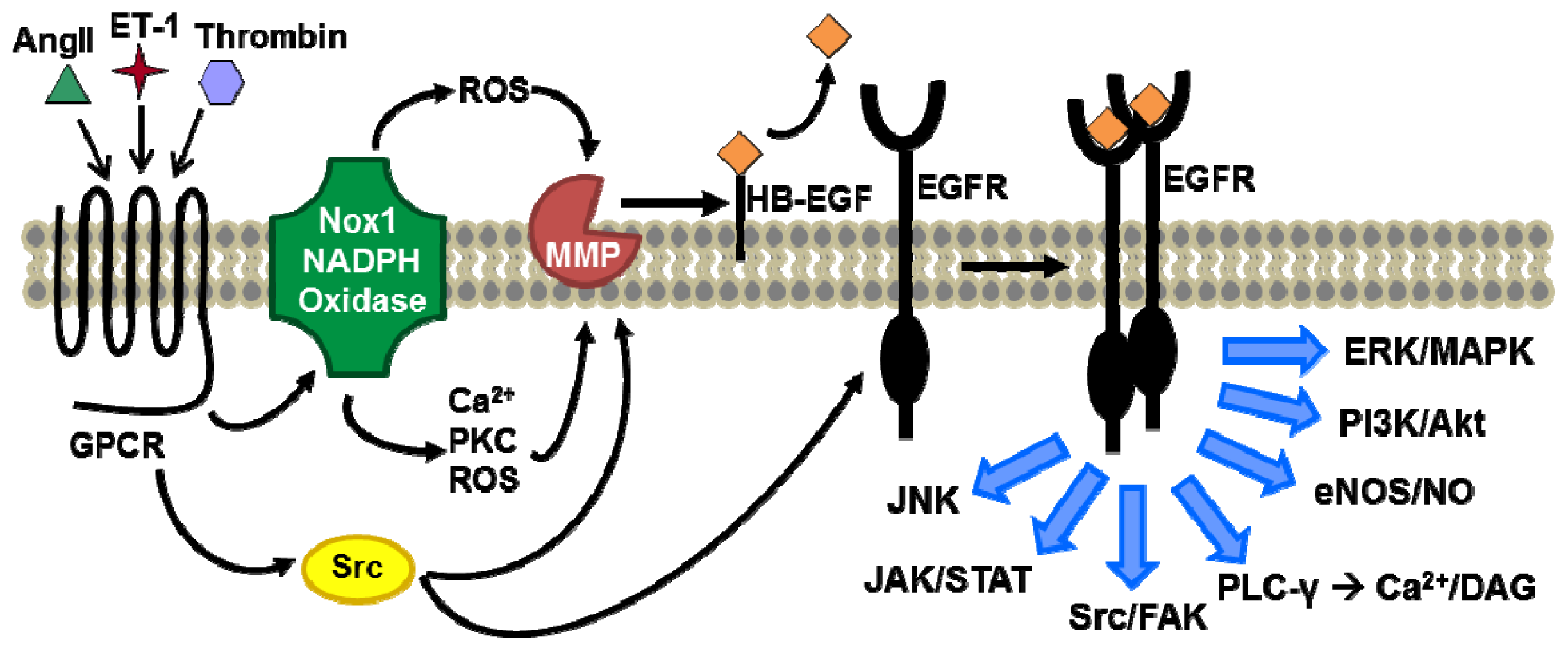{kind=link}
{kind=link}
1. Introduction
2. EGFR in Cardiac Development
3. EGFR and Ligands in the Blood Vessel
3.1. Vascular Smooth Muscle Cells (VSMCs)
3.2. Endothelial Cells
4. EGFR and Its Ligands in Cardiovascular Disease
4.1. Blood Pressure Regulation
4.2. Angiogenesis
4.3. Restenosis
4.4. Atherosclerosis
4.5. Cardiac Injury and Remodeling
4.6. Diabetes Mellitus
5. Therapeutic Implications
6. Conclusions

Acknowledgments
Conflicts of Interest
References
- Holbro, T.; Hynes, N.E. ErbB receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol 2004, 44, 195–217. [Google Scholar]
- Hackel, P.O.; Zwick, E.; Prenzel, N.; Ullrich, A. Epidermal growth factor receptors: Critical mediators of multiple receptor pathways. Curr. Opin. Cell Biol 1999, 11, 184–189. [Google Scholar]
- Wetzker, R.; Bohmer, F.D. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell Biol 2003, 4, 651–657. [Google Scholar]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar]
- Prenzel, N.; Fischer, O.M.; Streit, S.; Hart, S.; Ullrich, A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr. Relat. Cancer 2001, 8, 11–31. [Google Scholar]
- Andreev, J.; Galisteo, M.L.; Kranenburg, O.; Logan, S.K.; Chiu, E.S.; Okigaki, M.; Cary, L.A.; Moolenaar, W.H.; Schlessinger, J, Sr. c and Pyk2 mediate G-protein-coupled receptor activation of epidermal growth factor receptor (EGFR) but are not required for coupling to the mitogen-activated protein (MAP) kinase signaling cascade. J. Biol. Chem 2001, 276, 20130–20135. [Google Scholar]
- Lin, S.Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell Biol 2001, 3, 802–808. [Google Scholar]
- Liao, H.J.; Carpenter, G. Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression. Mol. Biol. Cell 2007, 18, 1064–1072. [Google Scholar]
- Liao, H.J.; Carpenter, G. Regulated intramembrane cleavage of the EGF receptor. Traffic 2012, 13, 1106–1112. [Google Scholar]
- Wilken, J.A.; Perez-Torres, M.; Nieves-Alicea, R.; Cora, E.M.; Christensen, T.A.; Baron, A.T.; Maihle, N.J. Shedding of soluble epidermal growth factor receptor (sEGFR) is mediated by a metalloprotease/fibronectin/integrin axis and inhibited by cetuximab. Biochemistry 2013, 52, 4531–4540. [Google Scholar]
- Massague, J.; Pandiella, A. Membrane-anchored growth factors. Annu. Rev. Biochem 1993, 62, 515–541. [Google Scholar]
- Burgess, A.W. Epidermal growth factor and transforming growth factor alpha. Br. Med. Bull 1989, 45, 401–424. [Google Scholar]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar]
- Hinkle, C.L.; Mohan, M.J.; Lin, P.; Yeung, N.; Rasmussen, F.; Milla, M.E.; Moss, M.L. Multiple metalloproteinases process protransforming growth factor-alpha (proTGF-alpha). Biochemistry 2003, 42, 2127–2136. [Google Scholar]
- Yu, W.H.; Woessner, J.F., Jr.; McNeish, J.D.; Stamenkovic, I. CD44 anchors the assembly of matrilysin/MMP-7 with heparin-binding epidermal growth factor precursor and ErbB4 and regulates female reproductive organ remodeling. Genes Dev 2002, 16, 307–323. [Google Scholar]
- Suzuki, M.; Raab, G.; Moses, M.A.; Fernandez, C.A.; Klagsbrun, M. Matrix metalloproteinase-3 releases active heparin-binding EGF-like growth factor by cleavage at a specific juxtamembrane site. J. Biol. Chem 1997, 272, 31730–31737. [Google Scholar]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995, 269, 234–238. [Google Scholar]
- Threadgill, D.W.; Dlugosz, A.A.; Hansen, L.A.; Tennenbaum, T.; Lichti, U.; Yee, D.; LaMantia, C.; Mourton, T.; Herrup, K.; Harris, R.C.; et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science 1995, 269, 230–234. [Google Scholar]
- Kornblum, H.I.; Hussain, R.; Wiesen, J.; Miettinen, P.; Zurcher, S.D.; Chow, K.; Derynck, R.; Werb, Z. Abnormal astrocyte development and neuronal death in mice lacking the epidermal growth factor receptor. J. Neurosci. Res 1998, 53, 697–717. [Google Scholar]
- Sibilia, M.; Steinbach, J.P.; Stingl, L.; Aguzzi, A.; Wagner, E.F. A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J 1998, 17, 719–731. [Google Scholar]
- Luetteke, N.C.; Phillips, H.K.; Qiu, T.H.; Copeland, N.G.; Earp, H.S.; Jenkins, N.A.; Lee, D.C. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev 1994, 8, 399–413. [Google Scholar]
- Barrick, C.J.; Roberts, R.B.; Rojas, M.; Rajamannan, N.M.; Suitt, C.B.; O’Brien, K.D.; Smyth, S.S.; Threadgill, D.W. Reduced EGFR causes abnormal valvular differentiation leading to calcific aortic stenosis and left ventricular hypertrophy in C57BL/6J but not 129S1/SvImJ mice. Am. J. Physiol. Heart Circ. Physiol 2009, 297, H65–H75. [Google Scholar]
- Gassmann, M.; Casagranda, F.; Orioli, D.; Simon, H.; Lai, C.; Klein, R.; Lemke, G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995, 378, 390–394. [Google Scholar]
- Lee, K.F.; Simon, H.; Chen, H.; Bates, B.; Hung, M.C.; Hauser, C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995, 378, 394–398. [Google Scholar]
- McBride, K.L.; Zender, G.A.; Fitzgerald-Butt, S.M.; Seagraves, N.J.; Fernbach, S.D.; Zapata, G.; Lewin, M.; Towbin, J.A.; Belmont, J.W. Association of common variants in ERBB4 with congenital left ventricular outflow tract obstruction defects. Birth Defects Res. A 2011, 91, 162–168. [Google Scholar]
- Iwamoto, R.; Yamazaki, S.; Asakura, M.; Takashima, S.; Hasuwa, H.; Miyado, K.; Adachi, S.; Kitakaze, M.; Hashimoto, K.; Raab, G.; et al. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc. Natl. Acad. Sci. USA 2003, 100, 3221–3226. [Google Scholar]
- Hartmann, D.; de Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lubke, T.; Lena Illert, A.; von Figura, K.; Saftig, P. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet 2002, 11, 2615–2624. [Google Scholar]
- Kato, M.; Inazu, T.; Kawai, Y.; Masamura, K.; Yoshida, M.; Tanaka, N.; Miyamoto, K.; Miyamori, I. Amphiregulin is a potent mitogen for the vascular smooth muscle cell line, A7r5. Biochem. Biophys. Res. Commun 2003, 301, 1109–1115. [Google Scholar]
- Dreux, A.C.; Lamb, D.J.; Modjtahedi, H.; Ferns, G.A. The epidermal growth factor receptors and their family of ligands: Their putative role in atherogenesis. Atherosclerosis 2006, 186, 38–53. [Google Scholar]
- Stanic, B.; Pandey, D.; Fulton, D.J.; Miller, F.J., Jr. Increased epidermal growth factor-like ligands are associated with elevated vascular nicotinamide adenine dinucleotide phosphate oxidase in a primate model of atherosclerosis. Arterioscler. Thromb. Vasc. Biol 2012, 32, 2452–2460. [Google Scholar]
- Gospodarowicz, D.; Hirabayashi, K.; Giguere, L.; Tauber, J.P. Factors controlling the proliferative rate, final cell density, and life span of bovine vascular smooth muscle cells in culture. J. Cell Biol 1981, 89, 568–578. [Google Scholar]
- Schreier, B.; Dohler, M.; Rabe, S.; Schneider, B.; Schwerdt, G.; Ruhs, S.; Sibilia, M.; Gotthardt, M.; Gekle, M.; Grossmann, C. Consequences of epidermal growth factor receptor (ErbB1) loss for vascular smooth muscle cells from mice with targeted deletion of ErbB1. Arterioscler. Thromb. Vasc. Biol 2011, 31, 1643–1652. [Google Scholar]
- Mazak, I.; Fiebeler, A.; Muller, D.N.; Park, J.K.; Shagdarsuren, E.; Lindschau, C.; Dechend, R.; Viedt, C.; Pilz, B.; Haller, H.; et al. Aldosterone potentiates angiotensin II-induced signaling in vascular smooth muscle cells. Circulation 2004, 109, 2792–2800. [Google Scholar]
- Min, L.J.; Mogi, M.; Li, J.M.; Iwanami, J.; Iwai, M.; Horiuchi, M. Aldosterone and angiotensin II synergistically induce mitogenic response in vascular smooth muscle cells. Circ. Res 2005, 97, 434–442. [Google Scholar]
- Berk, B.C.; Brock, T.A.; Webb, R.C.; Taubman, M.B.; Atkinson, W.J.; Gimbrone, M.A., Jr.; Alexander, R.W. Epidermal growth factor, a vascular smooth muscle mitogen, induces rat aortic contraction. J. Clin. Invest 1985, 75, 1083–1086. [Google Scholar]
- Ushio-Fukai, M.; Griendling, K.K.; Becker, P.L.; Hilenski, L.; Halleran, S.; Alexander, R.W. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol 2001, 21, 489–495. [Google Scholar]
- Chen, C.H.; Cheng, T.H.; Lin, H.; Shih, N.L.; Chen, Y.L.; Chen, Y.S.; Cheng, C.F.; Lian, W.S.; Meng, T.C.; Chiu, W.T.; et al. Reactive oxygen species generation is involved in epidermal growth factor receptor transactivation through the transient oxidization of src homology 2-containing tyrosine phosphatase in endothelin-1 signaling pathway in rat cardiac fibroblasts. Mol. Pharmacol 2006, 69, 1347–1355. [Google Scholar]
- Yang, C.M.; Lin, M.I.; Hsieh, H.L.; Sun, C.C.; Ma, Y.H.; Hsiao, L.D. Bradykinin-induced p42/p44 MAPK phosphorylation and cell proliferation via Src, EGF receptors, and PI3-K/Akt in vascular smooth muscle cells. J. Cell Physiol 2005, 203, 538–546. [Google Scholar]
- Iwasaki, H.; Eguchi, S.; Marumo, F.; Hirata, Y. Endothelin-1 stimulates DNA synthesis of vascular smooth-muscle cells through transactivation of epidermal growth factor receptor. J. Cardiovasc. Pharmacol 1998, 31, S182–S184. [Google Scholar]
- Kawanabe, Y.; Masaki, T.; Hashimoto, N. Involvement of epidermal growth factor receptor-protein tyrosine kinase transactivation in endothelin-1-induced vascular contraction. J. Neurosurg 2004, 100, 1066–1071. [Google Scholar]
- Kanda, Y.; Mizuno, K.; Kuroki, Y.; Watanabe, Y. Thrombin-induced p38 mitogen-activated protein kinase activation is mediated by epidermal growth factor receptor transactivation pathway. Br. J. Pharmacol 2001, 132, 1657–1664. [Google Scholar]
- Jagadeesha, D.K.; Takapoo, M.; Banfi, B.; Bhalla, R.C.; Miller, F.J., Jr. Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovasc. Res 2012, 93, 406–413. [Google Scholar]
- Eguchi, S.; Inagami, T. Signal transduction of angiotensin II type 1 receptor through receptor tyrosine kinase. Regul. Pept 2000, 91, 13–20. [Google Scholar]
- Ohtsu, H.; Dempsey, P.J.; Eguchi, S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am. J. Physiol. Cell Physiol 2006, 291, C1–C10. [Google Scholar]
- Seals, D.F.; Courtneidge, S.A. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev 2003, 17, 7–30. [Google Scholar]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar]
- Takaguri, A.; Shirai, H.; Kimura, K.; Hinoki, A.; Eguchi, K.; Carlile-Klusacek, M.; Yang, B.; Rizzo, V.; Eguchi, S. Caveolin-1 negatively regulates a metalloprotease-dependent epidermal growth factor receptor transactivation by angiotensin II. J. Mol. Cell Cardiol 2011, 50, 545–551. [Google Scholar]
- Miller, F.J., Jr.; Chu, X.; Stanic, B.; Tian, X.; Sharma, R.V.; Davisson, R.L.; Lamb, F.S. A differential role for endocytosis in receptor-mediated activation of Nox1. Antioxid. Redox Signal 2010, 12, 583–593. [Google Scholar]
- Stanic, B.; Katsuyama, M.; Miller, F.J., Jr. An oxidized extracellular oxidation-reduction state increases Nox1 expression and proliferation in vascular smooth muscle cells via epidermal growth factor receptor activation. Arterioscler. Thromb. Vasc. Biol 2010, 30, 2234–2241. [Google Scholar]
- Hsieh, H.L.; Lin, C.C.; Chan, H.J.; Yang, C.M. c-Src-dependent EGF receptor transactivation contributes to ET-1-induced COX-2 expression in brain microvascular endothelial cells. J. Neuroinflammation 2012, 9, 152. [Google Scholar]
- Mehta, V.B.; Zhou, Y.; Radulescu, A.; Besner, G.E. HB-EGF stimulates eNOS expression and nitric oxide production and promotes eNOS dependent angiogenesis. Growth Factors 2008, 26, 301–315. [Google Scholar]
- Zhou, Y.; Brigstock, D.; Besner, G.E. Heparin-binding EGF-like growth factor is a potent dilator of terminal mesenteric arterioles. Microvasc. Res 2009, 78, 78–85. [Google Scholar]
- Schreier, B.; Rabe, S.; Schneider, B.; Bretschneider, M.; Rupp, S.; Ruhs, S.; Neumann, J.; Rueckschloss, U.; Sibilia, M.; Gotthardt, M.; Grossmann, C.; Gekle, M. Loss of epidermal growth factor receptor in vascular smooth muscle cells and cardiomyocytes causes arterial hypotension and cardiac hypertrophy. Hypertension 2013, 61, 333–340. [Google Scholar]
- Amin, A.H.; Abd Elmageed, Z.Y.; Partyka, M.; Matrougui, K. Mechanisms of myogenic tone of coronary arteriole: Role of down stream signaling of the EGFR tyrosine kinase. Microvasc. Res 2011, 81, 135–142. [Google Scholar]
- Chansel, D.; Ciroldi, M.; Vandermeersch, S.; Jackson, L.F.; Gomez, A.M.; Henrion, D.; Lee, D.C.; Coffman, T.M.; Richard, S.; Dussaule, J.C.; Tharaux, P.L. Heparin binding EGF is necessary for vasospastic response to endothelin. FASEB J 2006, 20, 1936–1938. [Google Scholar]
- Flamant, M.; Tharaux, P.L.; Placier, S.; Henrion, D.; Coffman, T.; Chatziantoniou, C.; Dussaule, J.C. Epidermal growth factor receptor trans-activation mediates the tonic and fibrogenic effects of endothelin in the aortic wall of transgenic mice. FASEB J 2003, 17, 327–329. [Google Scholar]
- Hao, L.; Du, M.; Lopez-Campistrous, A.; Fernandez-Patron, C. Agonist-induced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ. Res 2004, 94, 68–76. [Google Scholar]
- Grossmann, C.; Freudinger, R.; Mildenberger, S.; Krug, A.W.; Gekle, M. Evidence for epidermal growth factor receptor as negative-feedback control in aldosterone-induced Na+ reabsorption. Am. J. Physiol. Renal Physiol 2004, 286, F1226–F1231. [Google Scholar]
- Larsen, A.K.; Ouaret, D.; El Ouadrani, K.; Petitprez, A. Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol. Ther 2011, 131, 80–90. [Google Scholar]
- Chalothorn, D.; Moore, S.M.; Zhang, H.; Sunnarborg, S.W.; Lee, D.C.; Faber, J.E. Heparin-binding epidermal growth factor-like growth factor, collateral vessel development, and angiogenesis in skeletal muscle ischemia. Arterioscler. Thromb. Vasc. Biol 2005, 25, 1884–1890. [Google Scholar]
- Amin, A.H.; Abd Elmageed, Z.Y.; Nair, D.; Partyka, M.I.; Kadowitz, P.J.; Belmadani, S.; Matrougui, K. Modified multipotent stromal cells with epidermal growth factor restore vasculogenesis and blood flow in ischemic hind-limb of type II diabetic mice. Lab. Invest 2010, 90, 985–996. [Google Scholar]
- Lai, W.H.; Ho, J.C.; Chan, Y.C.; Ng, J.H.; Au, K.W.; Wong, L.Y.; Siu, C.W.; Tse, H.F. Attenuation of hind-limb ischemia in mice with endothelial-like cells derived from different sources of human stem cells. PLoS One 2013, 8, e57876. [Google Scholar]
- Recchia, A.G.; Filice, E.; Pellegrino, D.; Dobrina, A.; Cerra, M.C.; Maggiolini, M. Endothelin-1 induces connective tissue growth factor expression in cardiomyocytes. J. Mol. Cell Cardiol 2009, 46, 352–359. [Google Scholar]
- Sugimoto, M.; Cutler, A.; Shen, B.; Moss, S.E.; Iyengar, S.K.; Klein, R.; Folkman, J.; Anand-Apte, B. Inhibition of EGF signaling protects the diabetic retina from insulin-induced vascular leakage. Am. J. Pathol 2013, 183, 987–995. [Google Scholar]
- Hewing, N.J.; Weskamp, G.; Vermaat, J.; Farage, E.; Glomski, K.; Swendeman, S.; Chan, R.V.; Chiang, M.F.; Khokha, R.; Anand-Apte, B.; Blobel, C.P. Intravitreal injection of TIMP3 or the EGFR inhibitor erlotinib offers protection from oxygen-induced retinopathy in mice. Invest. Ophthalmol. Vis. Sci 2013, 54, 864–870. [Google Scholar]
- Chan, A.K.; Kalmes, A.; Hawkins, S.; Daum, G.; Clowes, A.W. Blockade of the epidermal growth factor receptor decreases intimal hyperplasia in balloon-injured rat carotid artery. J. Vasc. Surg 2003, 37, 644–649. [Google Scholar]
- Shafi, S.; Lamb, D.; Modjtahedi, H.; Ferns, G. Periadventitial delivery of anti-EGF receptor antibody inhibits neointimal macrophage accumulation after angioplasty in a hypercholesterolaemic rabbit. Int. J. Exp. Pathol 2010, 91, 224–234. [Google Scholar]
- Lee, M.Y.; Martin, A.S.; Mehta, P.K.; Dikalova, A.E.; Garrido, A.M.; Datla, S.R.; Lyons, E.; Krause, K.-H.; Banfi, B.; Lambeth, J.D.; et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol 2009, 29, 480–487. [Google Scholar]
- Takahashi, M.; Hayashi, K.; Yoshida, K.; Ohkawa, Y.; Komurasaki, T.; Kitabatake, A.; Ogawa, A.; Nishida, W.; Yano, M.; Monden, M.; Sobue, K. Epiregulin as a major autocrine/paracrine factor released from ERK- and p38MAPK-activated vascular smooth muscle cells. Circulation 2003, 108, 2524–2529. [Google Scholar]
- Matsumoto, S.; Kishida, K.; Shimomura, I.; Maeda, N.; Nagaretani, H.; Matsuda, M.; Nishizawa, H.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; et al. Increased plasma HB-EGF associated with obesity and coronary artery disease. Biochem. Biophys. Res. Commun 2002, 292, 781–786. [Google Scholar]
- Sanchez-Vizcaino, E.; Vehi, C.; Camprecios, G.; Morcillo, C.; Soley, M.; Ramirez, I. Heparin-binding EGF-like growth factor in human serum. Association with high blood cholesterol and heart hypertrophy. Growth Factors 2009, 28, 98–103. [Google Scholar]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal 2006, 8, 1865–1879. [Google Scholar]
- Van der Vorst, E.P.; Keijbeck, A.A.; de Winther, M.P.; Donners, M.M. A disintegrin and metalloproteases: Molecular scissors in angiogenesis, inflammation and atherosclerosis. Atherosclerosis 2012, 224, 302–308. [Google Scholar]
- Liu, X.; Gu, X.; Li, Z.; Li, X.; Li, H.; Chang, J.; Chen, P.; Jin, J.; Xi, B.; Chen, D.; et al. Neuregulin-1/erbB-activation improves cardiac function and survival in models of ischemic, dilated, and viral cardiomyopathy. J. Am. Coll. Cardiol 2006, 48, 1438–1447. [Google Scholar]
- Crone, S.A.; Zhao, Y.Y.; Fan, L.; Gu, Y.; Minamisawa, S.; Liu, Y.; Peterson, K.L.; Chen, J.; Kahn, R.; Condorelli, G.; et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med 2002, 8, 459–465. [Google Scholar]
- Sysa-Shah, P.; Xu, Y.; Guo, X.; Belmonte, F.; Kang, B.; Bedja, D.; Pin, S.; Tsuchiya, N.; Gabrielson, K. Cardiac-specific over-expression of epidermal growth factor receptor 2 (ErbB2) induces pro-survival pathways and hypertrophic cardiomyopathy in mice. PLoS One 2012, 7, e42805. [Google Scholar]
- Ozcelik, C.; Erdmann, B.; Pilz, B.; Wettschureck, N.; Britsch, S.; Hubner, N.; Chien, K.R.; Birchmeier, C.; Garratt, A.N. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 8880–8885. [Google Scholar]
- Nair, B.G.; Rashed, H.M.; Patel, T.B. Epidermal growth factor produces inotropic and chronotropic effects in rat hearts by increasing cyclic AMP accumulation. Growth Factors 1993, 8, 41–48. [Google Scholar]
- Patel, T.B. Single transmembrane spanning heterotrimeric g protein-coupled receptors and their signaling cascades. Pharmacol. Rev 2004, 56, 371–385. [Google Scholar]
- Forster, K.; Kuno, A.; Solenkova, N.; Felix, S.B.; Krieg, T. The delta-opioid receptor agonist DADLE at reperfusion protects the heart through activation of pro-survival kinases via EGF receptor transactivation. Am. J. Physiol. Heart Circ. Physiol 2007, 293, H1604–1608. [Google Scholar]
- Fujio, Y.; Nguyen, T.; Wencker, D.; Kitsis, R.N.; Walsh, K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 2000, 101, 660–667. [Google Scholar]
- Matsui, T.; Nagoshi, T.; Rosenzweig, A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle 2003, 2, 220–223. [Google Scholar]
- Rajagopalan, V.; Zucker, I.H.; Jones, J.A.; Carlson, M.; Ma, Y.J. Cardiac ErbB-1/ErbB-2 mutant expression in young adult mice leads to cardiac dysfunction. Am. J. Physiol. Heart Circ. Physiol 2008, 295, H543–H554. [Google Scholar]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol 2013, 9, 459–469. [Google Scholar]
- Grossmann, C.; Krug, A.W.; Freudinger, R.; Mildenberger, S.; Voelker, K.; Gekle, M. Aldosterone-induced EGFR expression: Interaction between the human mineralocorticoid receptor and the human EGFR promoter. Am. J. Physiol. Endocrinol. Metab 2007, 292, E1790–E1800. [Google Scholar]
- Montezano, A.C.; Callera, G.E.; Yogi, A.; He, Y.; Tostes, R.C.; He, G.; Schiffrin, E.L.; Touyz, R.M. Aldosterone and angiotensin II synergistically stimulate migration in vascular smooth muscle cells through c-Src-regulated redox-sensitive RhoA pathways. Arterioscler. Thromb. Vasc. Biol 2008, 28, 1511–1518. [Google Scholar]
- Moriguchi, Y.; Matsubara, H.; Mori, Y.; Murasawa, S.; Masaki, H.; Maruyama, K.; Tsutsumi, Y.; Shibasaki, Y.; Tanaka, Y.; Nakajima, T.; et al. Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-beta synthesis via transcriptional and posttranscriptional mechanisms. Circ. Res 1999, 84, 1073–1084. [Google Scholar]
- Zhai, P.; Galeotti, J.; Liu, J.; Holle, E.; Yu, X.; Wagner, T.; Sadoshima, J. An angiotensin II type 1 receptor mutant lacking epidermal growth factor receptor transactivation does not induce angiotensin II-mediated cardiac hypertrophy. Circ. Res 2006, 99, 528–536. [Google Scholar]
- Kagiyama, S.; Qian, K.; Kagiyama, T.; Phillips, M.I. Antisense to epidermal growth factor receptor prevents the development of left ventricular hypertrophy. Hypertension 2003, 41, 824–829. [Google Scholar]
- Messaoudi, S.; Zhang, A.D.; Griol-Charhbili, V.; Escoubet, B.; Sadoshima, J.; Farman, N.; Jaisser, F. The epidermal growth factor receptor is involved in angiotensin II but not aldosterone/salt-induced cardiac remodelling. PLoS One 2012, 7, e30156. [Google Scholar]
- Methner, C.; Donat, U.; Felix, S.B.; Krieg, T. Cardioprotection of bradykinin at reperfusion involves transactivation of the epidermal growth factor receptor via matrix metalloproteinase-8. Acta Physiol. (Oxf) 2009, 197, 265–271. [Google Scholar]
- Belmadani, S.; Palen, D.I.; Gonzalez-Villalobos, R.A.; Boulares, H.A.; Matrougui, K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes 2008, 57, 1629–1637. [Google Scholar]
- Benter, I.F.; Yousif, M.H.; Griffiths, S.M.; Benboubetra, M.; Akhtar, S. Epidermal growth factor receptor tyrosine kinase-mediated signalling contributes to diabetes-induced vascular dysfunction in the mesenteric bed. Br. J. Pharmacol 2005, 145, 829–836. [Google Scholar]
- Yousif, M.H.; Benter, I.F.; Akhtar, S. The role of tyrosine kinase-mediated pathways in diabetes-induced alterations in responsiveness of rat carotid artery. Auton. Autacoid Pharmacol 2005, 25, 69–78. [Google Scholar]
- Matrougui, K. Diabetes and microvascular pathophysiology: Role of epidermal growth factor receptor tyrosine kinase. Diabetes Metab. Res. Rev 2010, 26, 13–16. [Google Scholar]
- Benter, I.F.; Benboubetra, M.; Hollins, A.J.; Yousif, M.H.; Canatan, H.; Akhtar, S. Early inhibition of EGFR signaling prevents diabetes-induced up-regulation of multiple gene pathways in the mesenteric vasculature. Vascul. Pharmacol 2009, 51, 236–245. [Google Scholar]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med 2000, 6, 443–446. [Google Scholar]
- Di Cosimo, S. Heart to heart with trastuzumab: A review on cardiac toxicity. Target. Oncol 2011, 6, 189–195. [Google Scholar]
- Orphanos, G.S.; Ioannidis, G.N.; Ardavanis, A.G. Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol 2009, 48, 964–970. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Makki, N.; Thiel, K.W.; Miller, F.J., Jr. The Epidermal Growth Factor Receptor and Its Ligands in Cardiovascular Disease. Int. J. Mol. Sci. 2013, 14, 20597-20613. https://doi.org/10.3390/ijms141020597
Makki N, Thiel KW, Miller FJ Jr. The Epidermal Growth Factor Receptor and Its Ligands in Cardiovascular Disease. International Journal of Molecular Sciences. 2013; 14(10):20597-20613. https://doi.org/10.3390/ijms141020597
Chicago/Turabian StyleMakki, Nader, Kristina W. Thiel, and Francis J. Miller, Jr. 2013. "The Epidermal Growth Factor Receptor and Its Ligands in Cardiovascular Disease" International Journal of Molecular Sciences 14, no. 10: 20597-20613. https://doi.org/10.3390/ijms141020597