Interplay between Cartilage and Subchondral Bone Contributing to Pathogenesis of Osteoarthritis




Abstract
:1. Introduction
2. Onset and Progression of OA
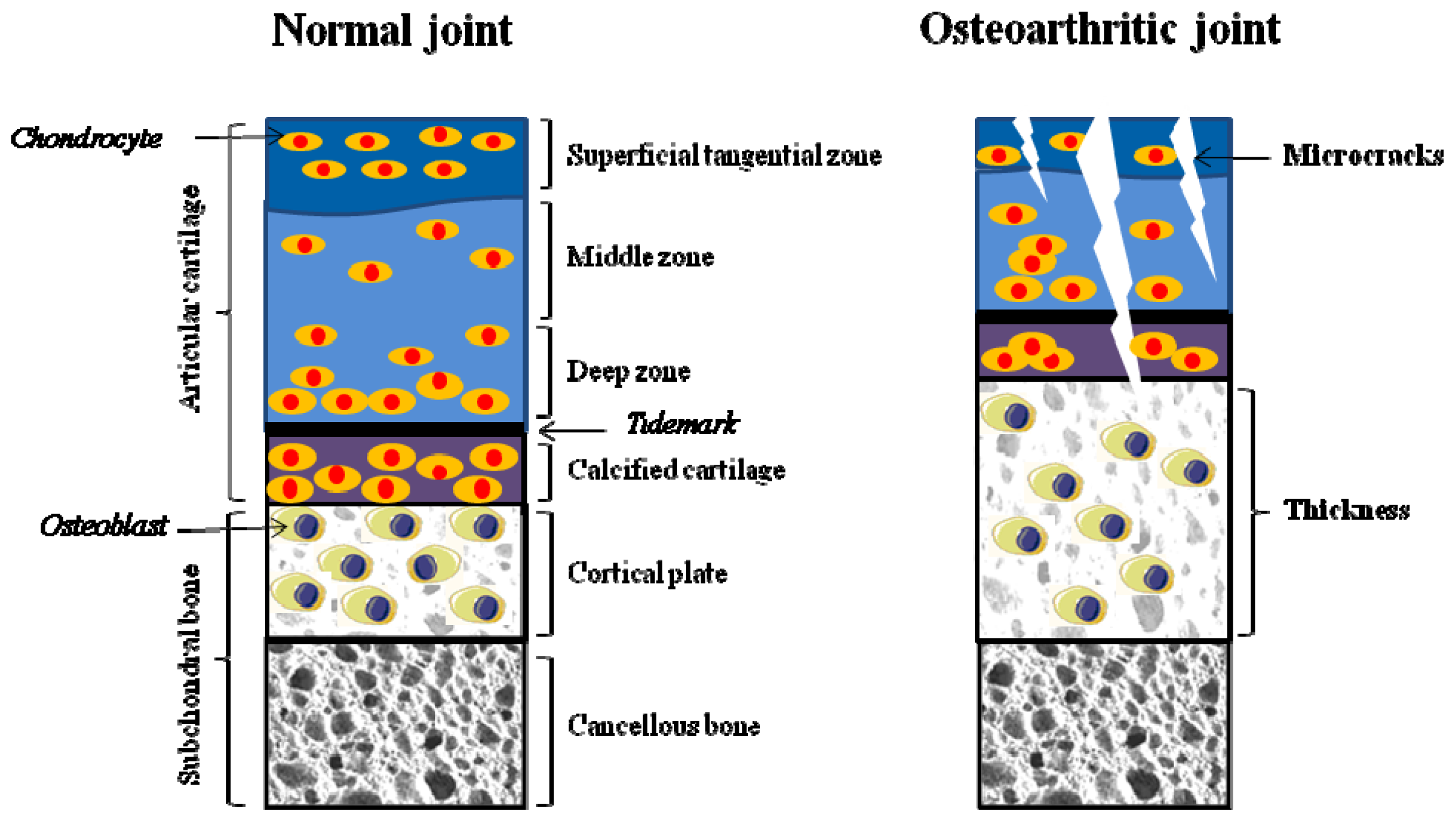
2.1. Alteration in Cartilage
2.2. Alteration in Subchondral Bone
3. Cross Talk between Articular Cartilage and Subchondral Bone
3.1. Biological Factors
3.2. Signaling Pathways
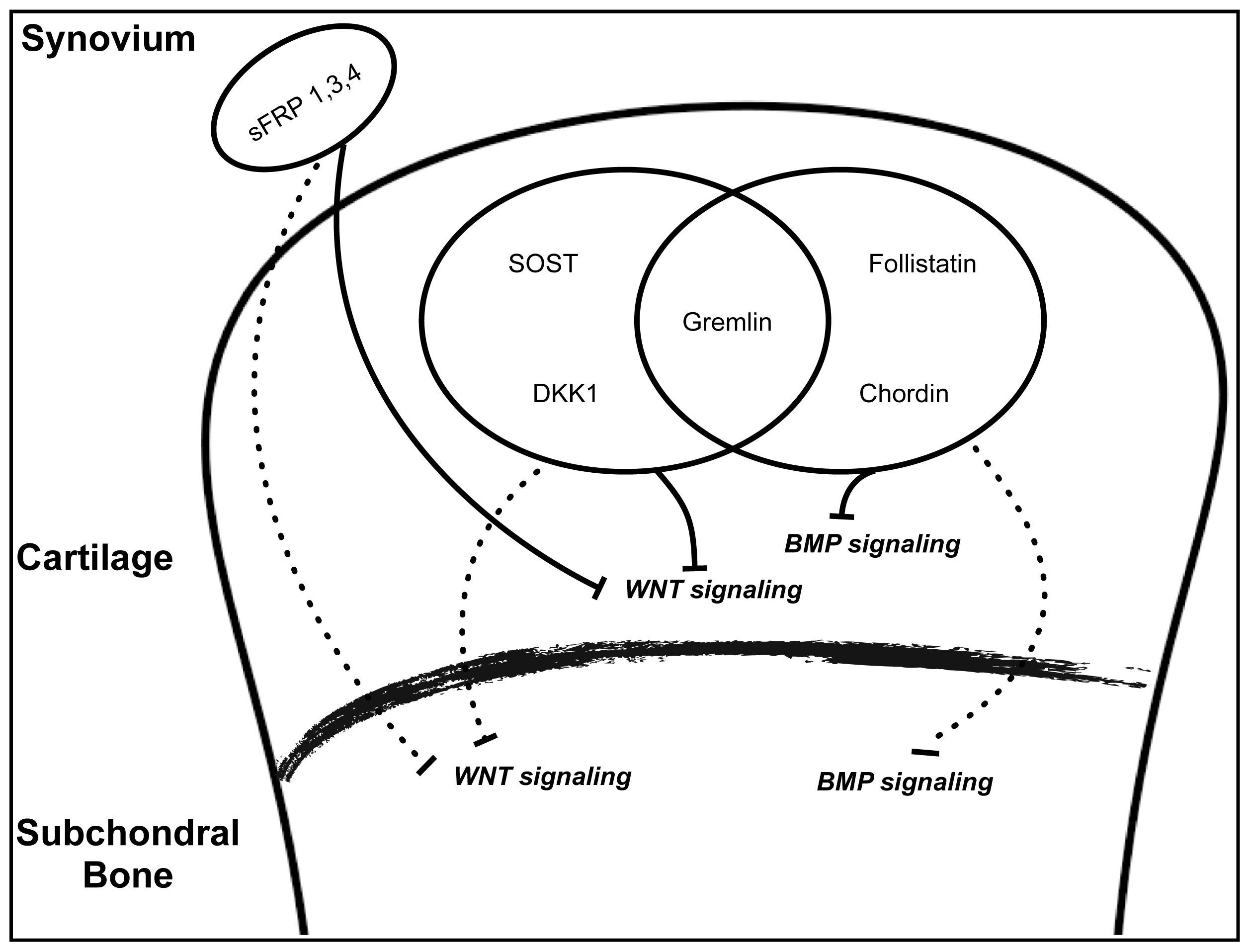
3.2.1. WNT Signaling
3.2.2. TGF-β/BMP Signaling
3.2.3. MAPK Signaling
4. Conclusions



{kind=link}
{kind=link}
| Biomarkers | Function in joint | During OA elevated expression represents | References |
|---|---|---|---|
| Biomarkers for Cartilage | |||
| Cartilage oligomeric matrix protein (COMP) | Help in inflammatory proliferation of synovial membrane, regulation of fibril assembly and to maintain the mature collagen network | Cartilage degradation | [28] |
| C-terminal telopeptide of type II collagen (CTX-II) | provide strength, integrity and maintain shape of tissue | Remodeling of calcified cartilage | [29] |
| Helical fragments (Helix-II and Coll 2-1, Coll 2-1 NO2) | Contribute in inflammatory processes and cartilage catabolism in the joint | Type II collagen degradation | [30] |
| Amino-terminal type II procollagen propeptide (PIINP) | One of the two propeptides of type II procollagen and rflect the rates of collagen type II synthesis | Cartilage degradation | [31] |
| Carboxy-terminal type II procollagen propeptide (PIICP) | One of the two propeptides of type II procollagen and reflect the rates of collagen type II synthesis | Cartilage degradation | [31] |
| YLK-40 Glycoprotein: noncollagenous protein | Have a vital role in creating or amending tissue inflammation, immunity and/or remodeling | Cartilage degradation | [32] |
| Keratan sulfate (KS-5D4) | Act as a cushion to absorb mechanical shock. | Aggrecan and cartilage degradation | [33] |
| Chondroitin sulfate 846 epitope (CS 846) | provides a hydrated gel structure (via its interaction with hyaluronan and link protein) that endows the cartilage with load-bearing properties | Cartilage turnover | [28] |
| Hyaluronic acid (HA) | Essential for viscoelasticity of synovium fluid and cartilage | Cartilage degradation | [34] |
| Biomarkers for Bone | |||
| N-terminal type I collagen telopeptides (NTX I) | Maintain bone remodeling process | Type I collagen degradation | [35] |
| C-terminal type I collagen (CTX I) or Serum CTX I | Crosslinking peptide of type I collagen and necessary for immunoreactivity | Increased osteoclastogenesis and Bone degradation | [35] |
| Amino-terminal procollagen propeptide of type I collagen (PINP) | One of the two propeptides of type I procollagen and represent synthesis rate of type I collagen | Bone degradation | [36] |
| Carboxy-terminal procollagen propeptide of type I collagen (PICP) | One of the two propeptides of type I procollagen and represent synthesis rate of type I collagen | Bone degradation | [36] |
| Osteocalcin (OC) | Essential for bone mineralization and recruitment of osteoblast and osteoclast at the site of bone formation | Anabolic bone turnover | [28] |
| Urinary total pyridinoline (PYD) | Contributes to stabilizing and reinforcing the whole structure of collagenous tissues as bone and cartilage | Catabolic bone turnover | [28,37] |
| Bone sialoprotein (BSP) | Necessary for mineralization at cartilage bone interface | Anabolic bone turnover | [37] |
Acknowledgments
Conflicts of Interest
References
- Castaneda, S.; Roman-Blas, J.A.; Largo, R.; Herrero-Beaumont, G. Subchondral bone as a key target for osteoarthritis treatment. Biochem. Pharmacol 2012, 83, 315–323. [Google Scholar]
- Lawrence, R.C.; Felson, D.T.; Helmick, C.G.; Arnold, L.M.; Choi, H.; Deyo, R.A.; Gabriel, S.; Hirsch, R.; Hochberg, M.C.; Hunder, G.G.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum 2008, 58, 26–35. [Google Scholar]
- Lawrence, J.S.; Bremner, J.M.; Bier, F. Osteo-arthrosis. Prevalence in the population and relationship between symptoms and X-ray changes. Ann. Rheum. Dis 1966, 25, 1–24. [Google Scholar]
- Felson, D.T. Clinical practice. Osteoarthritis of the knee. N. Engl. J. Med 2006, 354, 841–848. [Google Scholar]
- Felson, D.T.; Anderson, J.J.; Meenan, R.F. The comparative efficacy and toxicity of second-line drugs in rheumatoid arthritis. Results of two metaanalyses. Arthritis Rheum 1990, 33, 1449–1461. [Google Scholar]
- Roos, E.M. Joint injury causes knee osteoarthritis in young adults. Curr. Opin. Rheumatol 2005, 17, 195–200. [Google Scholar]
- Brandt, K.D.; Radin, E.L.; Dieppe, P.A.; van de Putte, L. Yet more evidence that osteoarthritis is not a cartilage disease. Ann. Rheum. Dis 2006, 65, 1261–1264. [Google Scholar]
- Kwan Tat, S.; Lajeunesse, D.; Pelletier, J.P.; Martel-Pelletier, J. Targeting subchondral bone for treating osteoarthritis: What is the evidence? Best Pract. Res. Clin. Rheumatol 2010, 24, 51–70. [Google Scholar]
- Karsdal, M.A.; Leeming, D.J.; Dam, E.B.; Henriksen, K.; Alexandersen, P.; Pastoureau, P.; Altman, R.D.; Christiansen, C. Should subchondral bone turnover be targeted when treating osteoarthritis? Osteoarthr. Cartil 2008, 16, 638–646. [Google Scholar]
- Wan, R.; Hu, J.; Zhou, Q.; Wang, J.; Liu, P.; Wei, Y. Application of co-expressed genes to articular cartilage: New hope for the treatment of osteoarthritis (review). Mol. Med. Rep 2012, 6, 16–18. [Google Scholar]
- Ansboro, S.; Greiser, U.; Barry, F.; Murphy, M. Strategies for improved targeting of therapeutic cells: Implications for tissue repair. Eur. Cells Mater 2012, 23, 310–319. [Google Scholar]
- Burr, D.B. The importance of subchondral bone in osteoarthrosis. Curr. Opin. Rheumatol 1998, 10, 256–262. [Google Scholar]
- Bailey, A.J.; Mansell, J.P. Do subchondral bone changes exacerbate or precede articular cartilage destruction in osteoarthritis of the elderly? Gerontology 1997, 43, 296–304. [Google Scholar]
- Clouet, J.; Vinatier, C.; Merceron, C.; Pot-vaucel, M.; Maugars, Y.; Weiss, P.; Grimandi, G.; Guicheux, J. From osteoarthritis treatments to future regenerative therapies for cartilage. Drug Discov. Today 2009, 14, 913–925. [Google Scholar]
- Poole, A.R.; Kojima, T.; Yasuda, T.; Mwale, F.; Kobayashi, M.; Laverty, S. Composition and structure of articular cartilage: A template for tissue repair. Clin. Orthop. Relat. Res 2001, 391, S26–S33. [Google Scholar]
- Burr, D.B. Anatomy and physiology of the mineralized tissues: Role in the pathogenesis of osteoarthrosis. Osteoarthr. Cartil 2004, 12, S20–S30. [Google Scholar]
- Servier Medical Art. Available online: http://www.servier.com/slidekit/?item=2 (accessed on 28 August 2013).
- Guilak, F. Biomechanical factors in osteoarthritis. Best Pract. Res. Clin. Rheumatol 2011, 25, 815–823. [Google Scholar]
- Hunter, D.J.; Felson, D.T. Osteoarthritis. BMJ 2006, 332, 639–642. [Google Scholar]
- Guilak, F.; Fermor, B.; Keefe, F.J.; Kraus, V.B.; Olson, S.A.; Pisetsky, D.S.; Setton, L.A.; Weinberg, J.B. The role of biomechanics and inflammation in cartilage injury and repair. Clin. Orthop. Relat. Res 2004, 423, 17–26. [Google Scholar]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol 2012, 8, 665–673. [Google Scholar]
- Goldring, M.B.; Goldring, S.R. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann. N. Y. Acad. Sci 2010, 1192, 230–237. [Google Scholar]
- Grimshaw, M.J.; Mason, R.M. Bovine articular chondrocyte function in vitro depends upon oxygen tension. Osteoarthr. Cartil 2000, 8, 386–392. [Google Scholar]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther 2009, 11, 224. [Google Scholar]
- Pulai, J.I.; Chen, H.; Im, H.J.; Kumar, S.; Hanning, C.; Hegde, P.S.; Loeser, R.F. NF-κB mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J. Immunol 2005, 174, 5781–5788. [Google Scholar]
- Floman, Y.; Eyre, D.R.; Glimcher, M.J. Induction of osteoarthrosis in the rabbit knee joint: Biochemical studies on the articular cartilage. Clin. Orthop. Relat. Res 1980, 147, 278–286. [Google Scholar]
- Eyre, D.R.; McDevitt, C.A.; Billingham, M.E.; Muir, H. Biosynthesis of collagen and other matrix proteins by articular cartilage in experimental osteoarthrosis. Biochem. J 1980, 188, 823–837. [Google Scholar]
- Kraus, V.B.; Burnett, B.; Coindreau, J.; Cottrell, S.; Eyre, D.; Gendreau, M.; Gardiner, J.; Garnero, P.; Hardin, J.; Henrotin, Y.; et al. Application of biomarkers in the development of drugs intended for the treatment of osteoarthritis. Osteoarthr. Cartil 2011, 19, 515–542. [Google Scholar]
- Rousseau, J.; Garnero, P. Biological markers in osteoarthritis. Bone 2012, 51, 265–277. [Google Scholar]
- Henrotin, Y.; Chevalier, X.; Deberg, M.; Balblanc, J.C.; Richette, P.; Mulleman, D.; Maillet, B.; Rannou, F.; Piroth, C.; Mathieu, P.; et al. Early decrease of serum biomarkers of type II collagen degradation (Coll 2–1 and joint inflammation (Coll 2–1 NO2 ) by hyaluronic acid intra-articular injections in patients with knee osteoarthritis: A research study part of the Biovisco study. J. Orthop. Res 2013, 31, 901–907. [Google Scholar]
- Nemirovskiy, O.V.; Sunyer, T.; Aggarwal, P.; Abrams, M.; Hellio, Le; Graverand, M.P.; Mathews, W.R. Discovery and development of the N-terminal procollagen type II (NPII) biomarker: A tool for measuring collagen type II synthesis. Osteoarthr. Cartil 2008, 16, 1494–1500. [Google Scholar]
- Lorenz, H.; Wenz, W.; Ivancic, M.; Steck, E.; Richter, W. Early and stable upregulation of collagen type II, collagen type I and YKL40 expression levels in cartilage during early experimental osteoarthritis occurs independent of joint location and histological grading. Arthritis Res. Ther 2005, 7, R156–R165. [Google Scholar]
- Matyas, J.R.; Atley, L.; Ionescu, M.; Eyre, D.R.; Poole, A.R. Analysis of cartilage biomarkers in the early phases of canine experimental osteoarthritis. Arthritis Rheum 2004, 50, 543–552. [Google Scholar]
- Pavelka, K.; Forejtova, S.; Olejarova, M.; Gatterova, J.; Senolt, L.; Spacek, P.; Braun, M.; Hulejova, M.; Stovickova, J.; Pavelkova, A. Hyaluronic acid levels may have predictive value for the progression of knee osteoarthritis. Osteoarthr. Cartil 2004, 12, 277–283. [Google Scholar]
- Bettica, P.; Cline, G.; Hart, D.J.; Meyer, J.; Spector, T.D. Evidence for increased bone resorption in patients with progressive knee osteoarthritis: Longitudinal results from the Chingford study. Arthritis Rheum 2002, 46, 3178–3184. [Google Scholar]
- Seibel, M.J. Biochemical markers of bone turnover. Part I: Biochemistry and variability. Clin. Biochem. Rev 2005, 26, 97–122. [Google Scholar]
- Senolt, L.; Pavelka, K. Molecular markers of osteoarthritis (in Czech). Acta Chir. Orthop. Traumatol. Cech 2005, 72, 191–196. [Google Scholar]
- Buckland-Wright, C. Subchondral bone changes in hand and knee osteoarthritis detected by radiography. Osteoarthr. Cartil 2004, 12, S10–S19. [Google Scholar]
- Thambyah, A.; Broom, N. On new bone formation in the pre-osteoarthritic joint. Osteoarthr. Cartil 2009, 17, 456–463. [Google Scholar]
- Brown, T.D.; Vrahas, M.S. The apparent elastic modulus of the juxtarticular subchondral bone of the femoral head. J. Orthop. Res 1984, 2, 32–38. [Google Scholar]
- Brandt, K.D.; Myers, S.L.; Burr, D.; Albrecht, M. Osteoarthritic changes in canine articular cartilage, subchondral bone, and synovium fifty-four months after transection of the anterior cruciate ligament. Arthritis Rheum 1991, 34, 1560–1570. [Google Scholar]
- Dedrick, D.K.; Goldstein, S.A.; Brandt, K.D.; O’Connor, B.L.; Goulet, R.W.; Albrecht, M. A longitudinal study of subchondral plate and trabecular bone in cruciate-deficient dogs with osteoarthritis followed up for 54 months. Arthritis Rheum 1993, 36, 1460–1467. [Google Scholar]
- Burr, D.B.; Schaffler, M.B. The involvement of subchondral mineralized tissues in osteoarthrosis: Quantitative microscopic evidence. Microsc. Res. Tech 1997, 37, 343–357. [Google Scholar]
- Bailey, A.J.; Mansell, J.P.; Sims, T.J.; Banse, X. Biochemical and mechanical properties of subchondral bone in osteoarthritis. Biorheology 2004, 41, 349–358. [Google Scholar]
- Sanchez, C.; Deberg, M.A.; Bellahcene, A.; Castronovo, V.; Msika, P.; Delcour, J.P.; Crielaard, J.M.; Henrotin, Y.E. Phenotypic characterization of osteoblasts from the sclerotic zones of osteoarthritic subchondral bone. Arthritis Rheum 2008, 58, 442–455. [Google Scholar]
- Truong, L.H.; Kuliwaba, J.S.; Tsangari, H.; Fazzalari, N.L. Differential gene expression of bone anabolic factors and trabecular bone architectural changes in the proximal femoral shaft of primary hip osteoarthritis patients. Arthritis Res. Ther 2006, 8, R188:1–R188:12. [Google Scholar]
- Hilal, G.; Martel-Pelletier, J.; Pelletier, J.P.; Ranger, P.; Lajeunesse, D. Osteoblast-like cells from human subchondral osteoarthritic bone demonstrate an altered phenotype in vitro: Possible role in subchondral bone sclerosis. Arthritis Rheum 1998, 41, 891–899. [Google Scholar]
- Day, J.S.; Ding, M.; van der Linden, J.C.; Hvid, I.; Sumner, D.R.; Weinans, H. A decreased subchondral trabecular bone tissue elastic modulus is associated with pre-arthritic cartilage damage. J. Orthop. Res 2001, 19, 914–918. [Google Scholar]
- Burr, D.B.; Radin, E.L. Microfractures and microcracks in subchondral bone: Are they relevant to osteoarthrosis? Rheum. Dis. Clin. N. Am 2003, 29, 675–685. [Google Scholar]
- Lajeunesse, D.; Reboul, P. Subchondral bone in osteoarthritis: A biologic link with articular cartilage leading to abnormal remodeling. Curr. Opin. Rheumatol 2003, 15, 628–633. [Google Scholar]
- Westacott, C.I.; Webb, G.R.; Warnock, M.G.; Sims, J.V.; Elson, C.J. Alteration of cartilage metabolism by cells from osteoarthritic bone. Arthritis Rheum 1997, 40, 1282–1291. [Google Scholar]
- Imhof, H.; Breitenseher, M.; Kainberger, F.; Rand, T.; Trattnig, S. Importance of subchondral bone to articular cartilage in health and disease. Top. Magn. Reson. Imaging 1999, 10, 180–192. [Google Scholar]
- Malinin, T.; Ouellette, E.A. Articular cartilage nutrition is mediated by subchondral bone: A long-term autograft study in baboons. Osteoarthr. Cartil 2000, 8, 483–491. [Google Scholar]
- Amin, A.K.; Huntley, J.S.; Simpson, A.H.; Hall, A.C. Chondrocyte survival in articular cartilage: The influence of subchondral bone in a bovine model. J. Bone Joint Surg. Br. Vol 2009, 91, 691–699. [Google Scholar]
- Jiao, K.; Niu, L.N.; Wang, M.Q.; Dai, J.; Yu, S.B.; Liu, X.D.; Wang, J. Subchondral bone loss following orthodontically induced cartilage degradation in the mandibular condyles of rats. Bone 2011, 48, 362–371. [Google Scholar]
- Johnstone, E.W.; Leane, P.B.; Byers, S.; Hopwood, J.J.; Foster, B.K. Metaphyseal factors promote calcium incorporation in physeal chondrocyte cultures. J. Orthop. Sci 2000, 5, 593–599. [Google Scholar]
- Moreno-Rubio, J.; Herrero-Beaumont, G.; Tardio, L.; Alvarez-Soria, M.A.; Largo, R. Nonsteroidal antiinflammatory drugs and prostaglandin E2 modulate the synthesis of osteoprotegerin and RANKL in the cartilage of patients with severe knee osteoarthritis. Arthritis Rheum 2010, 62, 478–488. [Google Scholar]
- Nurminskaya, M.; Magee, C.; Faverman, L.; Linsenmayer, T.F. Chondrocyte-derived transglutaminase promotes maturation of preosteoblasts in periosteal bone. Dev. Biol 2003, 263, 139–152. [Google Scholar]
- Benito, M.J.; Veale, D.J.; FitzGerald, O.; van den Berg, W.B.; Bresnihan, B. Synovial tissue inflammation in early and late osteoarthritis. Ann. Rheum. Dis 2005, 64, 1263–1267. [Google Scholar]
- Loeser, R.F. Molecular mechanisms of cartilage destruction: Mechanics, inflammatory mediators, and aging collide. Arthritis Rheum 2006, 54, 1357–1360. [Google Scholar]
- Brown, R.A.; Tomlinson, I.W.; Hill, C.R.; Weiss, J.B.; Phillips, P.; Kumar, S. Relationship of angiogenesis factor in synovial fluid to various joint diseases. Ann. Rheum. Dis 1983, 42, 301–307. [Google Scholar]
- Sanchez, C.; Deberg, M.A.; Piccardi, N.; Msika, P.; Reginster, J.Y.; Henrotin, Y.E. Osteoblasts from the sclerotic subchondral bone downregulate aggrecan but upregulate metalloproteinases expression by chondrocytes. This effect is mimicked by interleukin-6, -1β and oncostatin M pre-treated non-sclerotic osteoblasts. Osteoarthr. Cartil 2005, 13, 979–987. [Google Scholar]
- Botter, S.M.; Glasson, S.S.; Hopkins, B.; Clockaerts, S.; Weinans, H.; van Leeuwen, J.P.; van Osch, G.J. ADAMTS5−/− mice have less subchondral bone changes after induction of osteoarthritis through surgical instability: Implications for a link between cartilage and subchondral bone changes. Osteoarthr. Cartil 2009, 17, 636–645. [Google Scholar]
- Mansell, J.P.; Collins, C.; Bailey, A.J. Bone, not cartilage, should be the major focus in osteoarthritis. Nat. Clin. Pract. Rheumatol 2007, 3, 306–307. [Google Scholar]
- Upton, A.R.; Holding, C.A.; Dharmapatni, A.A.; Haynes, D.R. The expression of RANKL and OPG in the various grades of osteoarthritic cartilage. Rheumatol. Int 2012, 32, 535–540. [Google Scholar]
- Martinez-Calatrava, M.J.; Prieto-Potin, I.; Roman-Blas, J.A.; Tardio, L.; Largo, R.; Herrero-Beaumont, G. RANKL synthesized by articular chondrocytes contributes to juxta-articular bone loss in chronic arthritis. Arthritis Res. Ther 2012, 14, R149:1–R149:13. [Google Scholar]
- Sanchez, C.; Deberg, M.A.; Piccardi, N.; Msika, P.; Reginster, J.Y.; Henrotin, Y.E. Subchondral bone osteoblasts induce phenotypic changes in human osteoarthritic chondrocytes. Osteoarthr. Cartil 2005, 13, 988–997. [Google Scholar]
- Mansell, J.P.; Bailey, A.J. Abnormal cancellous bone collagen metabolism in osteoarthritis. J. Clin. Investig 1998, 101, 1596–603. [Google Scholar]
- Bellido, M.; Lugo, L.; Roman-Blas, J.A.; Castaneda, S.; Caeiro, J.R.; Dapia, S.; Calvo, E.; Largo, R.; Herrero-Beaumont, G. Subchondral bone microstructural damage by increased remodelling aggravates experimental osteoarthritis preceded by osteoporosis. Arthritis Res. Ther 2010, 12, R152:1–R152:11. [Google Scholar]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar]
- Hayami, T.; Pickarski, M.; Zhuo, Y.; Wesolowski, G.A.; Rodan, G.A.; Duong le, T. Characterization of articular cartilage and subchondral bone changes in the rat anterior cruciate ligament transection and meniscectomized models of osteoarthritis. Bone 2006, 38, 234–243. [Google Scholar]
- Hayami, T.; Pickarski, M.; Wesolowski, G.A.; McLane, J.; Bone, A.; Destefano, J.; Rodan, G.A.; Duong le, T. The role of subchondral bone remodeling in osteoarthritis: Reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheum 2004, 50, 1193–1206. [Google Scholar]
- Schett, G.; Stolina, M.; Bolon, B.; Middleton, S.; Adlam, M.; Brown, H.; Zhu, L.; Feige, U.; Zack, D.J. Analysis of the kinetics of osteoclastogenesis in arthritic rats. Arthritis Rheum 2005, 52, 3192–3201. [Google Scholar]
- Stolina, M.; Adamu, S.; Ominsky, M.; Dwyer, D.; Asuncion, F.; Geng, Z.; Middleton, S.; Brown, H.; Pretorius, J.; Schett, G.; et al. RANKL is a marker and mediator of local and systemic bone loss in two rat models of inflammatory arthritis. J. Bone Miner. Res 2005, 20, 1756–1765. [Google Scholar]
- Blanquaert, F.; Pereira, R.C.; Canalis, E. Cortisol inhibits hepatocyte growth factor/scatter factor expression and induces c-met transcripts in osteoblasts. Am. J. Physiol. Endocrinol. Metab 2000, 278, E509–E515. [Google Scholar]
- Guevremont, M.; Martel-Pelletier, J.; Massicotte, F.; Tardif, G.; Pelletier, J.P.; Ranger, P.; Lajeunesse, D.; Reboul, P. Human adult chondrocytes express hepatocyte growth factor (HGF) isoforms but not HgF: Potential implication of osteoblasts on the presence of HGF in cartilage. J. Bone Miner. Res 2003, 18, 1073–1081. [Google Scholar]
- Moldovan, F.; Pelletier, J.P.; Hambor, J.; Cloutier, J.M.; Martel-Pelletier, J. Collagenase-3 (matrix metalloprotease 13) is preferentially localized in the deep layer of human arthritic cartilage in situ: In vitro mimicking effect by transforming growth factor β. Arthritis Rheum 1997, 40, 1653–1661. [Google Scholar]
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Seol, J.W.; Park, S.Y. Sphingosine-1-phosphate inhibits interleukin-1β-induced inflammation in human articular chondrocytes. Int. J. Mol. Med 2012, 30, 1451–1458. [Google Scholar]
- Masuko, K.; Murata, M.; Beppu, M.; Nakamura, H.; Kato, T.; Yudoh, K. Sphingosine-1-phosphate modulates expression of vascular endothelial growth factor in human articular chondrocytes: A possible new role in arthritis. Int. J. Rheum. Dis 2012, 15, 366–373. [Google Scholar]
- Lories, R.J.; Luyten, F.P. The bone-cartilage unit in osteoarthritis. Nat. Rev. Rheumatol 2011, 7, 43–49. [Google Scholar]
- Logan, C.Y.; Nusse, R. The WNT signaling pathway in development and disease. Ann. Rev. Cell Dev. Biol 2004, 20, 781–810. [Google Scholar]
- MacDonald, B.T.; Tamai, K.; He, X. WNT/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar]
- Hall, C.L.; Kang, S.; MacDougald, O.A.; Keller, E.T. Role of WNTs in prostate cancer bone metastases. J. Cell. Biochem 2006, 97, 661–672. [Google Scholar]
- Lodewyckx, L.; Lories, R.J. WNT signaling in osteoarthritis and osteoporosis: What is the biological significance for the clinician? Curr. Rheumatol. Rep 2009, 11, 23–30. [Google Scholar]
- Zhu, M.; Tang, D.; Wu, Q.; Hao, S.; Chen, M.; Xie, C.; Rosier, R.N.; O’Keefe, R.J.; Zuscik, M.; Chen, D. Activation of β-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult β-catenin conditional activation mice. J. Bone Miner. Res 2009, 24, 12–21. [Google Scholar]
- Zhu, M.; Chen, M.; Zuscik, M.; Wu, Q.; Wang, Y.J.; Rosier, R.N.; O’Keefe, R.J.; Chen, D. Inhibition of β-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum 2008, 58, 2053–2064. [Google Scholar]
- Lodewyckx, L.; Luyten, F.P.; Lories, R.J. Genetic deletion of low-density lipoprotein receptor-related protein 5 increases cartilage degradation in instability-induced osteoarthritis. Rheumatology 2012, 51, 1973–1978. [Google Scholar]
- Hoang, B.; Moos, M., Jr.; Vukicevic, S.; Luyten, F.P. Primary structure and tissue distribution of FRZB, a novel protein related to Drosophila frizzled, suggest a role in skeletal morphogenesis. J. Biol. Chem 1996, 271, 26131–26137. [Google Scholar]
- Lories, R.J.; Peeters, J.; Bakker, A.; Tylzanowski, P.; Derese, I.; Schrooten, J.; Thomas, J.T.; Luyten, F.P. Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum 2007, 56, 4095–4103. [Google Scholar]
- Nalesso, G.; Sherwood, J.; Bertrand, J.; Pap, T.; Ramachandran, M.; de Bari, C.; Pitzalis, C.; Dell’accio, F. WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J. Cell Biol 2011, 193, 551–564. [Google Scholar] [Green Version]
- Ryu, J.H.; Chun, J.S. Opposing roles of WNT-5A and WNT-11 in interleukin-1beta regulation of type II collagen expression in articular chondrocytes. J. Biol. Chem 2006, 281, 22039–22047. [Google Scholar]
- Church, V.; Nohno, T.; Linker, C.; Marcelle, C.; Francis-West, P. WNT regulation of chondrocyte differentiation. J. Cell Sci 2002, 115, 4809–4818. [Google Scholar]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med 2013, 19, 179–192. [Google Scholar]
- Jenkins, Z.A.; van Kogelenberg, M.; Morgan, T.; Jeffs, A.; Fukuzawa, R.; Pearl, E.; Thaller, C.; Hing, A.V.; Porteous, M.E.; Garcia-Minaur, S.; et al. Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat. Genet 2009, 41, 95–100. [Google Scholar]
- Lyons, T.J.; McClure, S.F.; Stoddart, R.W.; McClure, J. The normal human chondro-osseous junctional region: Evidence for contact of uncalcified cartilage with subchondral bone and marrow spaces. BMC Musculoskelet. Disord 2006. [Google Scholar]
- Chan, B.Y.; Fuller, E.S.; Russell, A.K.; Smith, S.M.; Smith, M.M.; Jackson, M.T.; Cake, M.A.; Read, R.A.; Bateman, J.F.; Sambrook, P.N.; et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthr. Cartil 2011, 19, 874–885. [Google Scholar]
- Leijten, J.C.; Emons, J.; Sticht, C.; van Gool, S.; Decker, E.; Uitterlinden, A.; Rappold, G.; Hofman, A.; Rivadeneira, F.; Scherjon, S.; et al. Gremlin 1, frizzled-related protein, and Dkk-1 are key regulators of human articular cartilage homeostasis. Arthritis Rheum 2012, 64, 3302–3312. [Google Scholar]
- Ijiri, K.; Nagayoshi, R.; Matsushita, N.; Tsuruga, H.; Taniguchi, N.; Gushi, A.; Sakakima, H.; Komiya, S.; Matsuyama, T. Differential expression patterns of secreted frizzled related protein genes in synovial cells from patients with arthritis. J. Rheumatol 2002, 29, 2266–2270. [Google Scholar]
- Weng, L.H.; Wang, C.J.; Ko, J.Y.; Sun, Y.C.; Wang, F.S. Control of Dkk-1 ameliorates chondrocyte apoptosis, cartilage destruction, and subchondral bone deterioration in osteoarthritic knees. Arthritis Rheum 2010, 62, 1393–1402. [Google Scholar]
- Dell’accio, F.; de Bari, C.; Eltawil, N.M.; Vanhummelen, P.; Pitzalis, C. Identification of the molecular response of articular cartilage to injury, by microarray screening: WNT-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum 2008, 58, 1410–1421. [Google Scholar]
- Blom, A.B.; Brockbank, S.M.; van Lent, P.L.; van Beuningen, H.M.; Geurts, J.; Takahashi, N.; van der Kraan, P.M.; van de Loo, F.A.; Schreurs, B.W.; Clements, K.; et al. Involvement of the WNT signaling pathway in experimental and human osteoarthritis: Prominent role of WNT-induced signaling protein 1. Arthritis Rheum 2009, 60, 501–512. [Google Scholar]
- Kuliwaba, J.S.; Findlay, D.M.; Atkins, G.J.; Forwood, M.R.; Fazzalari, N.L. Enhanced expression of osteocalcin mRNA in human osteoarthritic trabecular bone of the proximal femur is associated with decreased expression of interleukin-6 and interleukin-11 mRNA. J. Bone Miner. Res 2000, 15, 332–341. [Google Scholar]
- Golovchenko, S.; Hattori, T.; Hartmann, C.; Gebhardt, M.; Gebhard, S.; Hess, A.; Pausch, F.; Schlund, B.; von der Mark, K. Deletion of beta catenin in hypertrophic growth plate chondrocytes impairs trabecular bone formation. Bone 2013, 55, 102–112. [Google Scholar]
- Blaney Davidson, E.N.; van der Kraan, P.M.; van den Berg, W.B. TGF-β and osteoarthritis. Osteoarthr. Cartil 2007, 15, 597–604. [Google Scholar]
- Goldring, M.B.; Tsuchimochi, K.; Ijiri, K. The control of chondrogenesis. J. Cell. Biochem 2006, 97, 33–44. [Google Scholar]
- Zhao, L.; Li, G.; Zhou, G.Q. SOX9 directly binds CREB as a novel synergism with the PKA pathway in BMP-2-induced osteochondrogenic differentiation. J. Bone Miner. Res 2009, 24, 826–836. [Google Scholar]
- Retting, K.N.; Song, B.; Yoon, B.S.; Lyons, K.M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093–1104. [Google Scholar]
- Chen, A.L.; Fang, C.; Liu, C.; Leslie, M.P.; Chang, E.; Di Cesare, P.E. Expression of bone morphogenetic proteins, receptors, and tissue inhibitors in human fetal, adult, and osteoarthritic articular cartilage. J. Orthop. Res 2004, 22, 1188–1192. [Google Scholar]
- Blaney Davidson, E.N.; Vitters, E.L.; van Lent, P.L.; van de Loo, F.A.; van den Berg, W.B.; van der Kraan, P.M. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Res. Ther 2007, 9, R102:1–R102:11. [Google Scholar]
- Lories, R.J.; Daans, M.; Derese, I.; Matthys, P.; Kasran, A.; Tylzanowski, P.; Ceuppens, J.L.; Luyten, F.P. Noggin haploinsufficiency differentially affects tissue responses in destructive and remodeling arthritis. Arthritis Rheum 2006, 54, 1736–1746. [Google Scholar]
- Wu, X.; Shi, W.; Cao, X. Multiplicity of BMP signaling in skeletal development. Ann. N. Y. Acad. Sci 2007, 1116, 29–49. [Google Scholar]
- Tchetina, E.V.; Squires, G.; Poole, A.R. Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J. Rheumatol 2005, 32, 876–886. [Google Scholar]
- Horiki, M.; Imamura, T.; Okamoto, M.; Hayashi, M.; Murai, J.; Myoui, A.; Ochi, T.; Miyazono, K.; Yoshikawa, H.; Tsumaki, N. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J. Cell Biol 2004, 165, 433–445. [Google Scholar]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; van den Berg, W.B. Bone morphogenetic proteins and articular cartilage: To serve and protect or a wolf in sheep clothing’s? Osteoarthr. Cartil 2010, 18, 735–741. [Google Scholar]
- Yamaguchi, A.; Komori, T.; Suda, T. Regulation of osteoblast differentiation mediated by bone morphogenetic proteins, hedgehogs, and Cbfa1. Endocr. Rev 2000, 21, 393–411. [Google Scholar]
- Daans, M.; Luyten, F.P.; Lories, R.J. GDF5 deficiency in mice is associated with instability-driven joint damage, gait and subchondral bone changes. Ann. Rheum. Dis 2011, 70, 208–213. [Google Scholar]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-β/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol 2001, 153, 35–46. [Google Scholar]
- Scharstuhl, A.; Glansbeek, H.L.; van Beuningen, H.M.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Inhibition of endogenous TGF-β during experimental osteoarthritis prevents osteophyte formation and impairs cartilage repair. J. Immunol 2002, 169, 507–514. [Google Scholar]
- Scharstuhl, A.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Reduction of osteophyte formation and synovial thickening by adenoviral overexpression of transforming growth factor beta/bone morphogenetic protein inhibitors during experimental osteoarthritis. Arthritis Rheum 2003, 48, 3442–3451. [Google Scholar]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med 2009, 15, 757–765. [Google Scholar]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med 2013, 19, 704–712. [Google Scholar]
- Tardif, G.; Pelletier, J.P.; Boileau, C.; Martel-Pelletier, J. The BMP antagonists follistatin and gremlin in normal and early osteoarthritic cartilage: An immunohistochemical study. Osteoarthr. Cartil 2009, 17, 263–270. [Google Scholar]
- Inkson, C.A.; Ono, M.; Kuznetsov, S.A.; Fisher, L.W.; Robey, P.G.; Young, M.F. TGF-β1 and WISP-1/CCN-4 can regulate each other’s activity to cooperatively control osteoblast function. J. Cell. Biochem 2008, 104, 1865–1878. [Google Scholar]
- Oh, C.D.; Chang, S.H.; Yoon, Y.M.; Lee, S.J.; Lee, Y.S.; Kang, S.S.; Chun, J.S. Opposing role of mitogen-activated protein kinase subtypes, ERK-1/2 and p38, in the regulation of chondrogenesis of mesenchymes. J. Biol. Chem 2000, 275, 5613–5619. [Google Scholar]
- Stanton, L.A.; Underhill, T.M.; Beier, F. MAP kinases in chondrocyte differentiation. Dev. Biol 2003, 263, 165–175. [Google Scholar]
- Greenblatt, M.B.; Shim, J.H.; Glimcher, L.H. Mitogen-activated protein kinase pathways in osteoblasts. Annu. Rev. Cell Dev. Biol 2013. [Google Scholar] [CrossRef]
- Sondergaard, B.C.; Schultz, N.; Madsen, S.H.; Bay-Jensen, A.C.; Kassem, M.; Karsdal, M.A. MAPKs are essential upstream signaling pathways in proteolytic cartilage degradation-divergence in pathways leading to aggrecanase and MMP-mediated articular cartilage degradation. Osteoarthr. Cartil 2010, 18, 279–288. [Google Scholar]
- Yang, C.M.; Chien, C.S.; Yao, C.C.; Hsiao, L.D.; Huang, Y.C.; Wu, C.B. Mechanical strain induces collagenase-3 (MMP-13) expression in MC3T3-E1 osteoblastic cells. J. Biol. Chem 2004, 279, 22158–22165. [Google Scholar]
- Prasadam, I.; Friis, T.; Shi, W.; van Gennip, S.; Crawford, R.; Xiao, Y. Osteoarthritic cartilage chondrocytes alter subchondral bone osteoblast differentiation via MAPK signalling pathway involving ERK1/2. Bone 2010, 46, 226–235. [Google Scholar]
- Prasadam, I.; van Gennip, S.; Friis, T.; Shi, W.; Crawford, R.; Xiao, Y. ERK-1/2 and p38 in the regulation of hypertrophic changes of normal articular cartilage chondrocytes induced by osteoarthritic subchondral osteoblasts. Arthritis Rheum 2010, 62, 1349–1360. [Google Scholar] [Green Version]
- Prasadam, I.; Crawford, R.; Xiao, Y. Aggravation of ADAMTS and matrix metalloproteinase production and role of ERK1/2 pathway in the interaction of osteoarthritic subchondral bone osteoblasts and articular cartilage chondrocytes—Possible pathogenic role in osteoarthritis. J. Rheumatol 2012, 39, 621–634. [Google Scholar]
- Hollenberg, M.D.; Compton, S.J. International union of pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol. Rev 2002, 54, 203–217. [Google Scholar]
- Boileau, C.; Amiable, N.; Martel-Pelletier, J.; Fahmi, H.; Duval, N.; Pelletier, J.P. Activation of proteinase-activated receptor 2 in human osteoarthritic cartilage upregulates catabolic and proinflammatory pathways capable of inducing cartilage degradation: A basic science study. Arthritis Res. Ther 2007, 9, R121:1–R121:10. [Google Scholar]
- Amiable, N.; Tat, S.K.; Lajeunesse, D.; Duval, N.; Pelletier, J.P.; Martel-Pelletier, J.; Boileau, C. Proteinase-activated receptor (PAR)-2 activation impacts bone resorptive properties of human osteoarthritic subchondral bone osteoblasts. Bone 2009, 44, 1143–1150. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sharma, A.R.; Jagga, S.; Lee, S.-S.; Nam, J.-S. Interplay between Cartilage and Subchondral Bone Contributing to Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2013, 14, 19805-19830. https://doi.org/10.3390/ijms141019805
Sharma AR, Jagga S, Lee S-S, Nam J-S. Interplay between Cartilage and Subchondral Bone Contributing to Pathogenesis of Osteoarthritis. International Journal of Molecular Sciences. 2013; 14(10):19805-19830. https://doi.org/10.3390/ijms141019805
Chicago/Turabian StyleSharma, Ashish R., Supriya Jagga, Sang-Soo Lee, and Ju-Suk Nam. 2013. "Interplay between Cartilage and Subchondral Bone Contributing to Pathogenesis of Osteoarthritis" International Journal of Molecular Sciences 14, no. 10: 19805-19830. https://doi.org/10.3390/ijms141019805