Development of Nonalcoholic Hepatopathy: Contributions of Oxidative Stress and Advanced Glycation End Products


Abstract
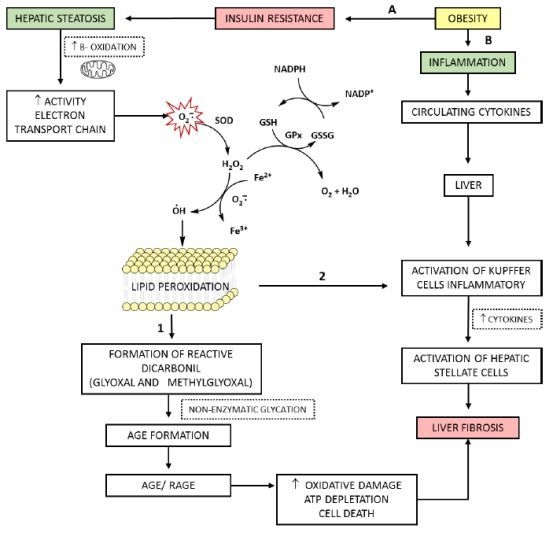
:
1. Introduction
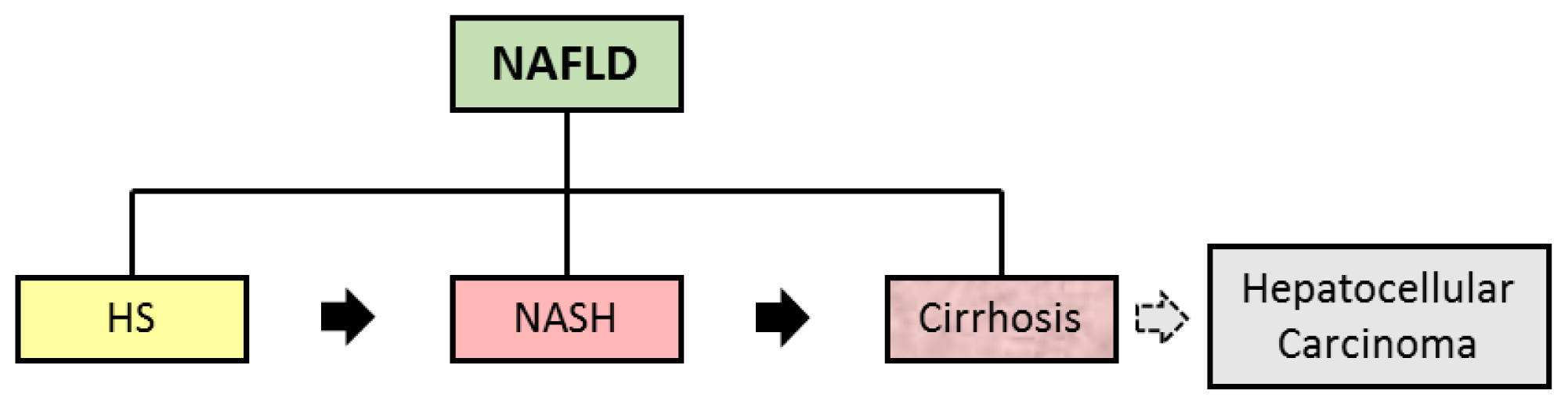
2. NAFLD, IR and MS
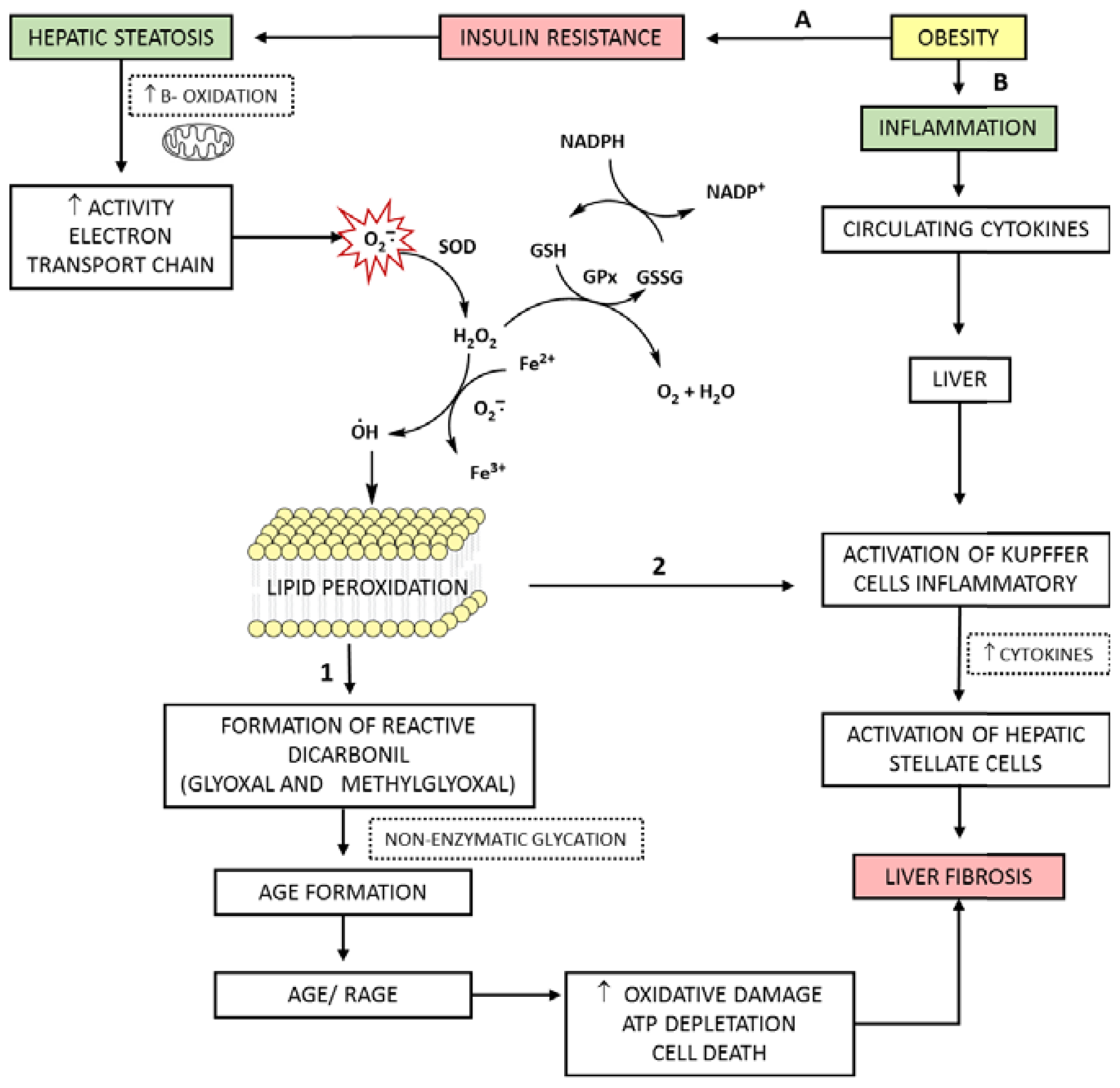
3. AGEs: Formation and Effects
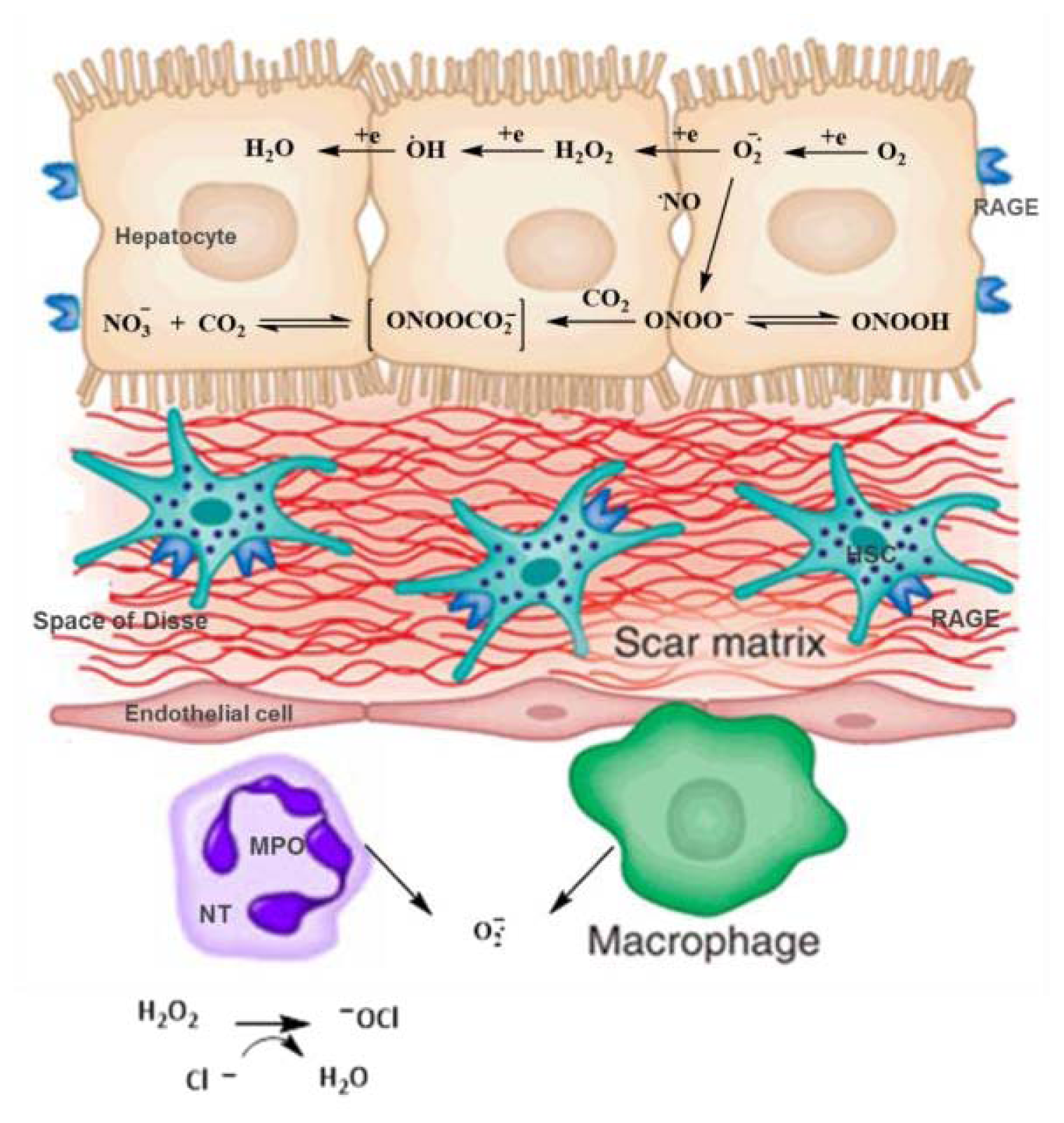
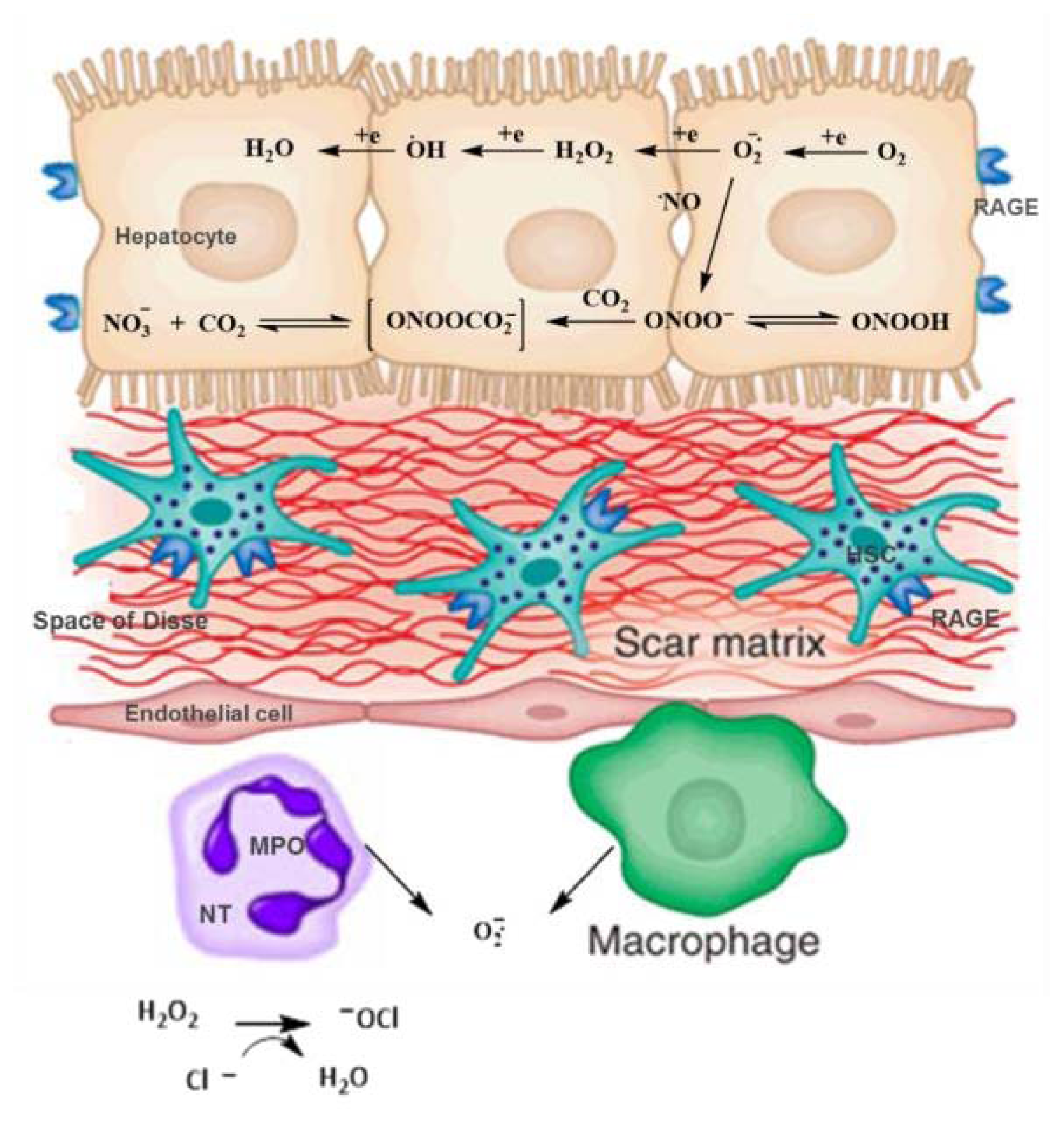
4. AGEs, RAGE and Increasing Oxidative Stress in Hepatic Disease
5. The Role of ROS and AGEs in the Evolution of NAFLD
6. Final Considerations
7. Conclusions
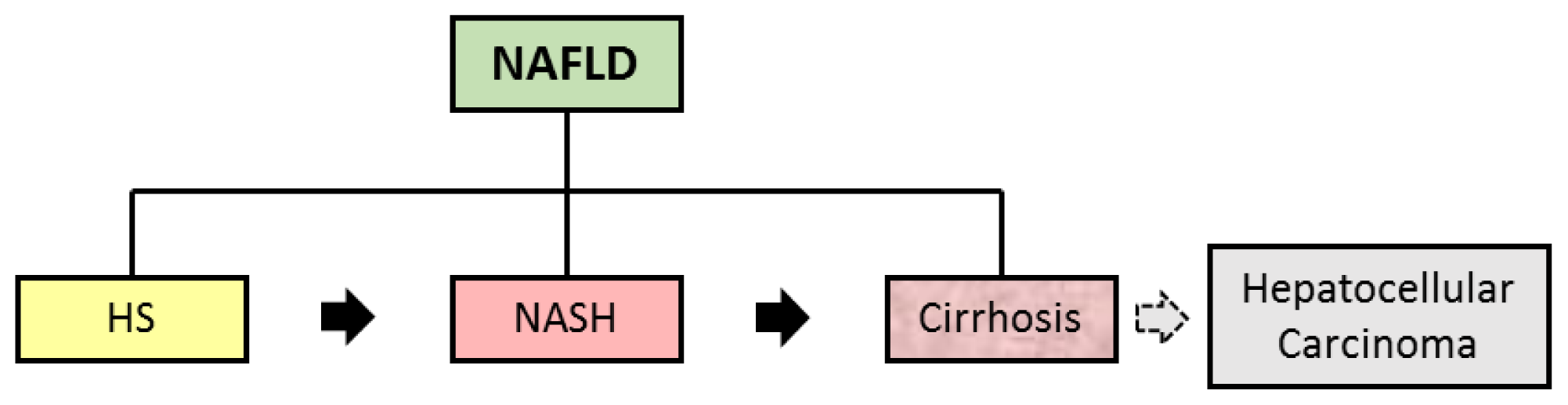
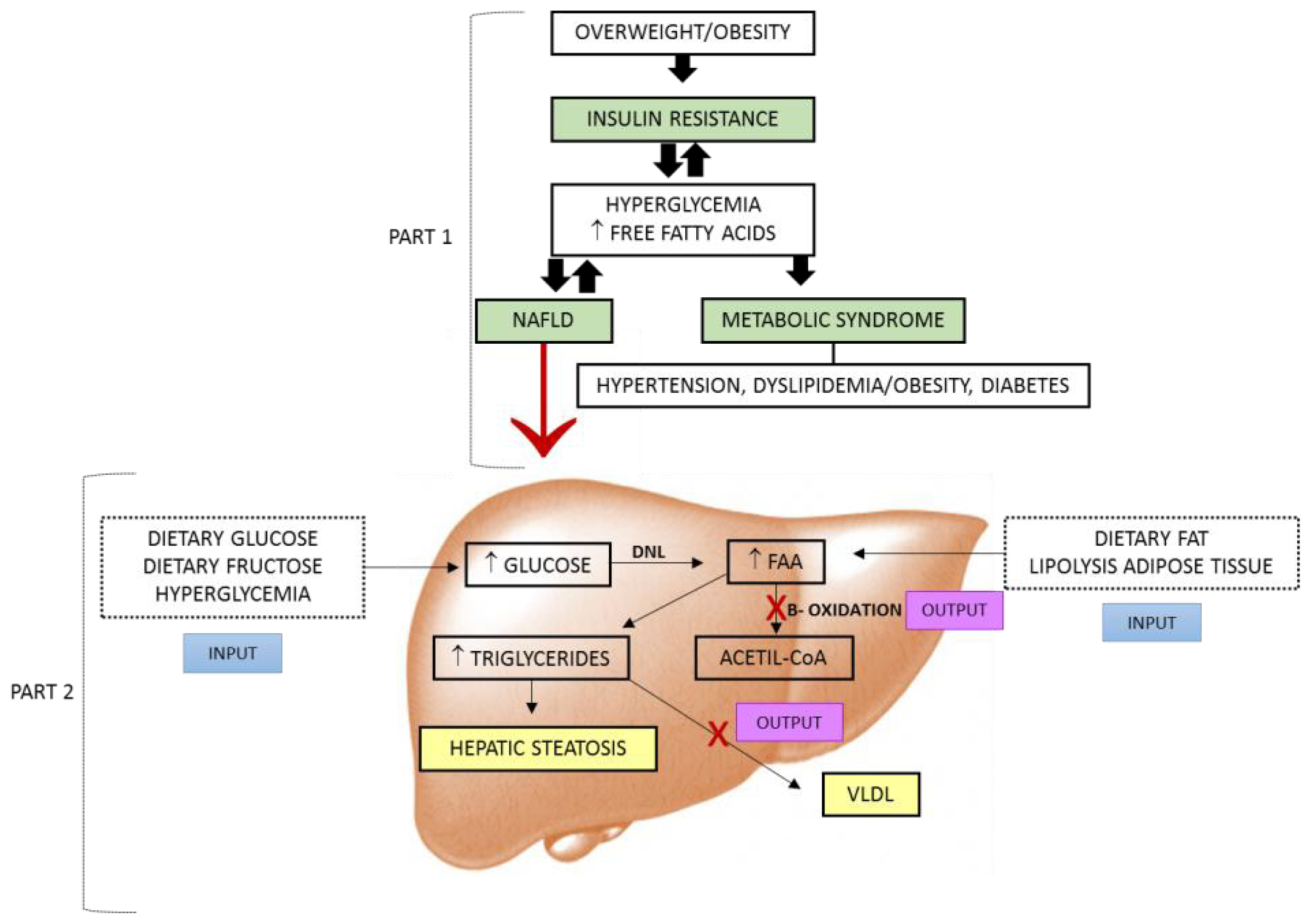
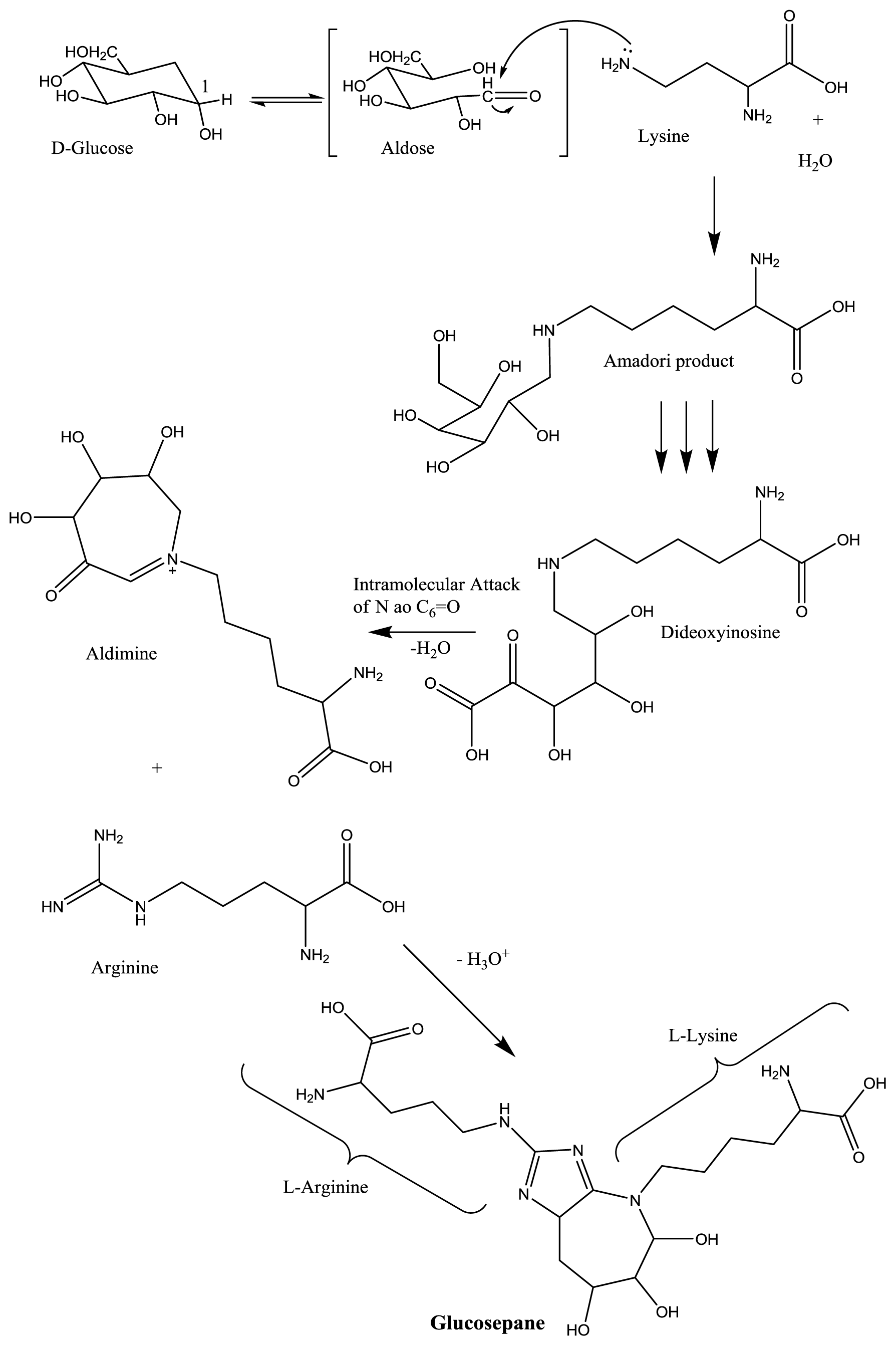



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Description | Diagnosis | Ref. |
|---|---|---|---|
| Metabolic Syndrome (MS) | Complex disorder represented by a set of cardiovascular risk factors, usually related to central fat deposition and IR | Abdominal obesity, defined by increased waist circumference (≥90 cm in men and ≥80 cm in women) and two or more of the following characteristics: blood pressure (≥130/85 mm/Hg), high levels of fasting glucose (≥100 mg/dL) or triglycerides (≥150 mg/dL) or low levels of HDL cholesterol (<40 mg/dL in men, <50 mg/dL in women) | [5–8] |
| Diabetes | Heterogeneous group of metabolic disorders that have in common the hyperglycemia, which is defective in insulin action, insulin secretion, or both | World Health Organization (WHO) criteria: fasting plasma glucose (FPG) ≥7.0 mmol/L (126 mg/dL) or 75 g oral glucose tolerance test (OGTT) with FPG ≥ 7.0 mmol/L (126 mg/dL) and/or 2 h plasma glucose ≥ 11.1 mmol/L (200 mg/dL) or, glycated haemoglobin (HbA1c) ≥6.5% /48 mmol/mol, or Random plasma glucose ≥11.1 mmol/L (200 mg/dL) in the presence of classical diabetes symptoms. | [5,6,8,9] |
| Nonalcoholic fatty liver disease (NAFLD) | Encompasses the entire spectra of fatty liver diseases in individuals without significant alcohol consumption, ranging from fatty liver to steatohepatitis and cirrhosis | (a) presence of hepatic steatosis by imaging or histology; (b) no significant alcohol consumption; (c) no competing etiologies for hepatic steatosis; and (d) no co-existing causes for chronic liver disease. | [5,7,10] |
| Hepatic Steatosis (HS) | Presence of hepatic steatosis with no evidence of hepatocellular injury in the form of ballooning of the hepatocytes or no evidence of fibrosis | Liver biopsy with histological analysis | [5] |
| Nonalcoholic Steatohepatiti s (NASH) | Presence of hepatic steatosis and inflammation with hepatocyte injury (ballooning) with or without fibrosis | Liver biopsy with histological analysis NAFLD Fibrosis Score * | [5] |
| Hepatic Cirrhosis | Presence of cirrhosis with no obvious etiology. Patients with cryptogenic cirrhosis are heavily enriched with metabolic risk factors such as obesity and metabolic syndrome | Liver biopsy with histological analysis NAFLD Fibrosis Score * | [5] |
Acknowledgments
Conflicts of Interest
References
- Basta, G.; Navarra, T.; Simone, P.; Turco, S.; Gastaldelli, A.; Filipponi, F. What is the role of the receptor for advanced glycation end products-ligand axis in liver injury? Liver Transpl 2011, 17, 633–640. [Google Scholar]
- Jaeschke, H.; Ramachandran, A. Reactive oxygen species in the normal and acutely injured liver. J. Hepatol 2011, 55, 227–228. [Google Scholar]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest 2004, 114, 147–152. [Google Scholar]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol 2006, 87, 1–16. [Google Scholar]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the american gastroenterological association, american association for the study of liver diseases, and american college of gastroenterology. Hepatology 2012, 55, 2005–2023. [Google Scholar]
- International Diabetes Federation. Available online: http://www.diabetesatlas.org (accessed on 1 June 2012).
- I Diretriz Brasileira de Diagnóstico e Tratamento da Síndrome Metabólica. Available online: http://www.scielo.br/scielo.php?script=sci_pdf&pid=S0066-782X2005000700001&lng=en&nrm=iso&tlng=pt (accessed on 22 September 2013).
- Souza, M.R.A.; Diniz, M.F.F.M.; Medeiros-Filho, J.E.M.; Araújo, M.S.T. Metabolic syndrome and risk factors for non-alcoholic fatty liver disease. Arq. Gastroenterol 2012, 49, 89–96. [Google Scholar]
- Negre-Salvayre, A.; Salvayre, R.; Auge, N.; Pamplona, R.; Portero-Otín, M. Hyperglycemia and glycation in diabetic complications. Antioxid. Redox Signal 2009, 11, 3071–3109. [Google Scholar]
- Kimura, Y.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Ishitobi, T.; Nabeshima, Y.; Arihiro, K.; Chayama, K. Atorvastatin decreases serum levels of advanced glycation end products (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J. Gastroenterol 2010, 45, 750–757. [Google Scholar]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar]
- Thornalley, P.J.; Battah, S.; Ahmed, N.; Agalou, S.; Babaei-jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation end products in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J 2003, 375, 581–592. [Google Scholar]
- Assis, M.A. Effect of heated hyperlipid diet and regular hyperlipid diet in the formation of glycation products and reactive oxygen species in wistar rats. Master’s Thesis, Federal University of Rio Grande do Sul, Porto Alegre (RS), 2009. [Google Scholar]
- Vlassara, H.; Palace, M.R. Diabetes and advanced glycation endproducts. J. Int. Med 2002, 251, 87–101. [Google Scholar]
- Gaens, K.H.J.; Niessen, P.M.G.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.M.; Driessen, A.; Wolfs, M.G.M.; Hofker, M.H.; Bloemen, J.G.; Dejong, C.H.; et al. Endogenous formation of Ne-(carboxymethyl)lysine is increased in fatty livers and induces inflammatory markers in an in vitro model of hepatic steatosis. J. Hepatol 2012, 56, 647–655. [Google Scholar]
- Vlassara, H.; Gary, E.S. The role of advanced glycation end-products in the etiology of IR and diabetes. US Endocrinol 2010, 6, 14–19. [Google Scholar]
- Busch, M.; Franke, S.; Ruster, C.; Wolf, G. Advanced glycation end-products and the kidney. Eur. J. Clin. Invest 2010, 40, 742–755. [Google Scholar]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Olmesartan blocks inflammatory reactions in endothelial cells evoked by advanced glycation end products by suppressing generation of reactive oxygen species. Ophthalmic Res 2008, 40, 10–15. [Google Scholar]
- Patel, R.; Baker, S.S.; Liu, W.; Desai, S.; Alkhouri, R.K.R.; Mastrandrea, L.; Sarfraz, A.; Cai, W.; Vlassara, H.; Patel, M.S.; et al. Effect of dietary advanced glycation end products on mouse liver. PLoS ONE 2012, 7, e35143. [Google Scholar]
- Edeas, M.; Attaf, D.; Mailfert, A.S.; Nasu, M.; Joubet, R. Maillard reaction, mitochondria and oxidative stress: Potential role of antioxidants. Pathol. Biol. (Paris) 2010, 58, 220–225. [Google Scholar]
- Vasconcelos, S.M.L.; Goulart, M.O.F.; Moura, J.B.F.; Benfato, V.M.M.S.B.; Kubota, L.T. Reactive oxygen and nitrogen species, antioxidants and markers of oxidative damage in human blood: The main analytical methods for their determination [in Portuguese]. Quim. Nova 2007, 30, 1323–1338. [Google Scholar]
- Bandeira, S.M.; Fonseca, L.J.S.; Guedes, G.S.; Rabelo, L.A.; Goulart, M.O.F.; Vasconcelos, S.M.L. Oxidative stress as an underlying contributor in the development of chronic complications in diabetes mellitus. Int. J. Mol. Sci 2013, 14, 3265–3284. [Google Scholar]
- Malaguarnera, M.; Rosa, M.D.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med 2009, 87, 679–695. [Google Scholar]
- Styskal, J.L.; Remmen, H.V.; Richardson, A.; Salmon, A.B. Oxidative stress and diabetes: What can we learn about insulin resistance from antioxidant mutant mouse models? Free Radic. Biol. Med 2012, 52, 46–58. [Google Scholar]
- Marra, F.; Gastaldelli, A.; Svegliati Baroni, G.; Tell, G.; Tiribelli, C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends. Mol. Med 2008, 14, 72–81. [Google Scholar]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with non-alcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, Jinhan; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter cd36 is a common target of lxr, pxr, and pparγ in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar]
- Pan, A.V.; Staub, H.L.; Chatkin, J.M.; Moretto, M.; Maggioni, L.; Rizzolli, J.; Mottin, C.C. Nonalcoholic fatty liver disease and the risk of cirrhosis [in Portuguese]. Sci. Med 2008, 18, 172–176. [Google Scholar]
- Caldwell, S.H.; Hylton, A.I. The clinical outcome of NAFLD including cryptogenic cirrhosis. In Fatty Liver Disease: NASH and Related Disorders; Farrell, G.C., George, J., Hall, P.M., McCullough, A.J., Eds.; Blackwell Publishing Ltd: Oxford, UK, 2005; pp. 168–180. [Google Scholar]
- Pacifico, L.; Nobili, V.; Anania, C.; Verdecchia, P.; Chiesa, C. Pediatric nonalcoholic fatty liver disease, metabolic syndrome and cardiovascular risk. World J. Gastroenterol 2011, 17, 3082–3091. [Google Scholar]
- Vannia, E.; Bugianesia, E.; Kotronenb, A.; de Minicisd, S.; Yki-Järvinenb, H.; Svegliati-Baronid, G. From the metabolic syndrome to NAFLD or vice versa? Dig. Liver Dis 2010, 42, 320–330. [Google Scholar]
- Seo, H.I.; Cho, Y.K.; Lee, W.Y.; Rhee, E.J.; Sung, K.C.; Kim, B.S.; Son, B.H.; Shin, J.H.; Joo, K.J.; Hong, H.P.; et al. Which metabolic syndrome criteria best predict the presence of non-alcoholic fatty liver disease? Diabetes Res. Clin. Pract 2012, 95, 19–24. [Google Scholar]
- Scheen, A.J.; Luyckx, F.H. Obesity and liver disease. Best Pract. Res. Clin. Endocrinol. Metab 2002, 16, 703–716. [Google Scholar]
- Utzschneider, K.M.; Kahn, S.E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab 2006, 1, 4753–4761. [Google Scholar]
- Liangpunsakul, S.; Chalasani, N. Unexplained elevations in alanine aminotransferase in individuals with the metabolic syndrome: Results from the third national health and nutrition survey (NHANES III). Am. J. Med. Sci 2005, 329, 111–116. [Google Scholar]
- Solís Herruzo, J.A.; García Ruiz, I.; Pérez Carreras, M.; Muñoz Yagüe, M.T. Non-alcoholic fatty liver disease. From insulin resistance to mitochondrial dysfunction. Rev. Esp. Enferm. Dig 2006, 98, 844–874. [Google Scholar]
- Leclercq, I.A.; Morais, A.S.; Schroyen, B.; Hul, N.V.; Geerts, A. Insulin resistance in hepatocytes and sinusoidal liver cells: Mechanisms and consequences. J. Hepatol 2007, 47, 142–156. [Google Scholar]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Wanless, I.R.; Shiota, K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: A four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin. Liver Dis 2004, 24, 99–106. [Google Scholar]
- Hyogo, H.; Yamagishi, S.; Iwamoto, K.; Arihiro, K.; Takeuchi, M.; Sato, T.; Ochi, H.; Nonaka, M.; Nabeshima, Y.; Inoue, M.; et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol 2007, 22, 1112–1119. [Google Scholar]
- Kumar, A.; Sharma, A.; Duseja, A.; Das, A.; Dhiman, R.K.; Chawla, Y.K.; Kohliy, K.K.; Bhansaliz, A. Patients with nonalcoholic fatty liver disease (NAFLD) have higher oxidative stress in comparison to chronic viral hepatitis. J. Clin. Exp. Hepatol 2013, 3, 12–18. [Google Scholar]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation end products: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. A Biol. Sci. Med. Sci 2007, 62, 427–433. [Google Scholar]
- Piarulli, F.; Sartore, G.; Lapolla, A. Glyco-oxidation and cardiovascular complications in type 2 diabetes: A clinical update. Acta Diabetol 2013, 50, 101–110. [Google Scholar]
- Yamagishi, S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) invascular damage in diabetes. Exp. Gerontol 2011, 46, 217–224. [Google Scholar]
- Schmidt, A.M.; Stern, D.M. RAGE: A new target for the prevention and treatment of the vascular and inflammatory complications of diabetes. Trends Endocrinol. Metab 2000, 11, 368–375. [Google Scholar]
- Gelain, D.P.; Pasquali, M.A.B.; Caregnato, F.F.; Moreira, J.C.F. Vitamin A (retinol) up-regulates the receptor for advanced glycation end products (RAGE) through p38 and Akt oxidant-dependent activation. Toxicology 2011, 289, 38–44. [Google Scholar]
- Vasques, L.M. Regulation of receptors for advanced glycation end products (RAGE) by vitamin A. Master’s Thesis, Federal University do Rio Grande do Sul, Porto Alegre (RS), Brazil, 2011. [Google Scholar]
- Kuhla, A.; Trieglaff, C.; Vollmar, B. Role of age and uncoupling protein-2 in oxidative stress, RAGE/AGE interaction and inflammatory liver injury. Exp. Gerontol 2011, 46, 868–876. [Google Scholar]
- Yilmaz, Y.; Ulukaya, E.; Gul, O.O.; Arabul, M.; Gul, C.M.; Atug, O.; Oral, A.Y.; Aker, S.; Dolar, E. Decreased plasma levels of soluble receptor for advanced glycation endproducts (sRAGE) in patients with nonalcoholic fatty liver disease. Clin. Biochem 2009, 42, 802–807. [Google Scholar]
- Hudson, B.I.; Harja, E.; Moser, B.; Schmidt, A.M. Soluble levels of receptor for advanced glycation endproducts (sRAGE) and coronary artery disease: The next C-reactive protein? Arterioscler. Thromb. Vasc. Biol 2005, 25, 879–882. [Google Scholar]
- Geroldi, D.; Falcone, C.; Emanuele, E. Soluble receptor for advanced glycation end products: From disease marker to potential therapeutic target. Curr. Med. Chem 2006, 13, 1971–1978. [Google Scholar]
- Friedman, S.L. Targeting siRNA to arrest fibrosis. Nat. Biotechnol 2008, 26, 399–400. [Google Scholar]
- Desic, S.D.; Aragon, I.B. The FAST index—A highly sensitive indicator of the heat impact on infant formula model. Food Chem 2011, 124, 1043–1049. [Google Scholar]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Wallenstein, S.; Striker, G.E.; Vlassara, H. Reduced oxidant stress and extended lifespan in mice exposed to a low glycotoxin diet: association with increased AGER1 expression. Am. J. Pathol 2007, 170, 1893–1902. [Google Scholar]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med 2012, 52, 59–69. [Google Scholar]
- Veteläinen, R.; van Vliet, A.; van Gulik, T.M. Essential pathogenic and metabolic differences in steatosis induced by choline or methione-choline deficient diets in a rat model. J. Gastroenterol. Hepatol 2007, 22, 1526–1533. [Google Scholar]
- Reis, J.S.; Veloso, C.A.; Mattos, R.T.; Purish, S.; Nogueira-Machado, J.A. Oxidative stress: Review of metabolic signaling in type 1 diabetes (in Portuguese). Arq. Bras. Endocrinol. Metab 2008, 52, 1096–1105. [Google Scholar]
- Nomura, K.; Yamanouchi, T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J. Nutr. Biochem 2012, 23, 203–208. [Google Scholar]
- Guimarães, E.L.M.; Empsen, C.; Geerts, A.; Grunsven, L.A.V. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J. Hepatol 2010, 52, 389–597. [Google Scholar]
- Trauner, M.; Arrese, M.; Wagner, M. Fatty liver and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 299–310. [Google Scholar]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol 2013, 59, 583–594. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Santos, J.C.d.F.; Valentim, I.B.; De Araújo, O.R.P.; Ataide, T.D.R.; Goulart, M.O.F. Development of Nonalcoholic Hepatopathy: Contributions of Oxidative Stress and Advanced Glycation End Products. Int. J. Mol. Sci. 2013, 14, 19846-19866. https://doi.org/10.3390/ijms141019846
Santos JCdF, Valentim IB, De Araújo ORP, Ataide TDR, Goulart MOF. Development of Nonalcoholic Hepatopathy: Contributions of Oxidative Stress and Advanced Glycation End Products. International Journal of Molecular Sciences. 2013; 14(10):19846-19866. https://doi.org/10.3390/ijms141019846
Chicago/Turabian StyleSantos, Juliana Célia de F., Iara B. Valentim, Orlando R. P. De Araújo, Terezinha Da R. Ataide, and Marília O. F. Goulart. 2013. "Development of Nonalcoholic Hepatopathy: Contributions of Oxidative Stress and Advanced Glycation End Products" International Journal of Molecular Sciences 14, no. 10: 19846-19866. https://doi.org/10.3390/ijms141019846
APA StyleSantos, J. C. d. F., Valentim, I. B., De Araújo, O. R. P., Ataide, T. D. R., & Goulart, M. O. F. (2013). Development of Nonalcoholic Hepatopathy: Contributions of Oxidative Stress and Advanced Glycation End Products. International Journal of Molecular Sciences, 14(10), 19846-19866. https://doi.org/10.3390/ijms141019846