Disease Animal Models of TDP-43 Proteinopathy and Their Pre-Clinical Applications

Abstract
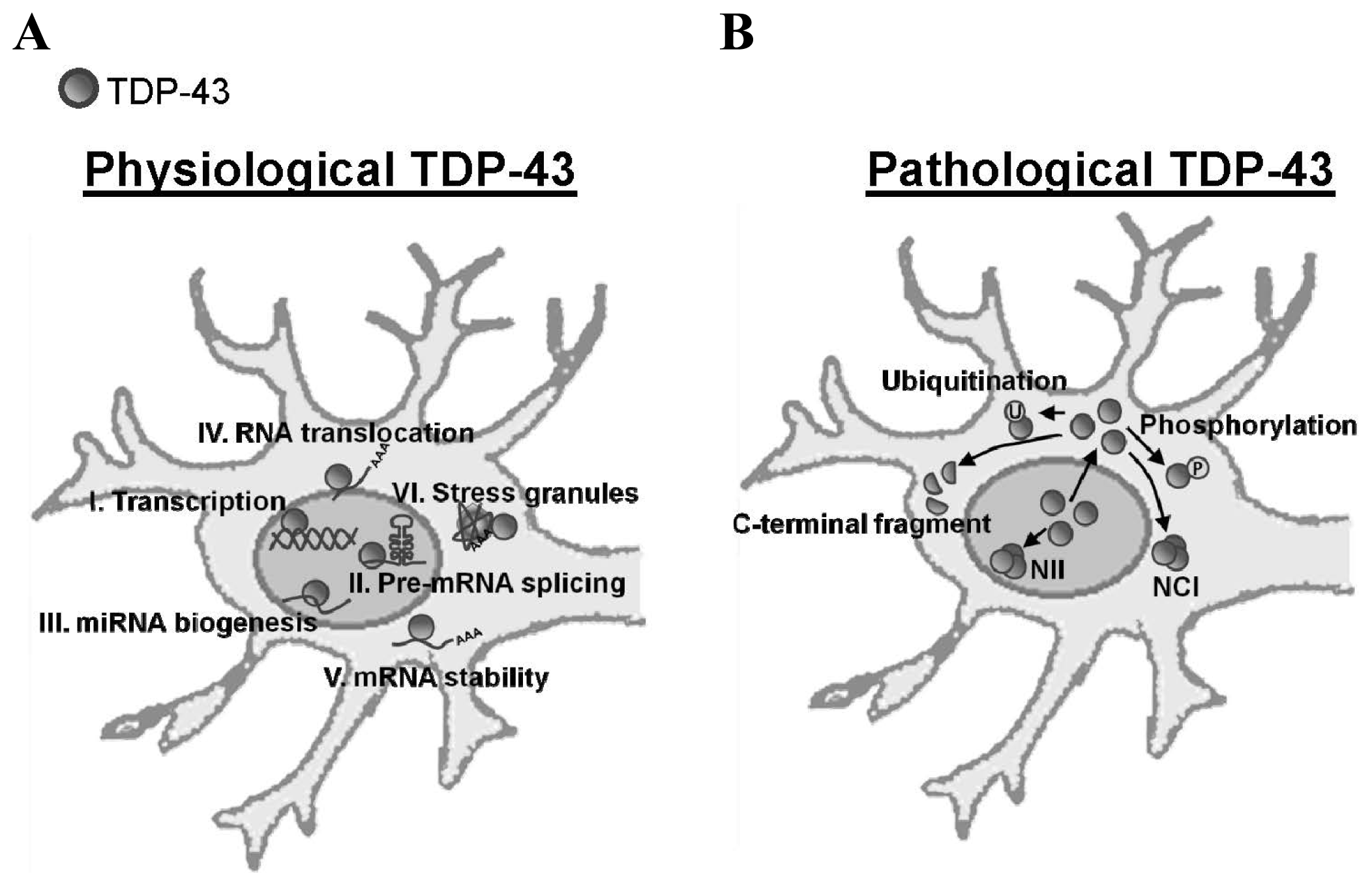
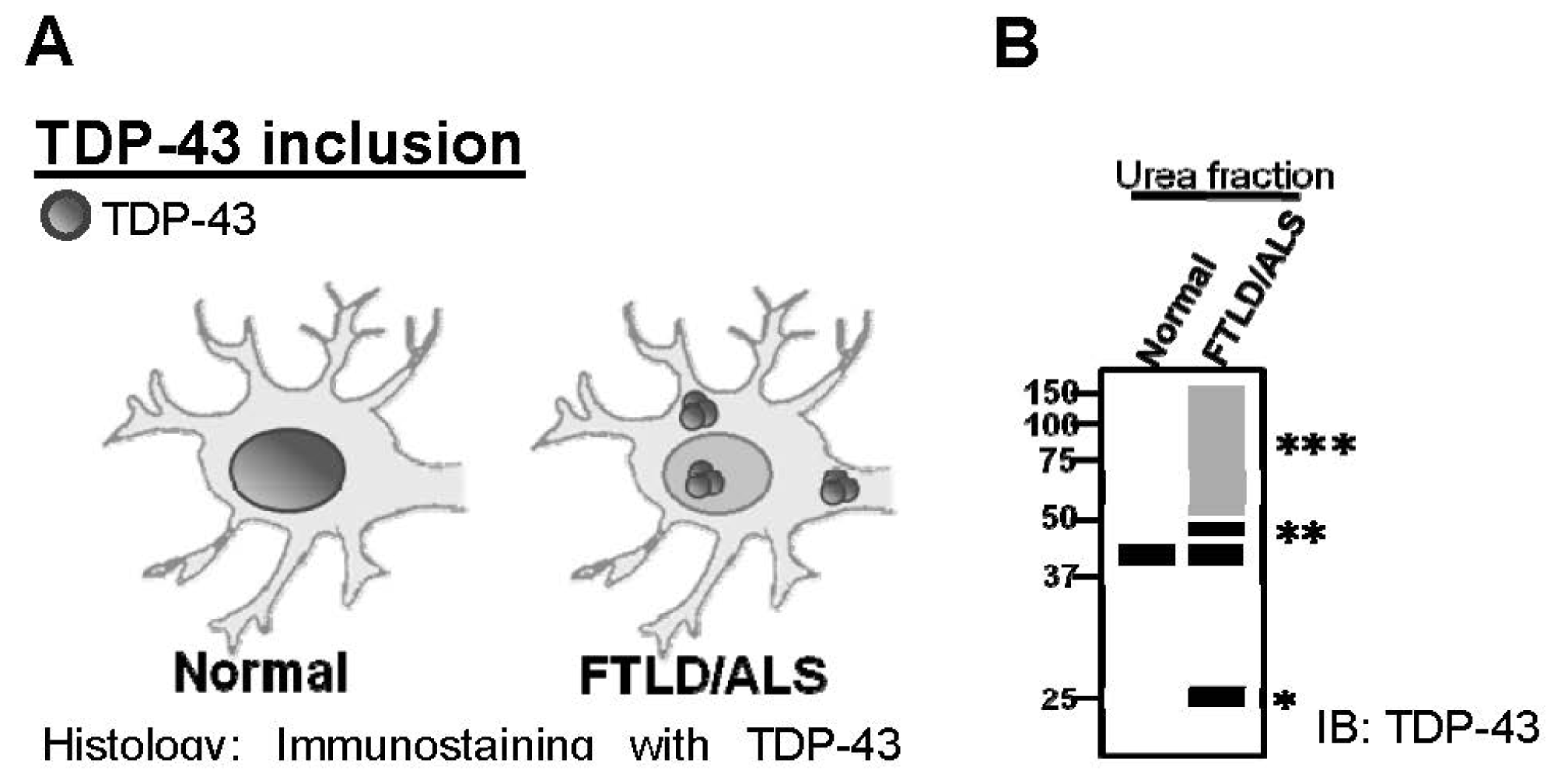
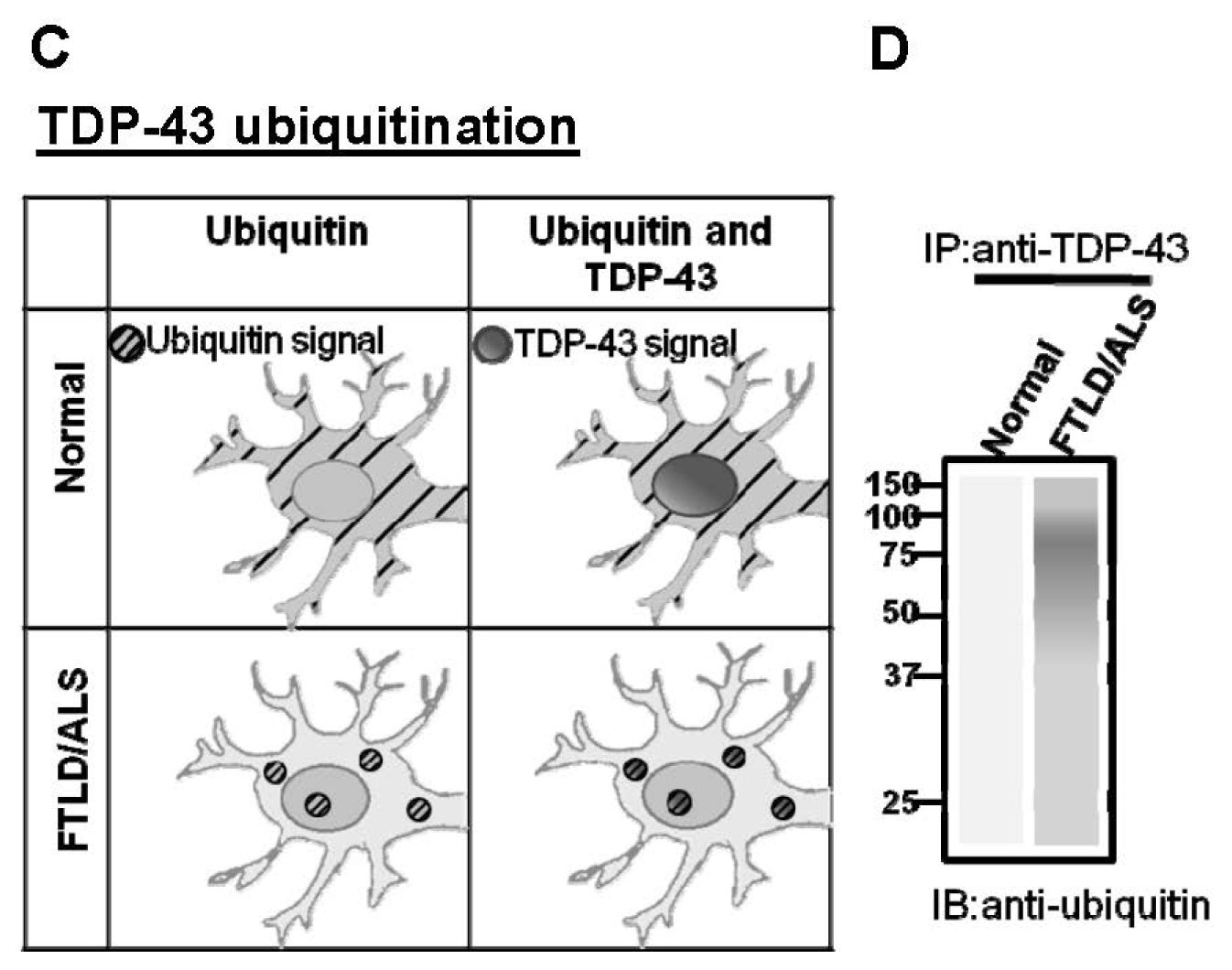
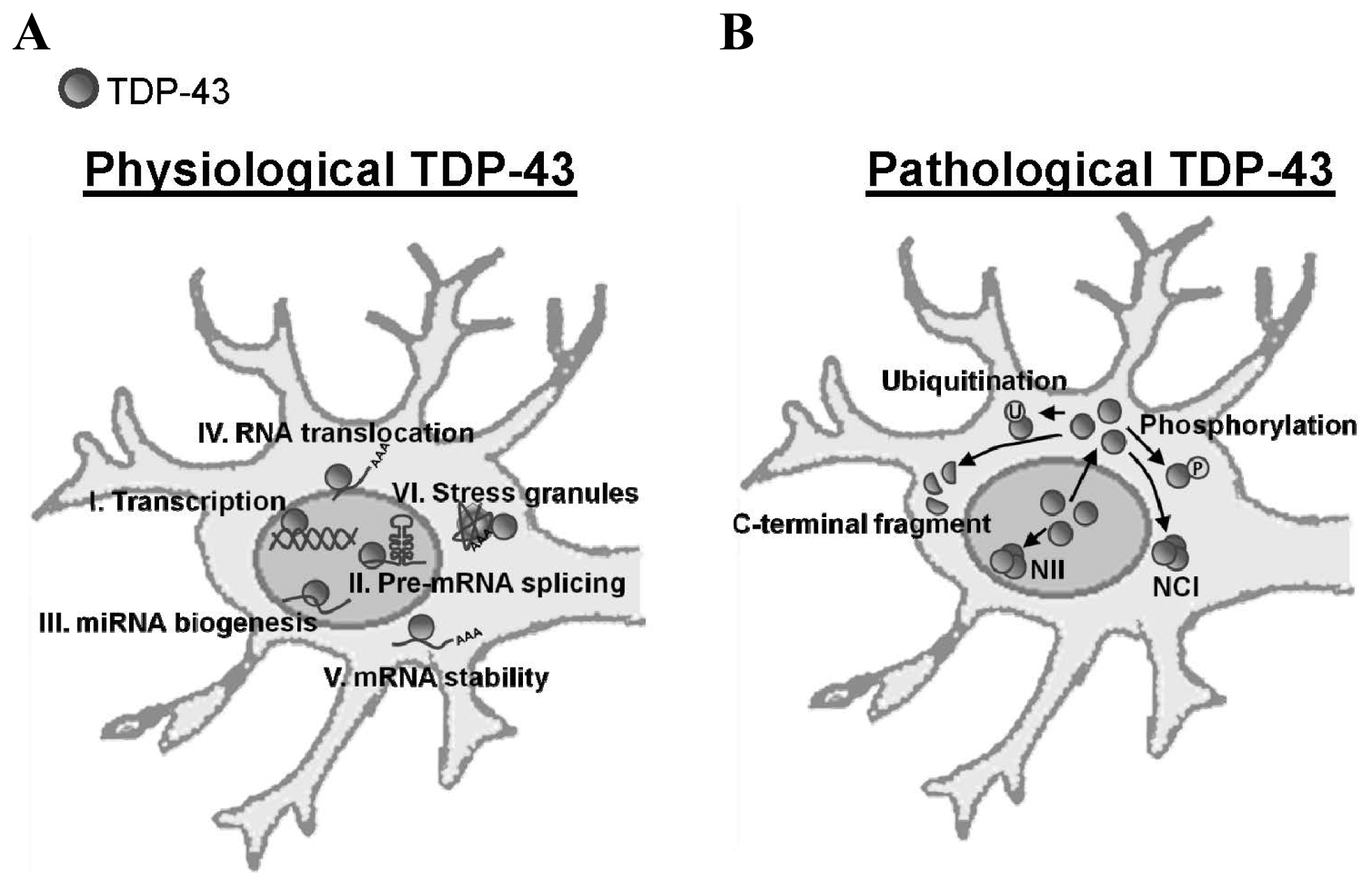
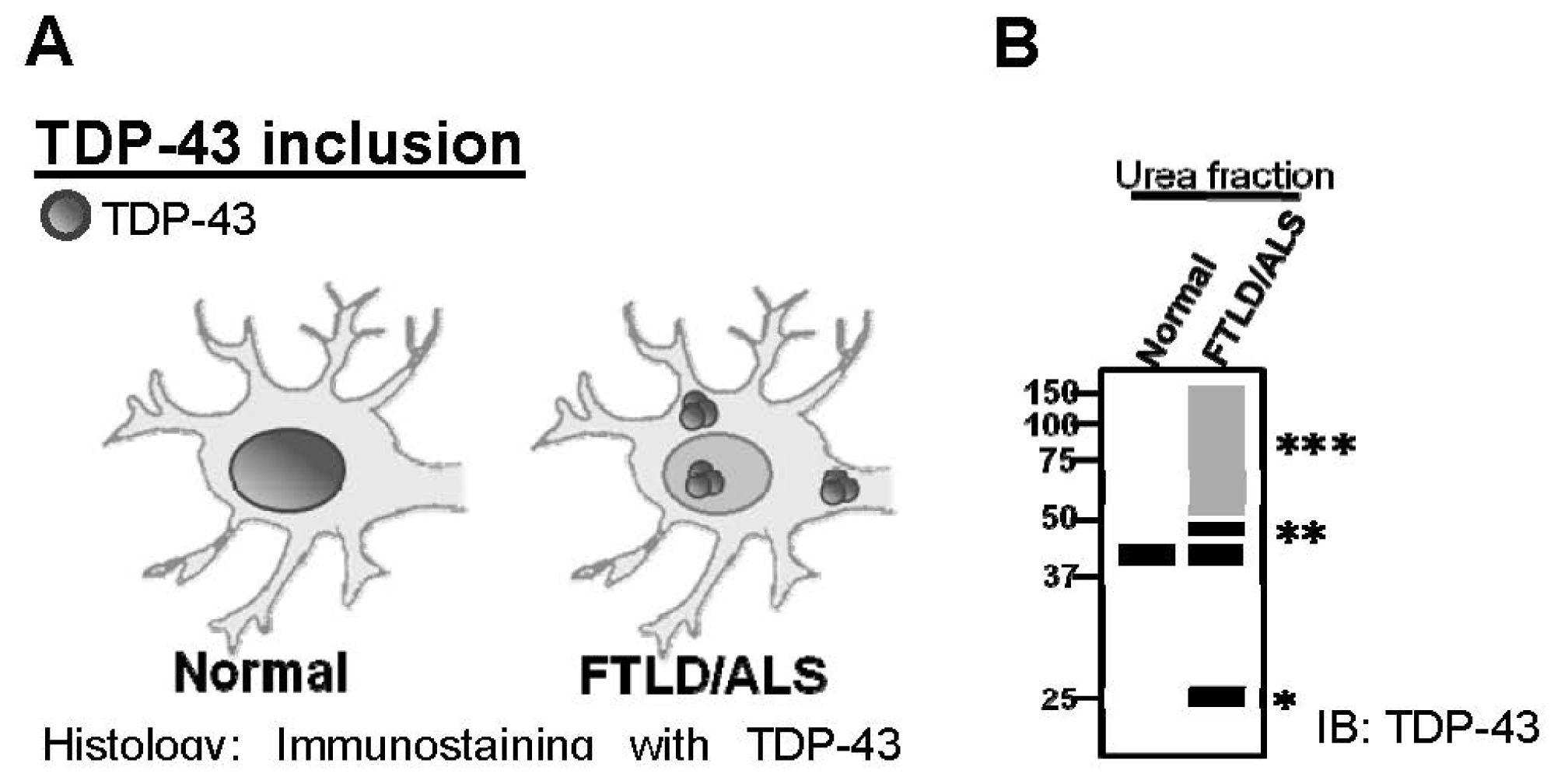
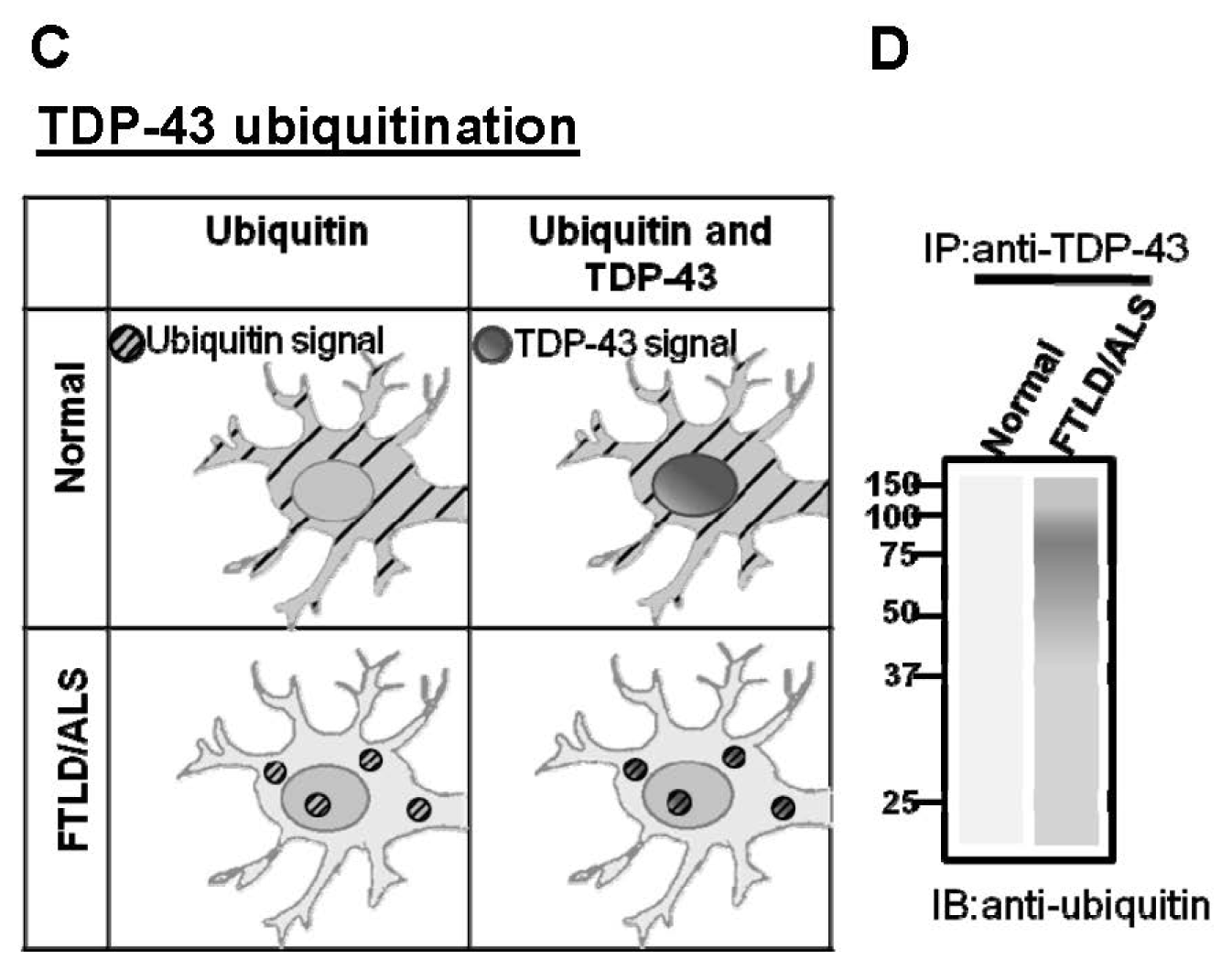
:1. Introduction
2. Disease Animal Models of TDP-43 Proteinopathy: Mammalian Models
2.1. Wild-Type or Mutant TDP-43 Overexpression by Transgenic Animals
2.1.1. Mouse Prion Promoter
2.1.2. Murine Thy-1.2 Promoter
2.1.3. CamKII Promoter
2.1.4. CamKII Promoter Combined with Tet-off System
2.1.5. Human Endogenous Promoter
2.1.6. Others
2.1.7. Brief Summary of Transgenic Mammals
2.2. Wild-Type or Mutant TDP-43 Overexpression by Virus Induced System
2.2.1. Non-Human Primate Model
2.2.2. Rat Models
2.2.3. Brief Summary of Viral-Modeling Mammalian Models
2.3. TDP-43 Knockout Animals
2.3.1. TDP-43 Knockout Mammalian Models
2.3.2. Brief Summary of Knockout Mammalian Models
3. Disease Animal Models of TDP-43 Proteinopathy: Non-Mammalian Models
3.1. Wild-Type or Mutant TDP-43 Expression by Transgenic Animals
3.1.1. Wild-Type or Mutant TDP-43 Expressed in C. elegans
3.1.2. Wild-Type or Mutant TDP-43 Expressed in Zebrafish
3.1.3. Wild-Type or Mutant TDP-43 Expressed in Sensory Neuron of Drosophila
3.1.4. Wild-Type or Mutant TDP-43 Expressed in Motor Neuron of Drosophila
3.1.5. Wild-Type or Mutant TDP-43 Expressed in Pan-Neuronal or Specific Neuron of Drosophila
3.1.6. Brief Summary of Non-Mammalian Transgenic Animal Models
3.2. TDP-43 Knockout Model
3.2.1. TDP-43 Knockout Non-Mammalian Models
3.2.2. Brief Summary of TDP-43 Knockout Non-Mammalian Models
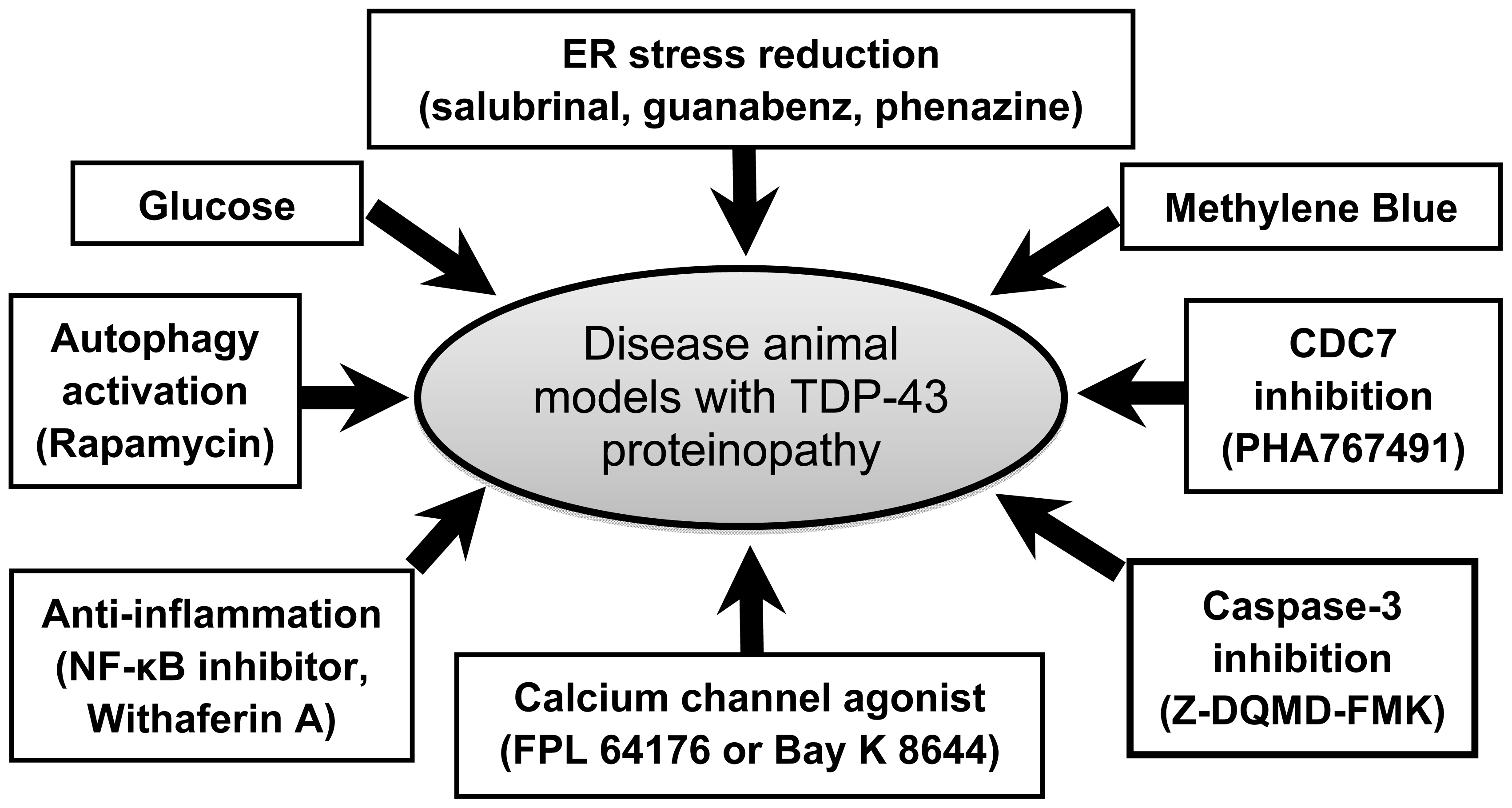
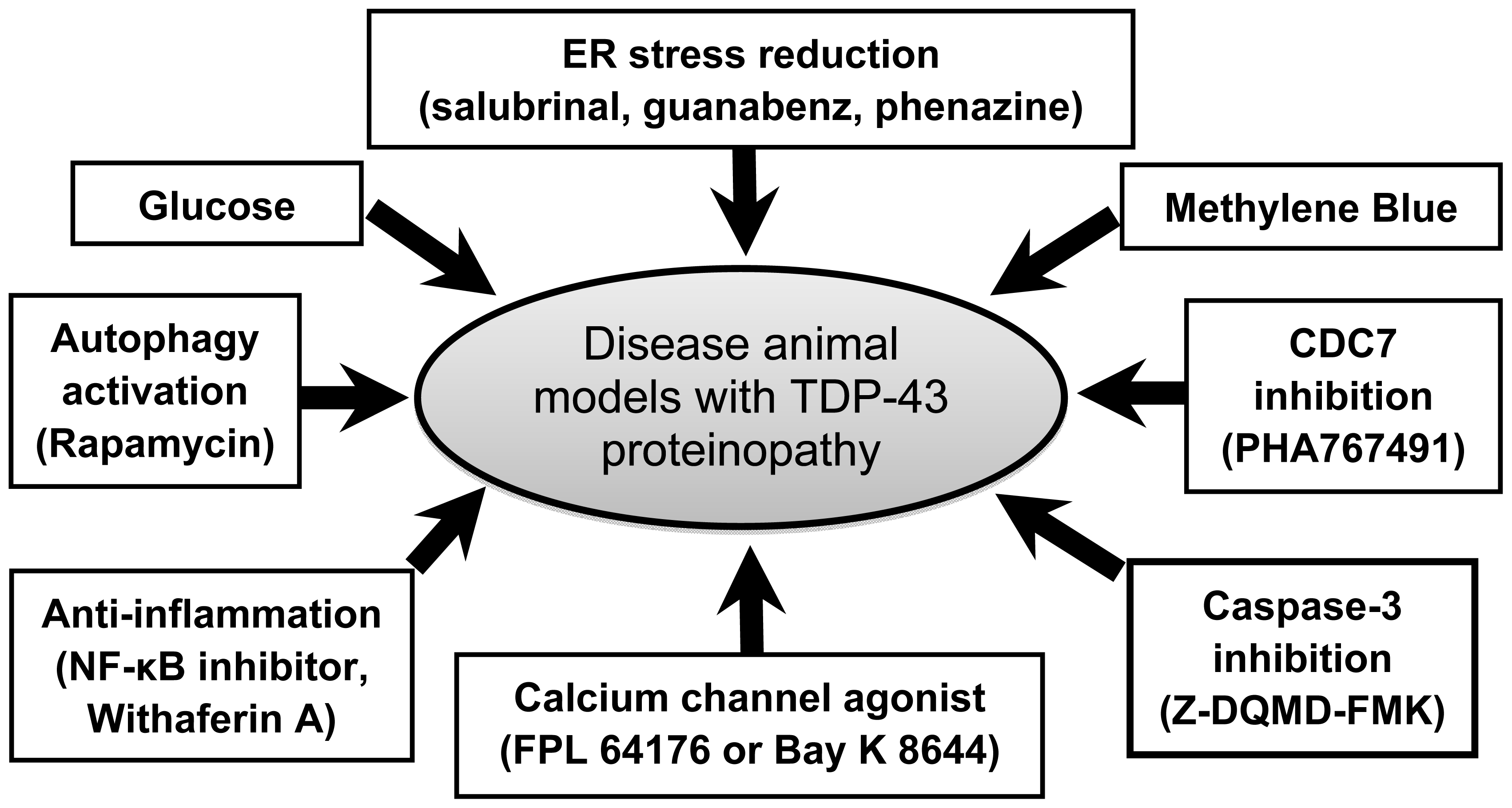
4. Recent Advances in Therapy
4.1. Glucose Enhancement
4.2. Methylene Blue Administration
4.3. Endoplasmic Reticulum Stress Reduction
4.4. CDC7 Inhibition
4.5. Anti-Inflammation
4.6. Autophagy Activation
4.7. Calcium Channel Activation
4.8. Caspase-3 Inhibition
5. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Line | Transgene | Promoter | TDP-43 proteinopathy | Phenotype | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TDP-43+ inclusion | ubiquitin+ and TDP-43+ inclusion | Cytoplasmic TDP-43 | Loss nuclear TDP-43 | Truncated TDP-43 (35 or 25 kDa) | Phosphorylated TDP-43 | Cognitive dysfunction | Motor dysfunction | |||||
| Mouse | Prp-TDP43A315T | Flag- A315T-hTDP-43 | mouse prion promoter | X | X | O | O | O | NA | NA | O | [15] |
| WT TDP-43 line 21 | WT-hTDP-43 | mouse prion promoter | X | X | O | NA | O | NA | NA | O | [16] | |
| A315 TDP-43 line 23 | A315T-hTDP-43 | mouse prion promoter | O | O (NCI, rare NII) | O | NA | O | O | NA | O | ||
| M337V TDP-43 line 39 | M337V-hTDP-43 | mouse prion promoter | NA | NA | O | NA | O | NA | NA | O | ||
| TDP-43prp | WT-hTDP-43 | mouse prion promoter | O | O (NCI and NII) | O | NA | O | O | NA | O | [17] | |
| TDP-43WT TAR4/4 | WT-hTDP-43 | murine Thy1.2 promoter | O | O (NCI and NII) | O | O | O | O | NA | O | [20] | |
| TDP-43 Tg W3 | WT-hTDP-43 | murine Thy1.2 promoter | O | O (NII) | NA | NA | NA | NA | NA | O | [21] | |
| CaMKII-TDP-43 Tg | WT-mTDP-43 | mouse CaMKII promoter | O | O (NCI) | O | O | O | NA | O | O | [24] | |
| hTDP-43-WT W12 | WT-hTDP-43 | mouse CamKII- tTA x tet off | O | O (rare NCI and NII) | O | X | X | O | NA | O | [25] | |
| hTDP-43-ΔNLS ΔNLS4 | ΔNLS-hTDP-43 | mouse CamKII- tTA x tet off | O | O (NCI) | O | O | X | O | NA | O | ||
| CAG-TDP-43 | WT-hTDP-43 | CAG | X | X | X | NA | X | NA | NA | X | [26] | |
| TDP-43 WT | WT-hTDP-43 | human endougenous promoter | X | X | X | X | X | NA | O | O | [27] | |
| TDP-43 A315T | A315T-hTDP-43 | human endougenous promoter | O | O (NCI) | O | O | O | NA | O | O | ||
| TDP-43 G348C | G348C-hTDP-43 | human endougenous promoter | O | O (NCI) | O | O | O | NA | O | O | ||
| hTDP-43M337V line 4 & 6 | M337V-hTDP-43 | mouse prion promoter | O (NCI) | X | O | NA | O | O | NA | O | [18] | |
| TgTDP-25 (B) and (F) | hTDP-25 | murine Thy1.2 promoter | X | X | O | NA | O | X | O | NA | [23] | |
| iTDP-43WT 5a | WT-hTDP-43 | mouse CamKII- tTA x tet off | O | O | O | NA | O | O | NA | NA | [28] | |
| TDP-43WT | myc-WT-hTDP-43 | mouse prion promoter | X | X | X | NA | NA | NA | NA | X | [19] | |
| TDP-43Q331K | myc-Q331K-hTDP-43 | mouse prion promoter | X | X | X | NA | NA | NA | NA | O | ||
| TDP-43M337V | myc-M337V-hTDP-43 | mouse prion promoter | X | X | X | NA | NA | NA | NA | O | ||
| p.M337V-hTDP-43 mt-TAR5/6 | M337V-hTDP-43 | Thy 1.2 | O | O | NA | O | O | O | NA | O | [22] | |
| Rat | miniTDP-43WT | WT-hTDP-43 | human endougenous promoter | X | X | O | NA | O (35 & 15 kDa) | O | NA | X | [29] |
| miniTDP43M337V | M337V-hTDP-43 | human endougenous promoter | NA | NA | O | NA | NA | NA | NA | O | ||
| TRE-TDP43M337V | M337V-hTDP-43 | CAG-tTA x tet off | O | NA | O | NA | O (35 & 15 kDa) | O | NA | O | ||
| NEF-tTA/TDP-43M337V | M337V-hTDP-43 | human NEF-tTA x tet off | X | X | NA | NA | NA | NA | NA | O | [30] | |
| ChAT–tTA-9/TDP-43M337V | M337V-hTDP-43 | mouse ChAT-tTA x tet off | O | O (NCI) | O | NA | NA | NA | NA | O | ||
| Species | Cynomolgus monkey | Rat | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Virus vector | AAV1 | AAV1 | AAV9 | AAV9 | AAV9 | Lenti virus | AAV9 | AAV9 | |
| Delivery gene | Flag- WT- hTDP-43 | Flag- WT- hTDP-43 | GFP-WT-hTDP43 | GFP-WT-hTDP43 | GFP-WT-hTDP43 | WT-hTDP-43 | hTDP-43-ΔNLS | TDP-25 | |
| Injection site | Spinal cord (C5–6) | Spinal cord (C6) | Substantia nigra (SN) | Intravenous (1-day-old pup) | Dorsal hippocampus | Motor cortex | Intravenous (1-day-old pup) | Intravenous (1-day-old pup) | |
| TDP-43 proteinopathy | TDP-43+ inclusion | O | X | X | NA | NA | O | X | X |
| ubiquitin+ and TDP-43+ inclusion | X | X | X | NA | NA | NA | X | X | |
| Cytoplasmic TDP-43 | O | X | O | NA | NA | O | O | O | |
| Truncated TDP-43 (35 or 25 kDa) | X | X | NA | NA | NA | O | NA | O | |
| Phosphorylated TDP-43 | O | X | NA | NA | NA | O | X | O | |
| Phenotype | Cognitive dysfunction | NA | NA | NA | NA | O | NA | NA | NA |
| Motor dysfunction | O | O | O | O | X | NA | O | O | |
| Ref. | [31] | [32] | [35] | [33] | [36] | [34] | |||
| Species | Line | Deletion | Deletion site | Embryonic lethality | TDP-43 proteinopathy | Histology hallmark | Phenotype | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Loss nuclear TDP-43 | Ubiquitin aggregate | Cognitive dysfunction | Motor dysfunction | ||||||
| Mouse | Tardbp-deficient | deleted exon 2 and 3 of Tardbp | ubiquitous | O | O | NA | NA | NA | [38] |
| Tardbp−/− | gene trap insertion of intron 2 and lead to in-frame fusion | ubiquitous | O | O | NA | NA | NA | [39] | |
| Tardbp−/− | Gene trap and insert β-geo after exon 2 of Tardbp | ubiquitous | O | O | NA | NA | NA | [40] | |
| Conditional Tardbp-KO | Er-Cre x TardbpF/F (floxed exon 3) | ubiquitous | X | O | NA | NA | NA | [41] | |
| HB9:Cre-Tardbplx/− | HB9-Cre x Tardbplx (floxed exon 2 and 3) | spinal cord motor neuron | X | O | O | NA | O | [42] | |
| TDP CKO | VAChT-Cre x TDP-43flox/flox (floxed exon 2) | motor neuron | X | O | NA | NA | O | [43] | |
| Species | Transgene | expression site (promoter) | TDP-43 proteinopathy | Phenotype | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| TDP-43+ inclusion | ubiquitin+ and TDP-43+ inclusion | Cytoplasmic TDP-43 | Loss nuclear TDP-43 | Truncated TDP-43 (35 or 25 kDa) | Phosphorylated TDP-43 | Motor dysfunction | ||||
| C. elegans | TDP-1 | ubiquitous (snb1) | NA | NA | X | X | NA | NA | O | [44] |
| WT-hTDP-43 | ubiquitous (snb1) | NA | X | X | X | NA | NA | O | ||
| WT-hTDP-43 | ubiquitous (snb1) | O | O | X | X | O | X | O | [45] | |
| A315T-hTDP-43 | ubiquitous (snb1) | O | O | X | X | O | O | O | ||
| G290A-hTDP-43 | ubiquitous (snb1) | O | NA | X | X | O | O | O | ||
| M337V-hTDP-43 | ubiquitous (snb1) | O | NA | X | X | O | O | O | ||
| WT-hTDP-43-YFP | ubiquitous (snb1) | O | NA | X | O | NA | NA | O | [46] | |
| Q331K-hTDP-43-YFP M337V-hTDP-43-YFP | ubiquitous (snb1) | NA | NA | NA | NA | NA | NA | O | ||
| C25-hTDP-43-YFP | ubiquitous (snb1) | O | NA | O | NA | NA | NA | O | ||
| WT-hTDP-43 | GABAergic neuron (unc-47) | X | NA | NA | NA | NA | NA | X | [47] | |
| A315T-hTDP-43 | GABAergic neuron (unc-47) | O | NA | NA | NA | NA | NA | O | ||
| Zebrafish | WT-hTDP-43 | ubiquitous (CMV) | NA | NA | NA | NA | NA | NA | X | [48] |
| A315T-hTDP-43 G384C-hTDP-43 A382T-hTDP-43 | ubiquitous (CMV) | NA | NA | NA | NA | NA | NA | O | ||
| Y220X | ubiquitous | NA | NA | NA | NA | NA | NA | NA | [49] | |
| Drosophila | dTDP-43 WT-hTDP-43 M337V-hTDP-43 Q331K-hTDP-43 209-414 a.a of hTDP-43 | sensory neuron | NA | NA | NA | NA | NA | NA | NA | [50] |
| WT-hTDP-43-RFP | eye (GMR) | O | X | O | NA | NA | NA | NA | [51] | |
| T202-hTDP-43-RFP | eye (GMR) | NA | NA | NA | NA | NA | NA | NA | ||
| WT-hTDP-43-RFP | mushroom body (OK107) | NA | NA | NA | NA | NA | NA | NA | ||
| WT-hTDP-43-RFP | motor neuron (OK371) | O | X | O | NA | NA | NA | O | ||
| WT-hTDP-43 | eye (GMR) | NA | NA | NA | NA | NA | NA | NA | [52] | |
| WT-hTDP-43 | motor neuron (D42) | O (Rare) | NA | O (Rare) | NA | NA | NA | NA | ||
| WT-hTDP-43 NES-mut-hTDP-43 NLS-mut-hTDP-43M337V-hTDP-43 | eye (GMR) | NA | NA | NA | NA | O | NA | NA | [53] | |
| WT-hTDP-43 | eye (GMR) | NA | NA | NA | NA | NA | NA | NA | [54] | |
| WT-hTDP-43 Q331K-hTDP-43 | motor neuron (D42) | NA | NA | NA | NA | NA | NA | O | ||
| WT-hTDP-43 A315T-hTDP-43 | pan neuronal (elav) or motor neuron (D42) | NA | NA | X | X | NA | NA | O (D42) | [55] | |
| G287S-hTDP-43 G348C-hTDP-43 A382T-hTDP-43 N390D-hTDP-43 | pan neuronal (elav) or motor neuron (D42) | NA | NA | X | X | NA | NA | NA | ||
| NLS-mut-hTDP-43 | pan neuronal (elav) or motor neuron (D42) | NA | NA | O | O | NA | NA | NA | ||
| CTF-hTDP-43 | NA | NA | O | O | NA | NA | O (D42) | |||
| FFLL-hTDP-43 | NA | NA | Oa | X | NA | NA | O (D42) | |||
| WT-hTDP-43 | eye (GMR) or pan neuronal (elav) | X | NA | O | X | O | O | NA | [56] | |
| NES-mut-hTDP-43 | O | NA | X | X | O | O | NA | |||
| NLS-mut-hTDP-43 | X | NA | O | O | O | O | NA | |||
| WT-hTDP-43 A315T-hTDP-43 | eye (GMR) or motor neuron (D42) | O (aggregate in axon) | NA | O | NA | NA | NA | O (D42) | [57] | |
| dTDP-43 | mushroom body (OK107) or motor neuron (D42) | O | NA | O | NA | NA | NA | O (D42) | [58] | |
| CTF-hTDP-43 | pan neuronal (elav) | O | NA | O | NA | NA | O | NA | [59] | |
| Mutant CTF of TDP-43 b | O | NA | O | NA | NA | X | NA | |||
| Mutant CTF of TDP-43C | X | NA | O | NA | NA | O | NA | |||
| WT-TBPH(dTDP-43) | motor neuron (D42) | NA | NA | O | NA | NA | NA | O | [60] | |
| UAS-dTDP-43-Flag | CCAP neuron (ccap) | O | O (NII) | O | X | NA | NA | NA | [61] | |
| WT-TBPH(dTDP-43) | pan-neuronal (elav) or upper motor neuron (EB1) or eye (GMR) | X (GMR) | X (GMR) | X (GMR) | X (GMR) | NA | NA | O (elav or EB1) | [62] | |
| Species | Line | Deletion | Deletion site | Embryonic lethality | TDP-43 proteinopathy | Phenotype | Ref. |
|---|---|---|---|---|---|---|---|
| Loss nuclear TDP-43 | Motor dysfunction | ||||||
| C. elegans | ok803 | deletion mutant which removes two RNA Recognize Motifs and the nucleare export signal of TDP-1 | ubiquitous | X | O | O | [63,64] |
| ok781 | deletion mutant which removes two RNA Recognize Motifs and the nucleare export signal of TDP-1 | ubiquitous | X | NA | O | [63] | |
| Zebrafish | TDP-43 AMO | an AMO sequence complimentary to tranlational start site of tardbp | ubiquitous | X | NA | O | [48] |
| TDP-43 AMO | tardbpl AMO | ubiquitous | X | NA | O | [49] | |
| tardbp−/− | genome editing with zinc finger nucleases, which target to tardbp | ubiquitous | X | NA | X | [65] | |
| tardbpl−/− | genome editing with zinc finger nucleases, which target to tardbpl | ubiquitous | X | NA | X | ||
| tardbpl−/− and tardbp−/− | genome editing with zinc finger nucleases, which target to tardbp and tardbpl | ubiquitous | X | NA | O | ||
| Drosophila | TBPH-KO | imprecise mobilization of TBPH transposone by transposase | ubiquitous | X | O | O | [66] |
| TBPH-null | use P-element mobilization to delete TBPH | ubiquitous | O | NA | NA | [67] | |
| dTDPex26 | use P-element mobilization to delete TBPH | ubiquitous | X | O | O | [58] | |
| TBPHDD100, TBPHDD96 | use imprecise P-element mobilization to delete TBPH | ubiquitous | X | NA | O | [62] |
Acknowledgments
Conflicts of Interest
References
- McMurtray, A.; Clark, D.G.; Christine, D.; Mendez, M.F. Early-onset dementia: Frequency and causes compared to late-onset dementia. Dement. Geriatr. Cogn. Disord 2006, 21, 59–64. [Google Scholar]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar]
- Sieben, A.; van Langenhove, T.; Engelborghs, S.; Martin, J.J.; Boon, P.; Cras, P.; de Deyn, P.P.; Santens, P.; van Broeckhoven, C.; Cruts, M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 2012, 124, 353–372. [Google Scholar] [Green Version]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol 2012, 124, 339–352. [Google Scholar]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar]
- Chen-Plotkin, A.S.; Lee, V.M.; Trojanowski, J.Q. TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol 2010, 6, 211–220. [Google Scholar]
- Yeung, C.W.; Woo, M.; Lee, K.; Greer, C.W. Characterization of the bacterial community structure of Sydney Tar Ponds sediment. Can. J. Microbiol 2011, 57, 493–503. [Google Scholar]
- Wang, H.Y.; Wang, I.F.; Bose, J.; Shen, C.K. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 2004, 83, 130–139. [Google Scholar]
- Wang, I.F.; Wu, L.S.; Chang, H.Y.; Shen, C.K. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem 2008, 105, 797–806. [Google Scholar]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci 2011, 14, 459–468. [Google Scholar]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS One 2010, 5, e13250. [Google Scholar]
- Bentmann, E.; Neumann, M.; Tahirovic, S.; Rodde, R.; Dormann, D.; Haass, C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem 2012, 287, 23079–23094. [Google Scholar]
- Cairns, N.J.; Neumann, M.; Bigio, E.H.; Holm, I.E.; Troost, D.; Hatanpaa, K.J.; Foong, C.; White, C.L., 3rd; Schneider, J.A.; Kretzschmar, H.A.; et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol 2007, 171, 227–240. [Google Scholar]
- Gitcho, M.A.; Bigio, E.H.; Mishra, M.; Johnson, N.; Weintraub, S.; Mesulam, M.; Rademakers, R.; Chakraverty, S.; Cruchaga, C.; Morris, J.C.; et al. TARDBP 3′-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol 2009, 118, 633–645. [Google Scholar]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar]
- Stallings, N.R.; Puttaparthi, K.; Luther, C.M.; Burns, D.K.; Elliott, J.L. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol. Dis 2010, 40, 404–414. [Google Scholar]
- Xu, Y.F.; Gendron, T.F.; Zhang, Y.J.; Lin, W.L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci 2010, 30, 10851–10859. [Google Scholar]
- Xu, Y.F.; Zhang, Y.J.; Lin, W.L.; Cao, X.; Stetler, C.; Dickson, D.W.; Lewis, J.; Petrucelli, L. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol. Neurodegener 2011, 6, 73. [Google Scholar]
- Arnold, E.S.; Ling, S.C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar]
- Wils, H.; Kleinberger, G.; Janssens, J.; Pereson, S.; Joris, G.; Cuijt, I.; Smits, V.; Ceuterick-de Groote, C.; van Broeckhoven, C.; Kumar-Singh, S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3858–3863. [Google Scholar]
- Shan, X.; Chiang, P.M.; Price, D.L.; Wong, P.C. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. USA 2010, 107, 16325–16330. [Google Scholar]
- Janssens, J.; Wils, H.; Kleinberger, G.; Joris, G.; Cuijt, I.; Ceuterick-de Groote, C.; van Broeckhoven, C.; Kumar-Singh, S. Overexpression of ALS-associated p.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol. Neurobiol 2013, 48, 22–35. [Google Scholar]
- Caccamo, A.; Majumder, S.; Oddo, S. Cognitive decline typical of frontotemporal lobar degeneration in transgenic mice expressing the 25-kDa C-terminal fragment of TDP-43. Am. J. Pathol 2012, 180, 293–302. [Google Scholar]
- Tsai, K.J.; Yang, C.H.; Fang, Y.H.; Cho, K.H.; Chien, W.L.; Wang, W.T.; Wu, T.W.; Lin, C.P.; Fu, W.M.; Shen, C.K. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J. Exp. Med 2010, 207, 1661–1673. [Google Scholar]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest 2011, 121, 726–738. [Google Scholar]
- Tian, T.; Huang, C.; Tong, J.; Yang, M.; Zhou, H.; Xia, X.G. TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Int. J. Biol. Sci 2011, 7, 234–243. [Google Scholar]
- Swarup, V.; Phaneuf, D.; Bareil, C.; Robertson, J.; Rouleau, G.A.; Kriz, J.; Julien, J.P. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 2011, 134, 2610–2626. [Google Scholar]
- Cannon, A.; Yang, B.; Knight, J.; Farnham, I.M.; Zhang, Y.; Wuertzer, C.A.; D’Alton, S.; Lin, W.L.; Castanedes-Casey, M.; Rousseau, L.; et al. Neuronal sensitivity to TDP-43 overexpression is dependent on timing of induction. Acta Neuropathol 2012, 123, 807–823. [Google Scholar]
- Zhou, H.; Huang, C.; Chen, H.; Wang, D.; Landel, C.P.; Xia, P.Y.; Bowser, R.; Liu, Y.J.; Xia, X.G. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet 2010, 6, e1000887. [Google Scholar]
- Huang, C.; Tong, J.; Bi, F.; Zhou, H.; Xia, X.G. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J. Clin. Invest 2012, 122, 107–118. [Google Scholar]
- Uchida, A.; Sasaguri, H.; Kimura, N.; Tajiri, M.; Ohkubo, T.; Ono, F.; Sakaue, F.; Kanai, K.; Hirai, T.; Sano, T.; et al. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain 2012, 135, 833–846. [Google Scholar]
- Tatom, J.B.; Wang, D.B.; Dayton, R.D.; Skalli, O.; Hutton, M.L.; Dickson, D.W.; Klein, R.L. Mimicking aspects of frontotemporal lobar degeneration and Lou Gehrig’s disease in rats via TDP-43 overexpression. Mol. Ther 2009, 17, 607–613. [Google Scholar]
- Dayton, R.D.; Wang, D.B.; Cain, C.D.; Schrott, L.M.; Ramirez, J.J.; King, M.A.; Klein, R.L. Frontotemporal lobar degeneration-related proteins induce only subtle memory-related deficits when bilaterally overexpressed in the dorsal hippocampus. Exp. Neurol 2012, 233, 807–814. [Google Scholar]
- Dayton, R.D.; Gitcho, M.A.; Orchard, E.A.; Wilson, J.D.; Wang, D.B.; Cain, C.D.; Johnson, J.A.; Zhang, Y.J.; Petrucelli, L.; Mathis, J.M.; et al. Selective forelimb impairment in rats expressing a pathological TDP-43 25 kDa C-terminal fragment to mimic amyotrophic lateral sclerosis. Mol. Ther 2013, 21, 1324–1334. [Google Scholar]
- Wang, D.B.; Dayton, R.D.; Henning, P.P.; Cain, C.D.; Zhao, L.R.; Schrott, L.M.; Orchard, E.A.; Knight, D.S.; Klein, R.L. Expansive gene transfer in the rat CNS rapidly produces amyotrophic lateral sclerosis relevant sequelae when TDP-43 is overexpressed. Mol. Ther 2010, 18, 2064–2074. [Google Scholar]
- Herman, A.M.; Khandelwal, P.J.; Rebeck, G.W.; Moussa, C.E. Wild type TDP-43 induces neuro-inflammation and alters APP metabolism in lentiviral gene transfer models. Exp. Neurol 2012, 235, 297–305. [Google Scholar]
- Pesiridis, G.S.; Tripathy, K.; Tanik, S.; Trojanowski, J.Q.; Lee, V.M. A “two-hit” hypothesis for inclusion formation by carboxyl-terminal fragments of TDP-43 protein linked to RNA depletion and impaired microtubule-dependent transport. J. Biol. Chem 2011, 286, 18845–18855. [Google Scholar]
- Wu, L.S.; Cheng, W.C.; Hou, S.C.; Yan, Y.T.; Jiang, S.T.; Shen, C.K. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis 2010, 48, 56–62. [Google Scholar]
- Sephton, C.F.; Good, S.K.; Atkin, S.; Dewey, C.M.; Mayer, P., 3rd; Herz, J.; Yu, G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem 2010, 285, 6826–6834. [Google Scholar]
- Kraemer, B.C.; Schuck, T.; Wheeler, J.M.; Robinson, L.C.; Trojanowski, J.Q.; Lee, V.M.; Schellenberg, G.D. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol 2010, 119, 409–419. [Google Scholar]
- Chiang, P.M.; Ling, J.; Jeong, Y.H.; Price, D.L.; Aja, S.M.; Wong, P.C. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 16320–16324. [Google Scholar]
- Wu, L.S.; Cheng, W.C.; Shen, C.K. Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J. Biol. Chem 2012, 287, 27335–27344. [Google Scholar]
- Iguchi, Y.; Katsuno, M.; Niwa, J.; Takagi, S.; Ishigaki, S.; Ikenaka, K.; Kawai, K.; Watanabe, H.; Yamanaka, K.; Takahashi, R.; et al. Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain 2013, 136, 1371–1382. [Google Scholar]
- Ash, P.E.; Zhang, Y.J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet 2010, 19, 3206–3218. [Google Scholar]
- Liachko, N.F.; Guthrie, C.R.; Kraemer, B.C. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci 2010, 30, 16208–16219. [Google Scholar]
- Zhang, T.; Mullane, P.C.; Periz, G.; Wang, J. TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum. Mol. Genet 2011, 20, 1952–1965. [Google Scholar]
- Vaccaro, A.; Tauffenberger, A.; Aggad, D.; Rouleau, G.; Drapeau, P.; Parker, J.A. Mutant TDP-43 and FUS cause age-dependent paralysis and neurodegeneration in C. elegans. PLoS One 2012, 7, e31321. [Google Scholar]
- Kabashi, E.; Lin, L.; Tradewell, M.L.; Dion, P.A.; Bercier, V.; Bourgouin, P.; Rochefort, D.; Bel Hadj, S.; Durham, H.D.; Vande Velde, C.; et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2010, 19, 671–683. [Google Scholar]
- Hewamadduma, C.A.; Grierson, A.J.; Ma, T.P.; Pan, L.; Moens, C.B.; Ingham, P.W.; Ramesh, T.; Shaw, P.J. Tardbpl splicing rescues motor neuron and axonal development in a mutant tardbp zebrafish. Hum. Mol. Genet 2013, 22, 2376–2386. [Google Scholar]
- Lu, Y.; Ferris, J.; Gao, F.B. Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol. Brain 2009, 2, 30. [Google Scholar]
- Li, Y.; Ray, P.; Rao, E.J.; Shi, C.; Guo, W.; Chen, X.; Woodruff, E.A., 3rd; Fushimi, K.; Wu, J.Y. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 3169–3174. [Google Scholar]
- Hanson, K.A.; Kim, S.H.; Wassarman, D.A.; Tibbetts, R.S. Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS). J. Biol. Chem 2010, 285, 11068–11072. [Google Scholar]
- Ritson, G.P.; Custer, S.K.; Freibaum, B.D.; Guinto, J.B.; Geffel, D.; Moore, J.; Tang, W.; Winton, M.J.; Neumann, M.; Trojanowski, J.Q.; et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci 2010, 30, 7729–7739. [Google Scholar]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar]
- Voigt, A.; Herholz, D.; Fiesel, F.C.; Kaur, K.; Muller, D.; Karsten, P.; Weber, S.S.; Kahle, P.J.; Marquardt, T.; Schulz, J.B. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS One 2010, 5, e12247. [Google Scholar]
- Miguel, L.; Frebourg, T.; Campion, D.; Lecourtois, M. Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol. Dis 2011, 41, 398–406. [Google Scholar]
- Estes, P.S.; Boehringer, A.; Zwick, R.; Tang, J.E.; Grigsby, B.; Zarnescu, D.C. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet 2011, 20, 2308–2321. [Google Scholar]
- Lin, M.J.; Cheng, C.W.; Shen, C.K. Neuronal function and dysfunction of Drosophila dTDP. PLoS One 2011, 6, e20371. [Google Scholar]
- Li, H.Y.; Yeh, P.A.; Chiu, H.C.; Tang, C.Y.; Tu, B.P. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS One 2011, 6, e23075. [Google Scholar]
- Hazelett, D.J.; Chang, J.C.; Lakeland, D.L.; Morton, D.B. Comparison of parallel high-throughput RNA sequencing between knockout of TDP-43 and its overexpression reveals primarily nonreciprocal and nonoverlapping gene expression changes in the central nervous system of Drosophila. G3 (Bethesda) 2012, 2, 789–802. [Google Scholar]
- Vanden Broeck, L.; Naval-Sanchez, M.; Adachi, Y.; Diaper, D.; Dourlen, P.; Chapuis, J.; Kleinberger, G.; Gistelinck, M.; van Broeckhoven, C.; Lambert, J.C.; et al. TDP-43 loss-of-function causes neuronal loss due to defective steroid receptor-mediated gene program switching in Drosophila. Cell Rep 2013, 3, 160–172. [Google Scholar]
- Diaper, D.C.; Adachi, Y.; Sutcliffe, B.; Humphrey, D.M.; Elliott, C.J.; Stepto, A.; Ludlow, Z.N.; Vanden Broeck, L.; Callaerts, P.; Dermaut, B.; et al. Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum. Mol. Genet 2013, 22, 1539–1557. [Google Scholar]
- Zhang, T.; Hwang, H.Y.; Hao, H.; Talbot, C., Jr.; Wang, J. Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. J. Biol. Chem 2012, 287, 8371–8382. [Google Scholar]
- Vaccaro, A.; Tauffenberger, A.; Ash, P.E.; Carlomagno, Y.; Petrucelli, L.; Parker, J.A. TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet 2012, 8, e1002806. [Google Scholar]
- Schmid, B.; Hruscha, A.; Hogl, S.; Banzhaf-Strathmann, J.; Strecker, K.; van der Zee, J.; Teucke, M.; Eimer, S.; Hegermann, J.; Kittelmann, M.; et al. Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc. Natl. Acad. Sci. USA 2013, 110, 4986–4991. [Google Scholar]
- Feiguin, F.; Godena, V.K.; Romano, G.; D’Ambrogio, A.; Klima, R.; Baralle, F.E. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett 2009, 583, 1586–1592. [Google Scholar]
- Fiesel, F.C.; Voigt, A.; Weber, S.S.; van den Haute, C.; Waldenmaier, A.; Gorner, K.; Walter, M.; Anderson, M.L.; Kern, J.V.; Rasse, T.M.; et al. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J 2010, 29, 209–221. [Google Scholar]
- Tauffenberger, A.; Vaccaro, A.; Aulas, A.; Vande Velde, C.; Parker, J.A. Glucose delays age-dependent proteotoxicity. Aging Cell 2012, 11, 856–866. [Google Scholar]
- Vaccaro, A.; Patten, S.A.; Ciura, S.; Maios, C.; Therrien, M.; Drapeau, P.; Kabashi, E.; Parker, J.A. Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PLoS One 2012, 7, e42117. [Google Scholar]
- Audet, J.N.; Soucy, G.; Julien, J.P. Methylene blue administration fails to confer neuroprotection in two amyotrophic lateral sclerosis mouse models. Neuroscience 2012, 209, 136–143. [Google Scholar]
- Vaccaro, A.; Patten, S.A.; Aggad, D.; Julien, C.; Maios, C.; Kabashi, E.; Drapeau, P.; Parker, J.A. Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 2013, 55, 64–75. [Google Scholar]
- Liachko, N.F.; McMillan, P.J.; Guthrie, C.R.; Bird, T.D.; Leverenz, J.B.; Kraemer, B.C. CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann. Neurol 2013, 74, 39–52. [Google Scholar]
- Swarup, V.; Phaneuf, D.; Dupre, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J. Exp. Med 2011, 208, 2429–2447. [Google Scholar]
- Wang, I.F.; Guo, B.S.; Liu, Y.C.; Wu, C.C.; Yang, C.H.; Tsai, K.J.; Shen, C.K. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar]
- Wang, I.F.; Tsai, K.J.; Shen, C.K. Autophagy activation ameliorates neuronal pathogenesis of FTLD-U mice: A new light for treatment of TARDBP/TDP-43 proteinopathies. Autophagy 2013, 9, 239–240. [Google Scholar]
- Armstrong, G.A.; Drapeau, P. Calcium channel agonists protect against neuromuscular dysfunction in a genetic model of TDP-43 mutation in ALS. J. Neurosci 2013, 33, 1741–1752. [Google Scholar]
- Kanazawa, M.; Kakita, A.; Igarashi, H.; Takahashi, T.; Kawamura, K.; Takahashi, H.; Nakada, T.; Nishizawa, M.; Shimohata, T. Biochemical and histopathological alterations in TAR DNA-binding protein-43 after acute ischemic stroke in rats. J. Neurochem 2011, 116, 957–965. [Google Scholar]
- Lavu, S.; Boss, O.; Elliott, P.J.; Lambert, P.D. Sirtuins—Novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov 2008, 7, 841–853. [Google Scholar]
- Brooks, M. MEthylene blue as antidote for cyanide and carbon monoxide poisoning. J. Am. Med. Assoc 1933, 100. [Google Scholar] [CrossRef]
- Schirmer, R.H.; Coulibaly, B.; Stich, A.; Scheiwein, M.; Merkle, H.; Eubel, J.; Becker, K.; Becher, H.; Muller, O.; Zich, T.; et al. Methylene blue as an antimalarial agent. Redox Rep 2003, 8, 272–275. [Google Scholar]
- Kwok, E.S.; Howes, D. Use of methylene blue in sepsis: A systematic review. J. Intensive Care Med 2006, 21, 359–363. [Google Scholar]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem 2011, 286, 16504–16515. [Google Scholar]
- Poteet, E.; Winters, A.; Yan, L.J.; Shufelt, K.; Green, K.N.; Simpkins, J.W.; Wen, Y.; Yang, S.H. Neuroprotective actions of methylene blue and its derivatives. PLoS One 2012, 7, e48279. [Google Scholar]
- Zhang, H.; Tan, C.F.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol 2008, 115, 115–122. [Google Scholar]
- Caccamo, A.; Majumder, S.; Deng, J.J.; Bai, Y.; Thornton, F.B.; Oddo, S. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J. Biol. Chem 2009, 284, 27416–27424. [Google Scholar]
- Davidson, Y.S.; Raby, S.; Foulds, P.G.; Robinson, A.; Thompson, J.C.; Sikkink, S.; Yusuf, I.; Amin, H.; DuPlessis, D.; Troakes, C.; et al. TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: Association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol 2011, 122, 703–713. [Google Scholar]
- Amador-Ortiz, C.; Lin, W.L.; Ahmed, Z.; Personett, D.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol 2007, 61, 435–445. [Google Scholar]
- Arai, T.; Mackenzie, I.R.; Hasegawa, M.; Nonoka, T.; Niizato, K.; Tsuchiya, K.; Iritani, S.; Onaya, M.; Akiyama, H. Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 2009, 117, 125–136. [Google Scholar]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 2007, 1184, 284–294. [Google Scholar]
- Rayaprolu, S.; Fujioka, S.; Traynor, S.; Soto-Ortolaza, A.I.; Petrucelli, L.; Dickson, D.W.; Rademakers, R.; Boylan, K.B.; Graff-Radford, N.R.; Uitti, R.J.; et al. TARDBP mutations in Parkinson’s disease. Parkinsonism Relat. Disord 2013, 19, 312–315. [Google Scholar]
- Zhang, Y.J.; Xu, Y.F.; Dickey, C.A.; Buratti, E.; Baralle, F.; Bailey, R.; Pickering-Brown, S.; Dickson, D.; Petrucelli, L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci 2007, 27, 10530–10534. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Y.-C.; Chiang, P.-M.; Tsai, K.-J. Disease Animal Models of TDP-43 Proteinopathy and Their Pre-Clinical Applications. Int. J. Mol. Sci. 2013, 14, 20079-20111. https://doi.org/10.3390/ijms141020079
Liu Y-C, Chiang P-M, Tsai K-J. Disease Animal Models of TDP-43 Proteinopathy and Their Pre-Clinical Applications. International Journal of Molecular Sciences. 2013; 14(10):20079-20111. https://doi.org/10.3390/ijms141020079
Chicago/Turabian StyleLiu, Yu-Chih, Po-Min Chiang, and Kuen-Jer Tsai. 2013. "Disease Animal Models of TDP-43 Proteinopathy and Their Pre-Clinical Applications" International Journal of Molecular Sciences 14, no. 10: 20079-20111. https://doi.org/10.3390/ijms141020079