Genetic Correction of Stem Cells in the Treatment of Inherited Diseases and Focus on Xeroderma Pigmentosum

Abstract
:1. Stem Cells
2. Stem Cells and the Microenvironment
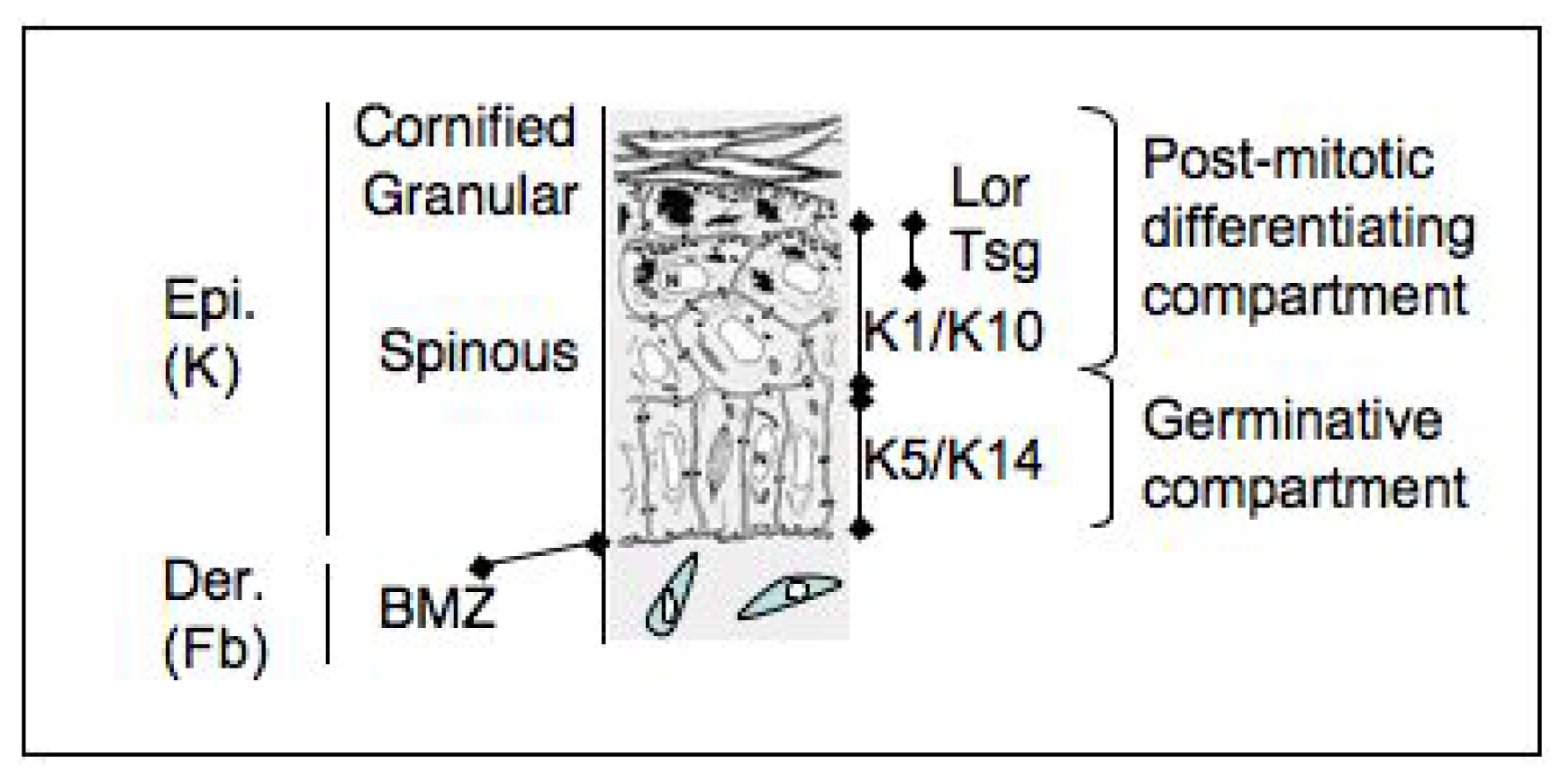
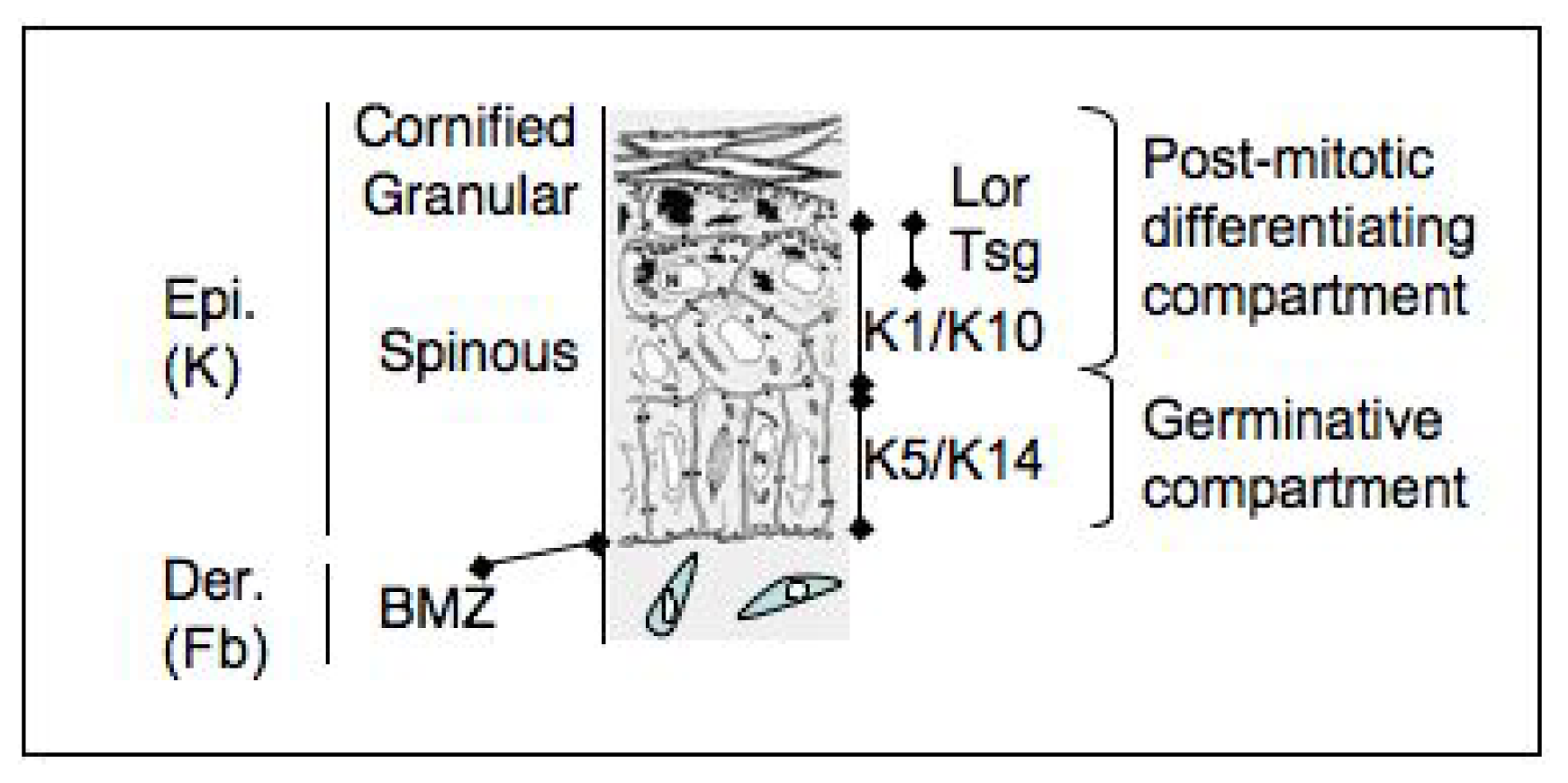
3. Epidermal Stem Cells
4. Stem Cells in the Organization of the Epidermis
5. Follicular and Interfollicular Stem Cells
6. How to Fish Them Out: Markers of Epidermal Stem Cells?
7. Stem Cells in Ex Vivo Gene Therapy
8. Candidate Diseases for Ex Vivo Genetic Correction
9. Specifications
9.1. Inheritance and Mutation Typology
9.2. Safety
9.3. Selection
10. Corrective Gene Transfer in Primary Epidermal Keratinocytes from Patients Suffering from Xeroderma Pigmentosum
10.1. Nucleotide Excision Repair of DNA and Related Diseases
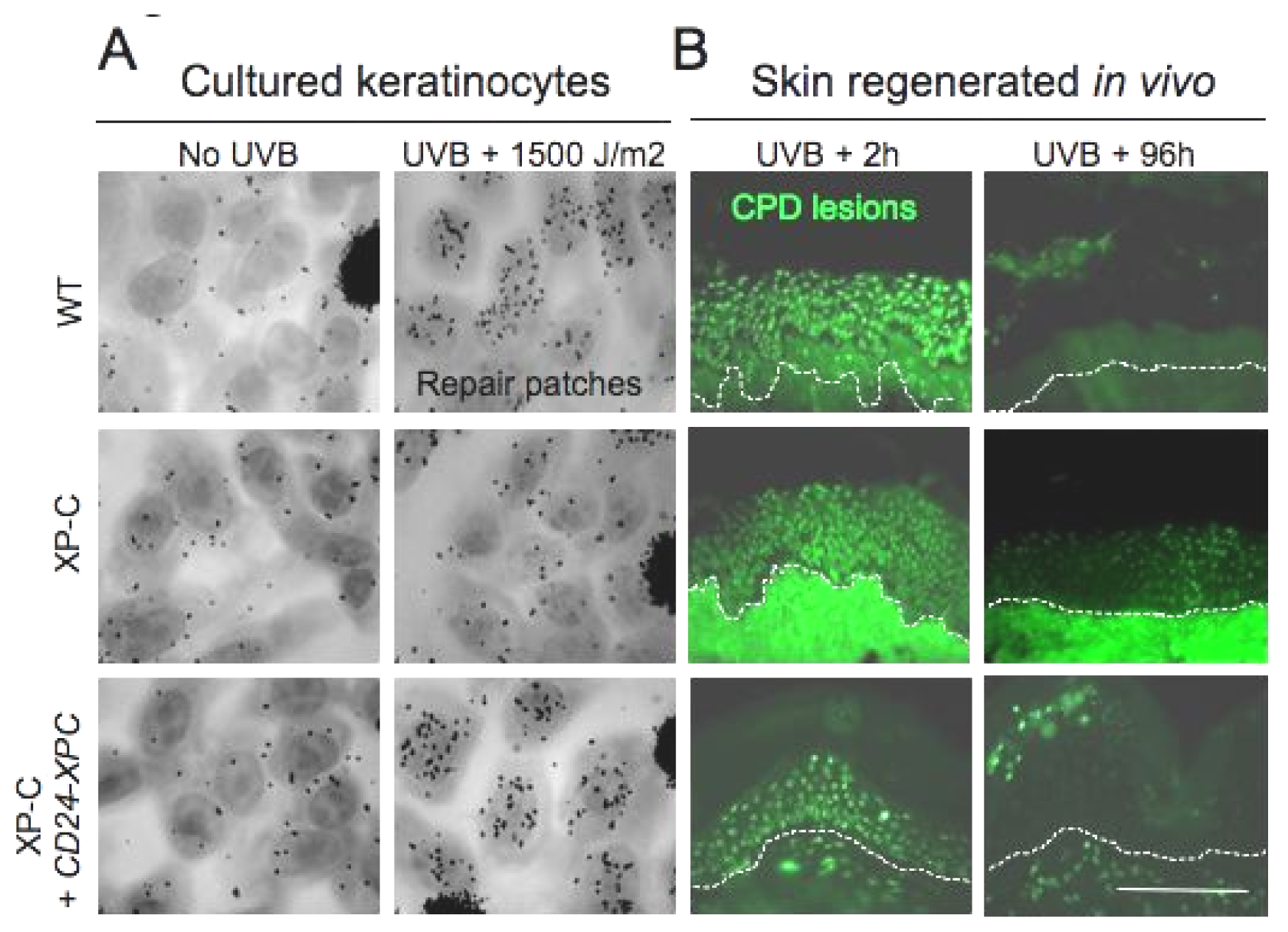
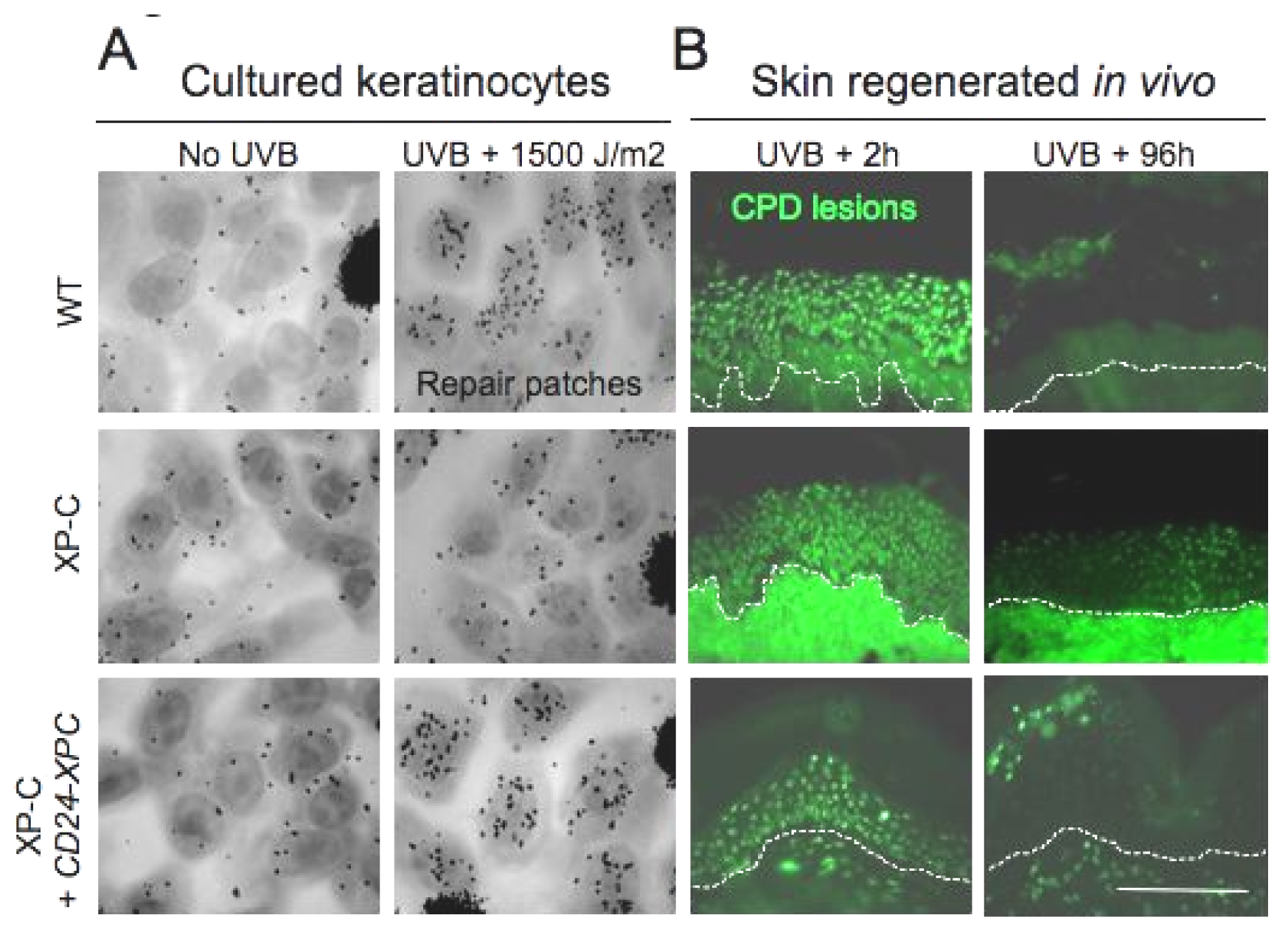
10.2. Genetic Correction of Xeroderma Pigmentosum
10.3. Mandatory Selection: Exploiting Differentiation/Proliferation Features of Human Epidermal Keratinocytes
11. Conclusions

{kind=link}
{kind=link}
| Marker | Function | Relative level in ESC | Reference |
|---|---|---|---|
| β1 integrin | Anchoring of basal keratinocytes | high (×2) | [31,37] |
| α6 integrin | Anchoring of basal keratinocytes | high | [32,33] |
| CD71 | Transferrin receptor | low | |
| Delta1 | Notch1 ligand | high (×2) | [38] |
| Desmogleine 3 | Desmosomal component | Low (/4) | [39] |
| EGF-R | EGF receptor | low | [34] |
| MCSP | Proteoglycan | high (×7) | [40] |
| Lrig 1 | EGF-R antagonist | high (×7) | [41,42] |
| Disease | Gene | Protein | Protein function | Site of expression | Symtoms |
|---|---|---|---|---|---|
| Netherton syndrome | SPINK 5 | LEKTI | Serine protease inhibitor | CL | Desquamation/barrier |
| Vulgaris ichtyosis | FLG | Filaggrin | CE/CL component | GL | Terminal differentiation/Barrier |
| Lamellar ichtyosis | TGM1 | Transglutaminase | Cross linking enzyme | GL | Terminal differentiation/Barrier |
| X-linked ichtyosis | STS | Aryl-C-steroid/sulfatase | Steroid sulfatase | GL | Terminal differentiation/Barrier |
| Epidermolysis Hyperkeratotis | KRT1/KRT10 | Keratins K1/K10 | IF | SPL | Mechano bullous disease |
| Epidermolysis Bullosa Simplex | KRT5/KRT14 | Keratins K5/K14 | IF | BL | Mechano bullous disease |
| Jonctional Epidermolysis Bullosa | ITGA6, ITGB4 | Integrins α6, Integrin β4 | Membrane receptors of HD | BMZ | Mechano bullous disease |
| Col17A1 | Collagen 17/BP180 | Anchoring filaments of HD | BMZ | Mechano bullous disease | |
| LAMA3, LAMB3, LAMC2 | Laminin 332 | BM components/Anchoring filaments of HD | BMZ | Mechano bullous disease | |
| Dystrophic Epidermolysis Bullosae | Col7A1 | Collagen 7 α1chain | Anchoring fibrils of HD | BMZ | Mechano bullous disease |
| Xeroderma pigmentosum | XPA to XPG | XPA to XPG | Repair of UV-induced DNA lesions | Ubiquitous | Photosensitivity/Skin cancer |
| Xeroderma pigmentosum variant | POLH | DNA polymerase η | Replicative translesion synthesis | Ubiquitous | Photosensitivity/Skin cancer |
Acknowledgments
Conflicts of Interest
References
- Lajtha, L.G. Stem cell concepts. Differentiation 1979, 14, 23–34. [Google Scholar]
- Kaur, J.; Tilkins, M.L. Methods for culturing human embryonic stem cells on feeders. Methods Mol. Biol 2013, 997, 93–113. [Google Scholar]
- Ogawa, M.; LaRue, A.C.; Mehrotra, M. Hematopoietic stem cells are pluripotent and not just “Hematopoietic”. Blood Cells Mol. Dis 2013, 51, 3–8. [Google Scholar]
- Oshima, H.; Rochat, A.; Kedzia, C.; Kobayashi, K.; Barrandon, Y. Morphogenesis and renewal of hair follicles from adult multipotent stem cells. Cell 2001, 104, 233–245. [Google Scholar]
- Rochat, A.; Kobayashi, K.; Barrandon, Y. Location of stem cells of human hair follicles by clonal analysis. Cell 1994, 76, 1063–1073. [Google Scholar]
- Spemann, H.; Mangold, H. Induction of embryonic primordia by implantation of organizers from a different species 1923. Int. J. Dev. Biol 2001, 45, 13–38. [Google Scholar]
- Reynolds, A.J.; Jahoda, C.A. Cultured dermal papilla cells induce follicle formation and hair growth by transdifferentiation of an adult epidermis. Development 1992, 115, 587–593. [Google Scholar]
- Pearton, D.J.; Yang, Y.; Dhouailly, D. Transdifferentiation of corneal epithelium into epidermis occurs by means of a multistep process triggered by dermal developmental signals. Proc. Natl. Acad. Sci. USA 2005, 102, 3714–3719. [Google Scholar]
- Bonfanti, P.; Claudinot, S.; Amici, A.W.; Farley, A.; Blackburn, C.C.; Barrandon, Y. Microenvironmental reprogramming of thymic epithelial cells to skin multipotent stem cells. Nature 2010, 466, 978–982. [Google Scholar]
- Mavilio, F.; Pellegrini, G.; Ferrari, S.; di Nunzio, F.; di Iorio, E.; Recchia, A.; Maruggi, G.; Ferrari, G.; Provasi, E.; Bonini, C.; et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med 2006, 12, 1397–1402. [Google Scholar]
- Rheinwald, J.G.; Green, H. Serial cultivation of strains of human epidermal keratinocytes: The formation of keratinizing colonies from single cells. Cell 1975, 6, 331–343. [Google Scholar]
- Rheinwald, J.G.; Green, H. Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature 1977, 265, 421–424. [Google Scholar]
- Barrandon, Y.; Green, H. Three clonal types of keratinocyte with different capacities for multiplication. Proc. Natl. Acad. Sci. USA 1987, 84, 2302–2306. [Google Scholar]
- Gallico, G.G., III; O’Connor, N.E.; Compton, C.C.; Kehinde, O.; Green, H. Permanent coverage of large burn wounds with autologous cultured human epithelium. N. Engl. J. Med 1984, 311, 448–451. [Google Scholar]
- Allen, T.D.; Potten, C.S. Fine-structural identification and organization of the epidermal proliferative unit. J. Cell Sci 1974, 15, 291–319. [Google Scholar]
- Potten, C.S. The epidermal proliferative unit: The possible role of the central basal cell. Cell Tissue Kinet 1974, 7, 77–88. [Google Scholar]
- Potten, C.S.; Allen, T.D. The fine structure and cell kinetics of mouse epidermis after wounding. J. Cell Sci 1975, 17, 413–447. [Google Scholar]
- Ghazizadeh, S.; Taichman, L.B. Organization of stem cells and their progeny in human epidermis. J. Invest. Dermatol 2005, 124, 367–372. [Google Scholar]
- Kolodka, T.M.; Garlick, J.A.; Taichman, L.B. Evidence for keratinocyte stem cells in vitro: Long term engraftment and persistence of transgene expression from retrovirus-transduced keratinocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 4356–4361. [Google Scholar]
- Clayton, E.; Doupe, D.P.; Klein, A.M.; Winton, D.J.; Simons, B.D.; Jones, P.H. A single type of progenitor cell maintains normal epidermis. Nature 2007, 446, 185–189. [Google Scholar]
- Cotsarelis, G.; Sun, T.T.; Lavker, R.M. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 1990, 61, 1329–1337. [Google Scholar]
- Claudinot, S.; Nicolas, M.; Oshima, H.; Rochat, A.; Barrandon, Y. Long-term renewal of hair follicles from clonogenic multipotent stem cells. Proc. Natl. Acad. Sci. USA 2005, 102, 14677–14682. [Google Scholar]
- Morris, R.J.; Liu, Y.; Marles, L.; Yang, Z.; Trempus, C.; Li, S.; Lin, J.S.; Sawicki, J.A.; Cotsarelis, G. Capturing and profiling adult hair follicle stem cells. Nat. Biotechnol 2004, 22, 411–417. [Google Scholar]
- Taylor, G.; Lehrer, M.S.; Jensen, P.J.; Sun, T.T.; Lavker, R.M. Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell 2000, 102, 451–461. [Google Scholar]
- Liu, Y.; Lyle, S.; Yang, Z.; Cotsarelis, G. Keratin 15 promoter targets putative epithelial stem cells in the hair follicle bulge. J. Invest. Dermatol 2003, 121, 963–968. [Google Scholar]
- Trempus, C.S.; Morris, R.J.; Bortner, C.D.; Cotsarelis, G.; Faircloth, R.S.; Reece, J.M.; Tennant, R.W. Enrichment for living murine keratinocytes from the hair follicle bulge with the cell surface marker CD34. J. Invest. Dermatol 2003, 120, 501–511. [Google Scholar]
- Ohyama, M. Hair follicle bulge: A fascinating reservoir of epithelial stem cells. J. Dermatol. Sci 2007, 46, 81–89. [Google Scholar]
- Ohyama, M.; Terunuma, A.; Tock, C.L.; Radonovich, M.F.; Pise-Masison, C.A.; Hopping, S.B.; Brady, J.N.; Udey, M.C.; Vogel, J.C. Characterization and isolation of stem cell-enriched human hair follicle bulge cells. J. Clin. Invest 2006, 116, 249–260. [Google Scholar]
- Lavker, R.M.; Sun, T.T. Heterogeneity in epidermal basal keratinocytes: Morphological and functional correlations. Science 1982, 215, 1239–1241. [Google Scholar]
- Adams, J.C.; Watt, F.M. Changes in keratinocyte adhesion during terminal differentiation: Reduction in fibronectin binding precedes alpha 5 beta 1 integrin loss from the cell surface. Cell 1990, 63, 425–435. [Google Scholar]
- Jones, P.H.; Watt, F.M. Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell 1993, 73, 713–724. [Google Scholar]
- Kaur, P.; Li, A. Adhesive properties of human basal epidermal cells: An analysis of keratinocyte stem cells, transit amplifying cells, and postmitotic differentiating cells. J. Invest. Dermatol 2000, 114, 413–420. [Google Scholar]
- Li, A.; Simmons, P.J.; Kaur, P. Identification and isolation of candidate human keratinocyte stem cells based on cell surface phenotype. Proc. Natl. Acad. Sci. USA 1998, 95, 3902–3907. [Google Scholar]
- Fortunel, N.O.; Hatzfeld, J.A.; Rosemary, P.A.; Ferraris, C.; Monier, M.N.; Haydont, V.; Longuet, J.; Brethon, B.; Lim, B.; Castiel, I.; et al. Long-term expansion of human functional epidermal precursor cells: Promotion of extensive amplification by low tgf-beta1 concentrations. J. Cell. Sci 2003, 116, 4043–4052. [Google Scholar]
- Tani, H.; Morris, R.J.; Kaur, P. Enrichment for murine keratinocyte stem cells based on cell surface phenotype. Proc. Natl. Acad. Sci. USA 2000, 97, 10960–10965. [Google Scholar]
- Ruetze, M.; Gallinat, S.; Wenck, H.; Deppert, W.; Knott, A. In situ localization of epidermal stem cells using a novel multi epitope ligand cartography approach. Integr. Biol 1039, 2, 241–249. [Google Scholar]
- Jones, P.H.; Harper, S.; Watt, F.M. Stem cell patterning and fate in human epidermis. Cell 1995, 80, 83–93. [Google Scholar]
- Lowell, S.; Jones, P.; le Roux, I.; Dunne, J.; Watt, F.M. Stimulation of human epidermal differentiation by delta-notch signalling at the boundaries of stem-cell clusters. Curr. Biol 2000, 10, 491–500. [Google Scholar]
- Wan, H.; Stone, M.G.; Simpson, C.; Reynolds, L.E.; Marshall, J.F.; Hart, I.R.; Hodivala-Dilke, K.M.; Eady, R.A. Desmosomal proteins, including desmoglein 3, serve as novel negative markers for epidermal stem cell-containing population of keratinocytes. J. Cell Sci 2003, 116, 4239–4248. [Google Scholar]
- Legg, J.; Jensen, U.B.; Broad, S.; Leigh, I.; Watt, F.M. Role of melanoma chondroitin sulphate proteoglycan in patterning stem cells in human interfollicular epidermis. Development 2003, 130, 6049–6063. [Google Scholar]
- Jensen, K.B.; Collins, C.A.; Nascimento, E.; Tan, D.W.; Frye, M.; Itami, S.; Watt, F.M. Lrig1 expression defines a distinct multipotent stem cell population in mammalian epidermis. Cell Stem Cell 2009, 4, 427–439. [Google Scholar]
- Jensen, K.B.; Watt, F.M. Single-cell expression profiling of human epidermal stem and transit-amplifying cells: Lrig1 is a regulator of stem cell quiescence. Proc. Natl. Acad. Sci. USA 2006, 103, 11958–11963. [Google Scholar]
- Barinaga, M. Fetal neuron grafts pave the way for stem cell therapies. Science 2000, 287, 1421–1422. [Google Scholar]
- Dyson, S.C.; Barker, R.A. Cell-based therapies for Parkinson’s disease. Expert Rev. Neurother 2011, 11, 831–844. [Google Scholar]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar]
- Takagi, Y.; Takahashi, J.; Saiki, H.; Morizane, A.; Hayashi, T.; Kishi, Y.; Fukuda, H.; Okamoto, Y.; Koyanagi, M.; Ideguchi, M.; et al. Dopaminergic neurons generated from monkey embryonic stem cells function in a parkinson primate model. J. Clin. Invest 2005, 115, 102–109. [Google Scholar]
- Solter, D. From teratocarcinomas to embryonic stem cells and beyond: A history of embryonic stem cell research. Nat. Rev. Genet 2006, 7, 319–327. [Google Scholar]
- Cooke, M.J.; Stojkovic, M.; Przyborski, S.A. Growth of teratomas derived from human pluripotent stem cells is influenced by the graft site. Stem Cells Dev 2006, 15, 254–259. [Google Scholar]
- Prokhorova, T.A.; Harkness, L.M.; Frandsen, U.; Ditzel, N.; Schroder, H.D.; Burns, J.S.; Kassem, M. Teratoma formation by human embryonic stem cells is site dependent and enhanced by the presence of matrigel. Stem Cells Dev 2009, 18, 47–54. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilic, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol 2008, 26, 1276–1284. [Google Scholar]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar]
- Gerrard, A.J.; Hudson, D.L.; Brownlee, G.G.; Watt, F.M. Towards gene therapy for haemophilia b using primary human keratinocytes. Nat. Genet 1993, 3, 180–183. [Google Scholar]
- Callejas, D.; Mann, C.J.; Ayuso, E.; Lage, R.; Grifoll, I.; Roca, C.; Andaluz, A.; Ruiz-de Gopegui, R.; Montane, J.; Munoz, S.; et al. Treatment of diabetes and long-term survival after insulin and glucokinase gene therapy. Diabetes 2013, 62, 1718–1729. [Google Scholar]
- Larcher, F.; del Rio, M.; Serrano, F.; Segovia, J.C.; Ramirez, A.; Meana, A.; Page, A.; Abad, J.L.; Gonzalez, M.A.; Bueren, J.; et al. A cutaneous gene therapy approach to human leptin deficiencies: Correction of the murine ob/ob phenotype using leptin-targeted keratinocyte grafts. FASEB J 2001, 15, 1529–1538. [Google Scholar]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.L.; et al. Gene therapy of human severe combined immunodeficiency (scid)-x1 disease. Science 2000, 288, 669–672. [Google Scholar]
- Aiuti, A.; Bachoud-Levi, A.C.; Blesch, A.; Brenner, M.K.; Cattaneo, F.; Chiocca, E.A.; Gao, G.; High, K.A.; Leen, A.M.; Lemoine, N.R.; et al. Progress and prospects: Gene therapy clinical trials (part 2). In Gene Ther; 2007; Volume 14, pp. 1555–1563. [Google Scholar]
- Fischer, A.; Cavazzana-Calvo, M. Gene therapy of inherited diseases. Lancet 2008, 371, 2044–2047. [Google Scholar]
- Gaspar, H.B.; Parsley, K.L.; Howe, S.; King, D.; Gilmour, K.C.; Sinclair, J.; Brouns, G.; Schmidt, M.; von Kalle, C.; Barington, T.; et al. Gene therapy of x-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet 2004, 364, 2181–2187. [Google Scholar]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. Lmo2-associated clonal T cell proliferation in two patients after gene therapy for scid-x1. Science 2003, 302, 415–419. [Google Scholar]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in x-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with wiskott-aldrich syndrome. Science 2013, 341. [Google Scholar] [CrossRef]
- Andressoo, J.O.; Weeda, G.; de Wit, J.; Mitchell, J.R.; Beems, R.B.; van Steeg, H.; van der Horst, G.T.; Hoeijmakers, J.H. An xpb mouse model for combined xeroderma pigmentosum and cockayne syndrome reveals progeroid features upon further attenuation of DNA repair. Mol. Cell Biol 2009, 29, 1276–1290. [Google Scholar]
- Clarkson, S.G. The xpg story. Biochimie 2003, 85, 1113–1121. [Google Scholar]
- De Boer, J.; Donker, I.; de Wit, J.; Hoeijmakers, J.H.; Weeda, G. Disruption of the mouse xeroderma pigmentosum group d DNA repair/basal transcription gene results in preimplantation lethality. Cancer Res 1998, 58, 89–94. [Google Scholar]
- Greenhaw, G.A.; Hebert, A.; Duke-Woodside, M.E.; Butler, I.J.; Hecht, J.T.; Cleaver, J.E.; Thomas, G.H.; Horton, W.A. Xeroderma pigmentosum and cockayne syndrome: Overlapping clinical and biochemical phenotypes. Am. J. Hum. Genet 1992, 50, 677–689. [Google Scholar]
- Herlin, C.; Sauniere, D.; Huertas, D. Xeroderma pigmentosum: Radical therapeutic procedure on the face using artificial skin. Ann. Chir. Plast. Esthet 2009, 54, 594–599. [Google Scholar]
- Carreau, M.; Quilliet, X.; Eveno, E.; Salvetti, A.; Danos, O.; Heard, J.M.; Mezzina, M.; Sarasin, A. Functional retroviral vector for gene therapy of xeroderma pigmentosum group d patients. Hum. Gene Ther 1995, 6, 1307–1315. [Google Scholar]
- Quilliet, X.; Chevallier-Lagente, O.; Eveno, E.; Stojkovic, T.; Destee, A.; Sarasin, A.; Mezzina, M. Long-term complementation of DNA repair deficient human primary fibroblasts by retroviral transduction of the xpd gene. Mutat. Res 1996, 364, 161–169. [Google Scholar]
- Zeng, L.; Quilliet, X.; Chevallier-Lagente, O.; Eveno, E.; Sarasin, A.; Mezzina, M. Retrovirus-mediated gene transfer corrects DNA repair defect of xeroderma pigmentosum cells of complementation groups a, b and c. Gene Ther 1997, 4, 1077–1084. [Google Scholar]
- Hadj-Rabia, S.; Oriot, D.; Soufir, N.; Dufresne, H.; Bourrat, E.; Mallet, S.; Poulhalon, N.; Ezzedine, E.; Grandchamp, B.; Taieb, A.; et al. Unexpected extradermatological findings in 31 patients with xeroderma pigmentosum type c. Br. J. Dermatol 2013, 168, 1109–1113. [Google Scholar]
- Frechet, M.; Warrick, E.; Vioux, C.; Chevallier, O.; Spatz, A.; Benhamou, S.; Sarasin, A.; Bernerd, F.; Magnaldo, T. Overexpression of matrix metalloproteinase 1 in dermal fibroblasts from DNA repair-deficient/cancer-prone xeroderma pigmentosum group c patients. Oncogene 2008, 27, 5223–5232. [Google Scholar]
- Gache, Y.; Pin, D.; Gagnoux-Palacios, L.; Carozzo, C.; Meneguzzi, G. Correction of dog dystrophic epidermolysis bullosa by transplantation of genetically modified epidermal autografts. J. Invest. Dermatol 2011, 131, 2069–2078. [Google Scholar]
- Arnaudeau-Begard, C.; Brellier, F.; Chevallier-Lagente, O.; Hoeijmakers, J.; Bernerd, F.; Sarasin, A.; Magnaldo, T. Genetic correction of DNA repair-deficient/cancer-prone xeroderma pigmentosum group c keratinocytes. Hum. Gene Ther 2003, 14, 983–996. [Google Scholar]
- Magnaldo, T.; Barrandon, Y. CD24 (heat stable antigen, nectadrin), a novel keratinocyte differentiation marker, is preferentially expressed in areas of the hair follicle containing the colony-forming cells. J. Cell Sci 1996, 109, 3035–3045. [Google Scholar]
- Bergoglio, V.; Larcher, F.; Chevallier-Lagente, O.; Bernheim, A.; Danos, O.; Sarasin, A.; Rio, M.D.; Magnaldo, T. Safe selection of genetically manipulated human primary keratinocytes with very high growth potential using CD24. Mol. Ther 2007, 15, 2186–2193. [Google Scholar]
- Warrick, E.; Garcia, M.; Chagnoleau, C.; Chevallier, O.; Bergoglio, V.; Sartori, D.; Mavilio, F.; Angulo, J.F.; Avril, M.F.; Sarasin, A.; et al. Preclinical corrective gene transfer in xeroderma pigmentosum human skin stem cells. Mol. Ther 2012, 20, 798–807. [Google Scholar]
- Mathor, M.B.; Ferrari, G.; Dellambra, E.; Cilli, M.; Mavilio, F.; Cancedda, R.; de Luca, M. Clonal analysis of stably transduced human epidermal stem cells in culture. Proc. Natl. Acad. Sci. USA 1996, 93, 10371–10376. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rouanet, S.; Warrick, E.; Gache, Y.; Scarzello, S.; Avril, M.-F.; Bernerd, F.; Magnaldo, T. Genetic Correction of Stem Cells in the Treatment of Inherited Diseases and Focus on Xeroderma Pigmentosum. Int. J. Mol. Sci. 2013, 14, 20019-20036. https://doi.org/10.3390/ijms141020019
Rouanet S, Warrick E, Gache Y, Scarzello S, Avril M-F, Bernerd F, Magnaldo T. Genetic Correction of Stem Cells in the Treatment of Inherited Diseases and Focus on Xeroderma Pigmentosum. International Journal of Molecular Sciences. 2013; 14(10):20019-20036. https://doi.org/10.3390/ijms141020019
Chicago/Turabian StyleRouanet, Sophie, Emilie Warrick, Yannick Gache, Sabine Scarzello, Marie-Françoise Avril, Françoise Bernerd, and Thierry Magnaldo. 2013. "Genetic Correction of Stem Cells in the Treatment of Inherited Diseases and Focus on Xeroderma Pigmentosum" International Journal of Molecular Sciences 14, no. 10: 20019-20036. https://doi.org/10.3390/ijms141020019
APA StyleRouanet, S., Warrick, E., Gache, Y., Scarzello, S., Avril, M.-F., Bernerd, F., & Magnaldo, T. (2013). Genetic Correction of Stem Cells in the Treatment of Inherited Diseases and Focus on Xeroderma Pigmentosum. International Journal of Molecular Sciences, 14(10), 20019-20036. https://doi.org/10.3390/ijms141020019