Mechanisms of Chemical Carcinogenesis in the Kidneys


Abstract
:1. Cancer of the Kidney
2. Renal Cell Carcinoma
3. Chemical Carcinogenicity
4. Susceptibility of the Kidneys to Chemical Carcinogenesis
4.1. Concentrating Effect
4.2. Xenobiotic Metabolizing Capability—Bio-Activation of Pro-Carcinogens
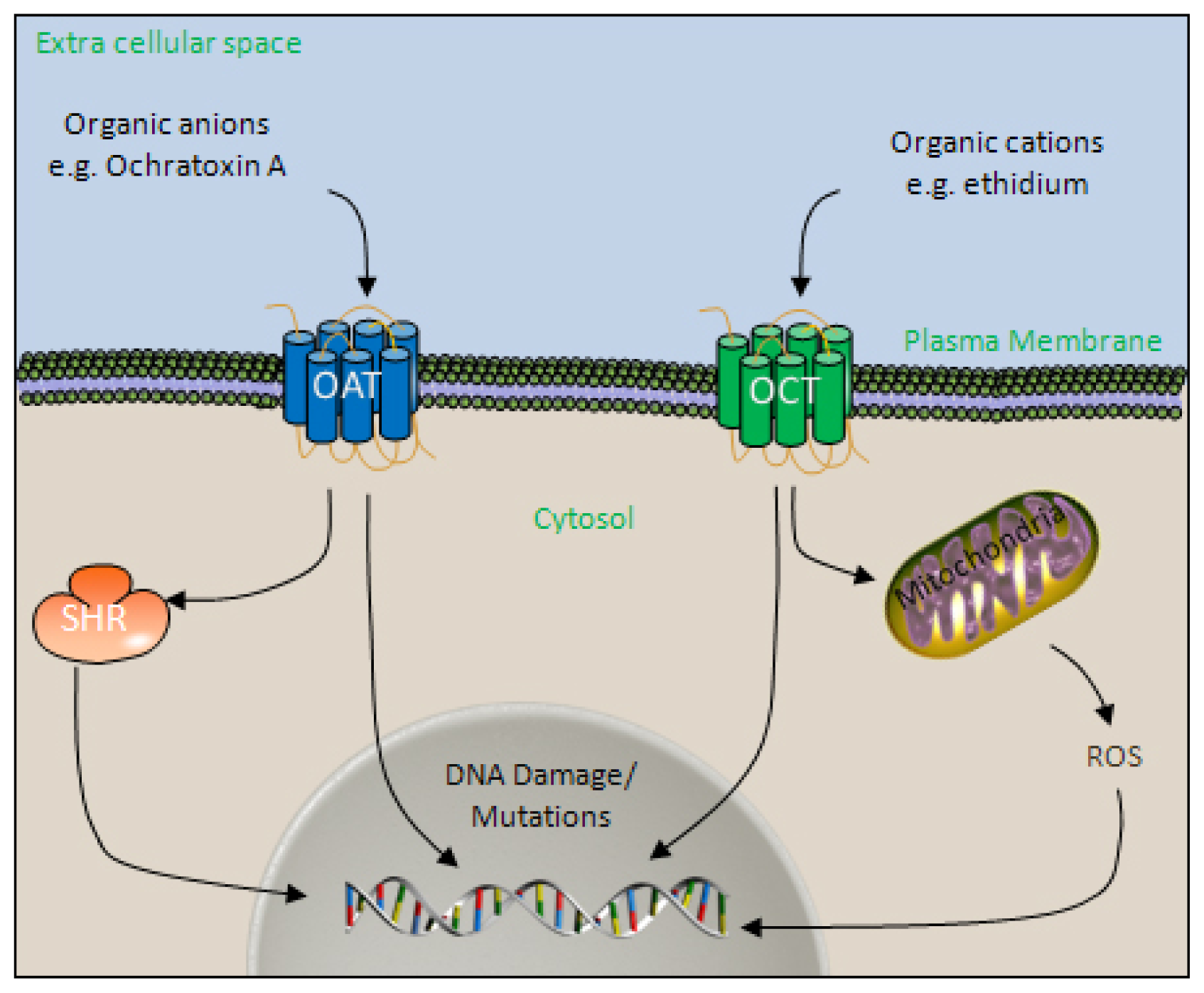
4.3. The Organic Ion Transport Systems
5. Mechanisms of Chemical Carcinogenicity
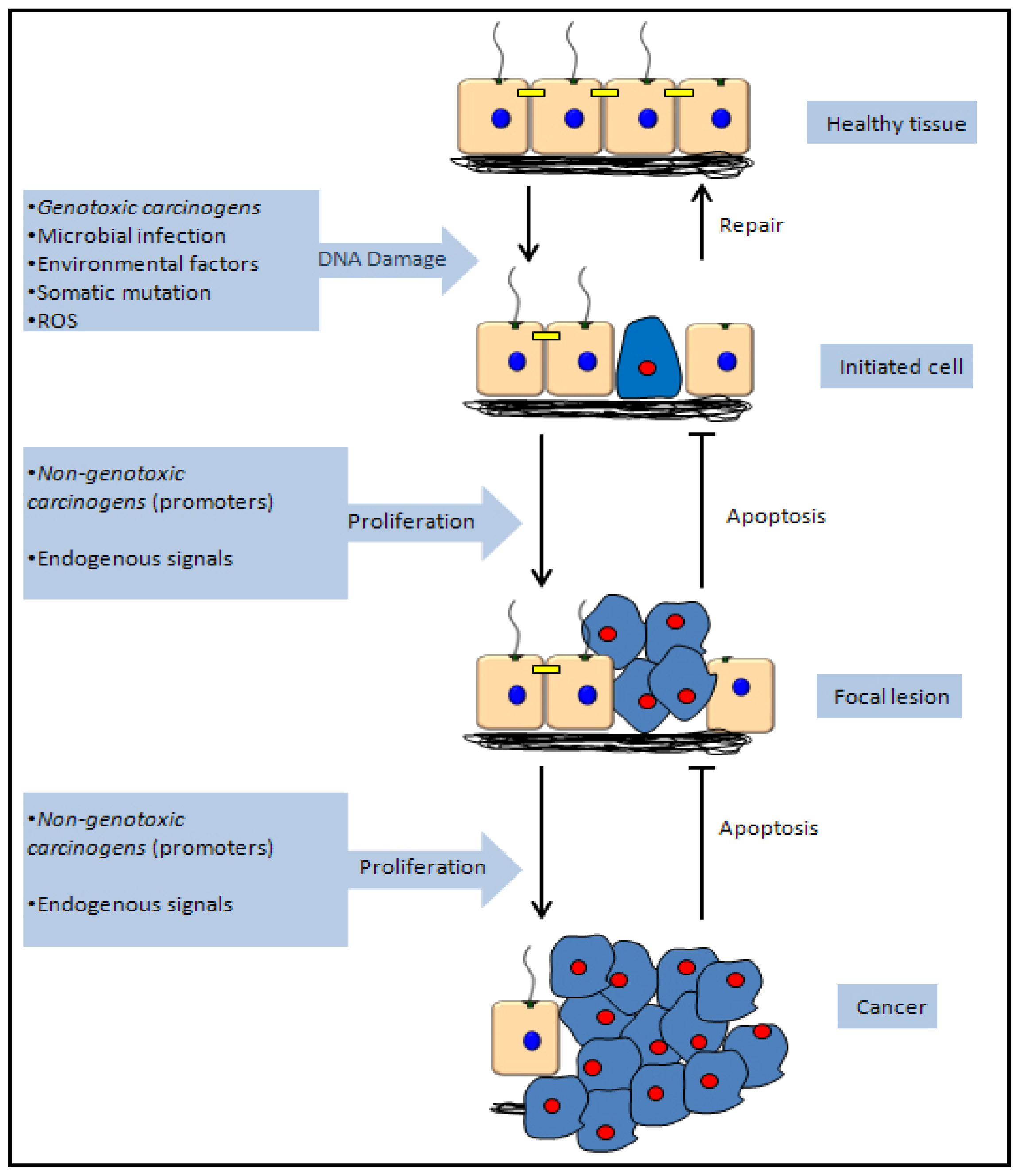
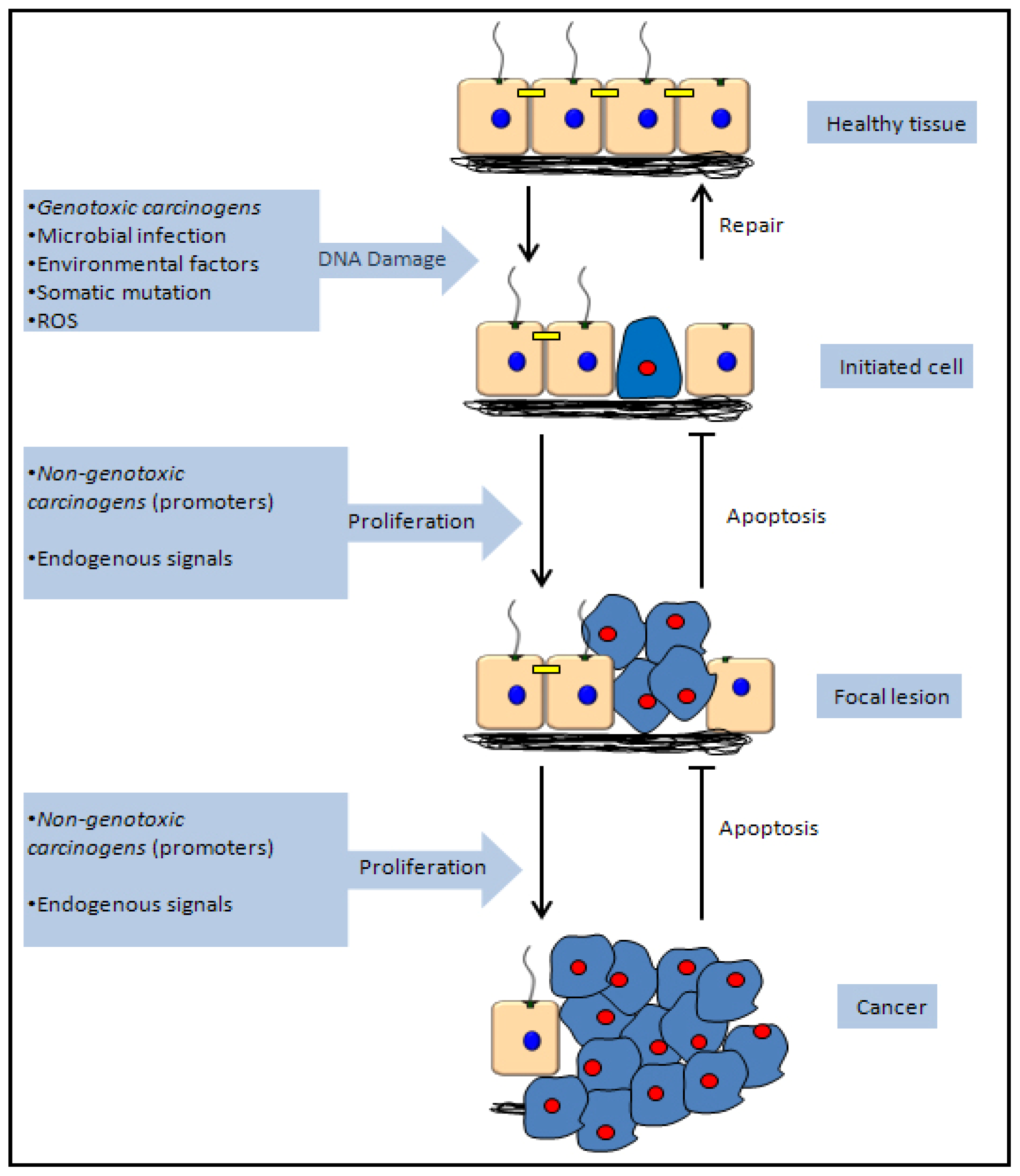
5.1. Genotoxic Carcinogenicity
5.2. Non-Genotoxic Carcinogenicity
6. Renal Carcinogens
6.1. Ochratoxin-A
6.2. Potassium Bromate
6.3. Aristolochic Acid
7. Screening for Chemical Carcinogens
8. Conclusions and Future Studies

| Site | Compound | Possible source of exposure | IARC Classification |
|---|---|---|---|
| Kidney | 2-Nitrofluorene | By-product of combustion | 2B |
| Aristolochic Acid | Use of aristolochia in herbal medicine | 1 | |
| Arsenic | Contamination of drinking water | 2A | |
| Benzo[a]pyrene | Motor vehicle exhaust fumes | 2A | |
| Bromodichloromethane | Chlorination of drinking water | 2B | |
| Cadmium | Cigarette smoking, industrial activity | 2A | |
| Chlorothalonil | Fungicide | 2A | |
| N-Nitrosomorpholine | Rubber manufacturing | 2B | |
| Ochratoxin A | Fungal contamination | 2B | |
| Potassium Bromate | By-product of water bromination | 2B | |
| Streptozotocin | Fungal contamination | 2B | |
| Renal pelvis and ureter | Aristolochic acid | Aristolochia species | 1 |
| Phenacetin (and Phenacetin containing mixtures) | Non-steroidal anti-inflammatory medication | 2A | |
| Urinary bladder | 2-Napthylamine | Cigarette smoking, industrial exposure | 1 |
| 4-Aminobiphenyl | Cigarette smoking, industrial exposure | 1 | |
| Arsenic | Contamination of drinking water | 1 | |
| Benzidine | Benzidine based dyes | 1 | |
| Chlornaphazine | Discontinued pharmaceutical agent | 1 | |
| Cyclophosphamide | Chemotheraputic pharmaceutical | 1 | |
| Ortho-Toluidine | Industrial/laboratory exposure | 1 | |

{kind=link}
{kind=link}
Acknowledgments
Conflicts of Interest
References
- Ljungberg, B.; Campbell, S.C.; Choi, H.Y.; Jacqmin, D.; Lee, J.E.; Weikert, S.; Kiemeney, L.A. The epidemiology of renal cell carcinoma. Eur. Urol 2011, 60, 615–621. [Google Scholar]
- Levi, F.; Lucchini, F.; Negri, E.; La Vecchia, C. Declining mortality from kidney cancer in Europe. Ann. Oncol 2004, 15, 1130–1135. [Google Scholar]
- Ng, C.S.; Wood, C.G.; Silverman, P.M.; Tannir, N.M.; Tamboli, P.; Sandler, C.M. Renal cell carcinoma: Diagnosis, staging, and surveillance. AJR Am. J. Roentgenol 2008, 191, 1220–1232. [Google Scholar]
- Eble, J.N. Pathology and genetics of tumours of the urinary system and male genital organs. BJU Int 2004, 94, 675. [Google Scholar]
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G., Jr. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 1995, 269, 1444–1446. [Google Scholar]
- Pause, A.; Lee, S.; Worrell, R.A.; Chen, D.Y.T.; Burgess, W.H.; Linehan, W.M.; Klausner, R.D. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 2156–2161. [Google Scholar]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar]
- Wiesener, M.S.; Münchenhagen, P.M.; Berger, I.; Morgan, N.V.; Roigas, J.; Schwiertz, A.; Jürgensen, J.S.; Gruber, G.; Maxwell, P.H.; Löning, S.A.; et al. Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1alpha in clear cell renal carcinomas. Cancer Res 2001, 61, 5215–5222. [Google Scholar]
- Krieg, M.; Haas, R.; Brauch, H.; Acker, T.; Flamme, I.; Plate, K.H. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 2000, 19, 5435–5443. [Google Scholar]
- Pavlovich, C.P.; Schmidt, L.S. Searching for the hereditary causes of renal-cell carcinoma. Nat. Rev. Cancer 2004, 4, 381–393. [Google Scholar]
- Zbar, B.; Klausner, R.; Linehan, W.M. Studying cancer families to identify kidney cancer genes. Annu. Rev. Med 2003, 54, 217–233. [Google Scholar]
- Giménez-Bachs, J.M.; Salinas-Sánchez, A.S.; Sánchez-Sánchez, F.; Lorenzo-Romero, J.G.; Donate-Moreno, M.J.; Pastor-Navarro, H.; Carrión-López, P.; Escribano-Martínez, J.; Virseda-Rodríguez, J.A. VHL protein alterations in sporadic renal cell carcinoma. Clin. Oncol 2007, 19, 784–789. [Google Scholar]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar]
- Firth, J.D.; Ebert, B.L.; Ratcliffe, P.J. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J. Biol. Chem 1995, 270, 21021–21027. [Google Scholar]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem 2006, 281, 9030–9037. [Google Scholar]
- Rozhin, J.; Sameni, M.; Ziegler, G.; Sloane, B.F. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Res 1994, 54, 6517–6525. [Google Scholar]
- Taipale, J.; Keski-Oja, J. Growth factors in the extracellular matrix. FASEB J 1997, 11, 51–59. [Google Scholar]
- Dallas, S.L.; Rosser, J.L.; Mundy, G.R.; Bonewald, L.F. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem 2002, 277, 21352–21360. [Google Scholar]
- European Commission Health and Consumer Directorate-General. Draft Report on Alternative (Non-animal) Methods for Cosmetics Testing: Current Status and Future Prospects—2010. Available online: http://ec.europa.eu/consumers/sectors/cosmetics/files/pdf/animal_testing/introduction_public_consultation_at_en.pdf (accessed on 25 April 2013).
- Choudhary, D.; Jansson, I.; Stoilov, I.; Sarfarazi, M.; Schenkman, J.B. Expression patterns of mouse and human CYP orthologs (families 1–4 during development and in different adult tissues. Arch. Biochem. Biophys 2005, 436, 50–61. [Google Scholar]
- Uppstad, H.; Øvrebø, S.; Haugen, A.; Mollerup, S. Importance of CYP1A1 and CYP1B1 in bioactivation of benzo[a]pyrene in human lung cell lines. Toxicol. Lett 2010, 192, 221–228. [Google Scholar]
- Plottner, S.; Plöttner, S.; Borza, A.; Wolf, A.; Bolt, H.M.; Kuhlmann, J.; Föllmann, W. Evaluation of time dependence and interindividual differences in benzo[a]pyrene-mediated CYP1A1 induction and genotoxicity in porcine urinary bladder cell cultures. J. Toxicol. Environ. Health Part A 2008, 71, 969–975. [Google Scholar]
- Chen, M.; Gong, L.; Qi, X.; Xing, G.; Luan, Y.; Wu, Y.; Xiao, Y.; Yao, J.; Li, Y.; Xue, X. Inhibition of renal NQO1 activity by dicoumarol suppresses nitroreduction of aristolochic acid I and attenuates its nephrotoxicity. Toxicol. Sci 2011, 122, 288–296. [Google Scholar]
- Meinl, W.; Pabel, U.; Osterloh-Quiroz, M.; Hengstler, J.G.; Glatt, H. Human sulphotransferases are involved in the activation of aristolochic acids and are expressed in renal target tissue. Int. J. Cancer 2006, 118, 1090–1097. [Google Scholar]
- Berkhin, E.B.; Humphreys, M.H. Regulation of renal tubular secretion of organic compounds. Kidney Int 2001, 59, 17–30. [Google Scholar]
- Launay-Vacher, V.; Izzedine, H.; Karie, S.; Hulot, J.S.; Baumelou, A.; Deray, G. Renal tubular drug transporters. Nephron. Physiol 2006, 103, 97–106. [Google Scholar]
- Dickman, K.G.; Sweet, D.H.; Bonala, R.; Ray, T.; Wu, A. Physiological and molecular characterization of aristolochic acid transport by the kidney. J. Pharmacol. Exp. Ther 2011, 338, 588–597. [Google Scholar]
- Lee, W.-K.; Reichold, M.; Edemir, B.; Ciarimboli, G.; Warth, R.; Koepsell, H.; Thévenod, F. Organic cation transporters OCT1, 2, and 3 mediate high-affinity transport of the mutagenic vital dye ethidium in the kidney proximal tubule. Am. J. Physiol. Renal Physiol 2009, 296, F1504–F1513. [Google Scholar]
- Williams, G.M. Mechanisms of chemical carcinogenesis and application to human cancer risk assessment. Toxicology 2001, 166, 3–10. [Google Scholar]
- Enoch, S.J.; Cronin, M.T.D. A review of the electrophilic reaction chemistry involved in covalent DNA binding. Crit. Rev. Toxicol 2010, 40, 728–748. [Google Scholar]
- Casarett, L.J.; Doull, J.; Klaassen, C.D. Casarett and Doull’s Toxicology: The Basic Science of Poisons; McGraw Hill Professional: New York, NY, USA, 2008. [Google Scholar]
- Pomorski, L.; Bartos, M.; Okruszek, A.; Matejkowska, M.; Tazbir, J.; Kuzdak, K. Carcinogenic effect of combined administration of 2,4-diaminoanisole sulfate, 4,4′-thiodianiline and N,N′-diethylthiourea in male Wistar rats. Neoplasma 2002, 49, 247–250. [Google Scholar]
- Aubé, M.; Larochelle, C.; Ayotte, P. 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene (p,p′-DDE) disrupts the estrogen-androgen balance regulating the growth of hormone-dependent breast cancer cells. Breast Cancer Res 2008, 10, R16. [Google Scholar]
- Van Kesteren, P.C.E.; Beems, R.B.; Luijten, M.; Robinson, J.; de Vries, A.; van Steeg, H. DNA repair-deficient Xpa/p53 knockout mice are sensitive to the non-genotoxic carcinogen cyclosporine A: Escape of initiated cells from immunosurveillance? Carcinogenesis 2009, 30, 538–543. [Google Scholar]
- Xu, J.; Walsha, S.B.; Verneya, Z.M.; Kopelovichc, L.; Elmetsa, C.A.; Athar, M. Procarcinogenic effects of cyclosporine A are mediated through the activation of TAK1/TAB1 signaling pathway. Biochem. Biophys. Res. Commun 2011, 408, 363–368. [Google Scholar]
- Sai, K.; Kangb, K.; Hirosec, A.; Hasegawac, R.; Troskod, J.E.; Inoue, T. Inhibition of apoptosis by pentachlorophenol in v-myc-transfected rat liver epithelial cells: Relation to down-regulation of gap junctional intercellular communication. Cancer Lett 2001, 173, 163–174. [Google Scholar]
- Mally, A.; Chipman, J.K. Non-genotoxic carcinogens: Early effects on gap junctions, cell proliferation and apoptosis in the rat. Toxicology 2002, 180, 233–248. [Google Scholar]
- Hard, G.C. Mechanisms of chemically induced renal carcinogenesis in the laboratory rodent. Toxicol. Pathol 1998, 26, 104–112. [Google Scholar]
- Taniwaki, M.H.; Pitt, J.I.; Teixeira, A.A.; Iamanaka, B.T. The source of ochratoxin A in Brazilian coffee and its formation in relation to processing methods. Int. J. Food Microbiol 2003, 82, 173–179. [Google Scholar]
- Magnoli, C.; Violante, M.; Combina, M.; Palacio, G.; Dalcero, A. Mycoflora and ochratoxin-producing strains of Aspergillus section Nigri in wine grapes in Argentina. Lett. Appl. Microbiol 2003, 37, 179–184. [Google Scholar]
- Copetti, M.V.; Pereira, J.L.; Iamanaka, B.T.; Pitt, J.I.; Taniwaki, M.H. Ochratoxigenic fungi and ochratoxin A in cocoa during farm processing. Int. J. Food Microbiol 2010, 143, 67–70. [Google Scholar]
- Sørensen, L.M.; Mogensen, J.; Nielsen, K.F. Simultaneous determination of ochratoxin A, mycophenolic acid and fumonisin B(2) in meat products. Anal. Bioanal. Chem 2010, 398, 1535–1542. [Google Scholar]
- Juan, C.; Pena, A.; Lino, C.; Moltó, J.C.; Mañes, J. Levels of ochratoxin A in wheat and maize bread from the central zone of Portugal. Int. J. Food Microbiol 2008, 127, 284–289. [Google Scholar]
- Noonim, P.; Mahakarnchanakul, W.; Nielsen, K.F.; Frisvad, J.C.; Samson, R.A. Isolation, identification and toxigenic potential of ochratoxin A-producing Aspergillus species from coffee beans grown in two regions of Thailand. Int. J. Food Microbiol 2008, 128, 197–202. [Google Scholar]
- Martínez-Culebras, P.V.; Crespo-Semperea, A.; Sánchez-Hervása, M.; Elizaquivelb, P.; Aznarb, R.; Ramónb, D. Molecular characterization of the black Aspergillus isolates responsible for ochratoxin A contamination in grapes and wine in relation to taxonomy of Aspergillus section Nigri. Int. J. Food Microbiol 2009, 132, 33–41. [Google Scholar]
- Lock, E.A.; Hard, G.C. Chemically induced renal tubule tumors in the laboratory rat and mouse: Review of the NCI/NTP database and categorization of renal carcinogens based on mechanistic information. Crit. Rev. Toxicol 2004, 34, 211–299. [Google Scholar]
- NTP. Toxicology and Carcinogenesis Studies of Ochratoxin A (CAS No 303-47-9) in F344/N Rats (Gavage Studies). Natl. Toxicol. Program Tech. Rep. Ser 1989, 358, 1–142.
- Boorman, G.A.; McDonald, M.R.; Imoto, S.; Persing, R. Renal lesions induced by ochratoxin A exposure in the F344 rat. Toxicol. Pathol 1992, 20, 236–245. [Google Scholar]
- IARC. International Agency for Research on Cancer (IARC)—Summaries & Evaluations Ochratoxin A (Group 2B). 2003. Available online: http://www.inchem.org/documents/iarc/vol56/13-ochra.html (accessed on 25 April 2013).
- Zanic-Grubisić, T.; Zrinski, R.; Čepelak, I.; Petrik, J.; Radić, B.; Pepeljnjak, S. Studies of ochratoxin A-induced inhibition of phenylalanine hydroxylase and its reversal by phenylalanine. Toxicol. Appl. Pharmacol 2000, 167, 132–139. [Google Scholar]
- Creppy, E.E.; Schlegel, M.; Röschenthaler, R.; Dirheimer, G. Phenylalanine prevents acute poisoning by ochratoxina in mice. Toxicol. Lett 1980, 6, 77–80. [Google Scholar]
- Zepnik, H.; Völkel, W.; Dekant, W. Toxicokinetics of the mycotoxin ochratoxin A in F 344 rats after oral administration. Toxicol. Appl. Pharmacol 2003, 192, 36–44. [Google Scholar]
- Stein, A.F.; Phillips, T.D.; Kubena, L.F.; Harvey, R.B. Renal tubular secretion and reabsorption as factors in ochratoxicosis: Effects of probenecid on nephrotoxicity. J. Toxicol. Environ. Health 1985, 16, 593–605. [Google Scholar]
- Tsuda, M.; Sekine, T.; Takeda, M.; Cha, S.H.; Kanai, Y.; Kimura, M.; Endou, H. Transport of ochratoxin A by renal multispecific organic anion transporter 1. J. Pharmacol. Exp. Ther 1999, 289, 1301–1305. [Google Scholar]
- Dai, J.; Wright, M.W.; Manderville, R.A. Ochratoxin a forms a carbon-bonded c8-deoxyguanosine nucleoside adduct: Implications for c8 reactivity by a phenolic radical. J. Am. Chem. Soc 2003, 125, 3716–3717. [Google Scholar]
- Mantle, P.G.; Faucet-Marquis, V.; Manderville, R.A.; Squillaci, B.; Pfohl-Leszkowicz, A. Structures of covalent adducts between DNA and ochratoxin a: A new factor in debate about genotoxicity and human risk assessment. Chem. Res. Toxicol 2010, 23, 89–98. [Google Scholar]
- Gautier, J.; Richoz, J.; Welti, D.H.; Markovic, J.; Gremaud, E.; Guengerich, F.P.; Turesky, R.J. Metabolism of ochratoxin A: Absence of formation of genotoxic derivatives by human and rat enzymes. Chem. Res. Toxicol 2001, 14, 34–45. [Google Scholar]
- Delatour, T.; Mally, A.; Richoz, J.; Özden, S.; Dekant, W.; Ihmels, H.; Otto, D.; Gasparutto, D.; Marin-Kuan, M.; Schilter, B. Absence of 2′-deoxyguanosine-carbon 8-bound ochratoxin A adduct in rat kidney DNA monitored by isotope dilution LC-MS/MS. Mol. Nutr. Food Res 2008, 52, 472–482. [Google Scholar]
- Mally, A.; Zepnik, H.; Wanek, P.; Eder, E.; Dingley, K.; Ihmels, H.; Völkel, W.; Dekant, W; Ochratoxin, A. Lack of formation of covalent DNA adducts. Chem. Res. Toxicol 2004, 17, 234–242. [Google Scholar]
- Arbillaga, L.; Azqueta, A.; van Delft, J.H.; Lopez de Cerain, A. In vitro gene expression data supporting a DNA non-reactive genotoxic mechanism for ochratoxin A. Toxicol. Appl. Pharmacol 2007, 220, 216–224. [Google Scholar]
- Cavin, C.; Delatour, T.; Marin-Kuan, M.; Holzhäuser, D.; Higgins, L.; Bezençon, C.; Guignard, G.; Junod, S.; Richoz-Payot, J.; Gremaud, E.; Hayes, J.D. Reduction in antioxidant defenses may contribute to ochratoxin A toxicity and carcinogenicity. Toxicol. Sci 2007, 96, 30–39. [Google Scholar]
- Boesch-Saadatmandi, C.; Loboda, A.; Jozkowicz, A.; Huebbe, P.; Blank, R.; Wolffram, S.; Dulak, J.; Rimbach, G. Effect of ochratoxin A on redox-regulated transcription factors, antioxidant enzymes and glutathione-S-transferase in cultured kidney tubulus cells. Food Chem. Toxicol 2008, 46, 2665–2671. [Google Scholar]
- Cavin, C.; Delatour, T.; Marin-Kuan, M.; Fenaille, F.; Holzhäuser, D.; Guignard, G.; Bezençon, C.; Piguet, D.; Parisod, V.; Richoz-Payot, J.; et al. Ochratoxin A-mediated DNA and protein damage: Roles of nitrosative and oxidative stresses. Toxicol. Sci 2009, 110, 84–94. [Google Scholar]
- Cavanagh, J.E.; Weinberg, H.S.; Gold, A.; Sangaiah, R.; Marbury, D.; Glaze, W.H.; Collette, T.W.; Richardson, S.D.; Thruston, A.D., Jr. Ozonation byproducts: Identification of bromohydrins from the ozonation of natural waters with enhanced bromide levels. Environ. Sci. Technol 1992, 26, 1658–1662. [Google Scholar]
- IARC, Some Chemicals that Cause Tumours of the Kidney or Urinary Bladder in Rodents and Some Other Substances. In IARC Monographs of the Evaluation of Carcinogenic Risks to Humans; The International Agency for Research on Cancer: Lyon, France, 1999; Volume 73.
- El-Sokkary, G.H. Melatonin protects against oxidative stress induced by the kidney carcinogen KBrO3. Neuro Endocrinol. Lett 2000, 21, 461–468. [Google Scholar]
- Khan, N.; Sultana, S. Abrogation of potassium bromate-induced renal oxidative stress and subsequent cell proliferation response by soy isoflavones in Wistar rats. Toxicology 2004, 201, 173–184. [Google Scholar]
- Rahman, A.; Ahmed, S.; Khan, N.; Sultana, S.; Athar, M. Glyceryl trinitrate, a nitric oxide donor, suppresses renal oxidant damage caused by potassium bromate. Redox Rep 1999, 4, 263–269. [Google Scholar]
- Ballmaier, D.; Epe, B. Oxidative DNA damage induced by potassium bromate under cell-free conditions and in mammalian cells. Carcinogenesis 1995, 16, 335–342. [Google Scholar]
- Umemura, T.; Kanki, K.; Kuroiwa, Y.; Ishii, Y.; Okano, K.; Nohmi, T.; Nishikawa, A.; Hirose, M. In vivo mutagenicity and initiation following oxidative DNA lesion in the kidneys of rats given potassium bromate. Cancer Sci 2006, 97, 829–835. [Google Scholar]
- Cadenas, S.; Barja, G. Resveratrol, melatonin, vitamin E, and PBN protect against renal oxidative DNA damage induced by the kidney carcinogen KBrO3. Free Radic. Biol. Med 1999, 26, 1531–1537. [Google Scholar]
- Umemura, T.; Tasaki, M.; Kijima, A.; Okamura, T.; Inoue, T.; Ishii, Y.; Suzuki, Y.; Masui, N.; Nohmi, T.; Nishikawa, A. Possible participation of oxidative stress in causation of cell proliferation and in vivo mutagenicity in kidneys of gpt delta rats treated with potassium bromate. Toxicology 2009, 257, 46–52. [Google Scholar]
- IARC, Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. In IARC Monogr. Eval. Carcinog. Risks Hum; 2002; Volume 82, pp. 1–556.
- Lord, G.M.; Hollstein, M.; Arlt, V.M.; Roufosse, C.; Pusey, C.D.; Cook, T.; Schmeiser, H.H. DNA adducts and p53 mutations in a patient with aristolochic acid-associated nephropathy. Am. J. Kidney Dis 2004, 43, e11–e17. [Google Scholar]
- Stiborova, M.; Sopko, B.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Hudecek, J. The binding of aristolochic acid I to the active site of human cytochromes P450 1A1 and 1A2 explains their potential to reductively activate this human carcinogen. Cancer Lett 2005, 229, 193–204. [Google Scholar]
- Fernando, R.C.; Schmeiser, H.H.; Nicklas, W.; Wiessler, M. Detection and quantitation of dG-AAI and dA-AAI adducts by 32P-postlabeling methods in urothelium and exfoliated cells in urine of rats treated with aristolochic acid I. Carcinogenesis 1992, 13, 1835–1839. [Google Scholar]
- Stiborová, M.; Fernando, R.C.; Schmeiser, H.H.; Frei, E.; Pafau, W.; Wiessler, M. Characterization of DNA adducts formed by aristolochic acids in the target organ (forestomach) of rats by 32P-postlabelling analysis using different chromatographic procedures. Carcinogenesis 1994, 15, 1187–1192. [Google Scholar]
- Arlt, V.M.; Schmeiser, H.H.; Pfeifer, G.P. Sequence-specific detection of aristolochic acid-DNA adducts in the human p53 gene by terminal transferase-dependent PCR. Carcinogenesis 2001, 22, 133–140. [Google Scholar]
- Schmeiser, H.H.; Janssen, J.W.G.; Lyons, J.; Scherf, H.R.; Pfau, W.; Buchmann, A.; Bartram, C.R.; Wiessler, M. Aristolochic acid activates ras genes in rat tumors at deoxyadenosine residues. Cancer Res 1990, 50, 5464–5469. [Google Scholar]
- Wang, Y.; Meng, F.; Arlt, V.M.; Mei, N.; Chen, T.; Parsons, B.L. Aristolochic acid-induced carcinogenesis examined by ACB-PCR quantification of H-Ras and K-Ras mutant fraction. Mutagenesis 2011, 26, 619–628. [Google Scholar]
- Duarte, S.C.; Lino, C.M.; Pena, A. Ochratoxin A in feed of food-producing animals: An undesirable mycotoxin with health and performance effects. Veter. Microbiol 2011, 154, 1–13. [Google Scholar]
- ToxBank. Potassium Bromate. 2012. Available online: http://wiki.toxbank.net/w/index.php/Potassium_Bromate (accessed on 25 April 2013).
- West Coast Analytical Service Aristolochic Acid. Available online: http://www.wcaslab.com/tech/Aristolochic_Acid.htm (accessed on 22 May 2013).
- About Chemistry Chemcial Structure of Chlorform. Available online: http://chemistry.about.com/od/factsstructures/ig/Chemical-Structures---C/Chloroform-Chemical-Structure.htm (accessed on 22 May 2013).
- IARC. Preamble to the IARC Monographs. Available online: http://monographs.iarc.fr (accessed on 25 April 2013).
- National Toxicology Program. NTP 11th Report on Carcinogens. Rep. Carcinog 2005, 44, A1–A32.
- Höfer, T.; Gerner, I.; Gundert-Remy, U.; Liebsch, M.; Schulte, A.; Spielmann, H.; Vogel, R.; Wettig, K. Animal testing and alternative approaches for the human health risk assessment under the proposed new European chemicals regulation. Arch. Toxicol 2004, 78, 549–564. [Google Scholar]
- Rogiers; Garthoff, B.; Webb, S.; Pauwels, M.; Muller, K.; Devolder, T.; Spielmann, H. The impact of REACH, The report of CONAM/ecopa Chemical Policy Working Group; 2007. [Google Scholar]
- Martin-Martin, N.; Slattery, C.; McMorrow, T.; Ryan, M.P. TGF-β1 mediates sirolimus and cyclosporine A-induced alteration of barrier function in renal epithelial cells via a noncanonical ERK1/2 signaling pathway. Am. J. Physiol. Renal Physiol 2011, 301, F1281–F1292. [Google Scholar]
- Martin-Martin, N.; Dan, Q.; Amoozadeh, Y.; Waheed, F.; McMorrow, T.; Ryan, M.P.; Szászi, K. RhoA and Rho kinase mediate cyclosporine A and sirolimus-induced barrier tightening in renal proximal tubular cells. Int. J. Biochem. Cell Biol 2012, 44, 178–188. [Google Scholar]
- Jennings, P.; Aydin, S.; Bennett, J.; McBride, R.; Weiland, C.; Tuite, N.; Gruber, L.N.; Perco, P.; Gaora, P. Ó.; Ellinger-Ziegelbauer, H. Inter-laboratory comparison of human renal proximal tubule (HK-2) transcriptome alterations due to Cyclosporine A exposure and medium exhaustion. Toxicol. In Vitro 2009, 23, 486–499. [Google Scholar]
- Radford, R.; Slattery, C.; Jennings, P.; Blaque, O.; Pfaller, W.; Gmuender, H.; van Delft, J.; Ryan, M.P.; McMorrow, T. Carcinogens induce loss of the primary cilium in human renal proximal tubular epithelial cells independently of effects on the cell cycle. Am. J. Physiol. Renal Physiol 2012, 302, F905–F916. [Google Scholar]
- Ellis, J.K.; Athersuch, T.J.; Cavill, R.; Radford, R.; Slattery, C.; Jennings, P.; McMorrow, T.; Ryan, M.P.; Ebbels, T.M.D.; Keun, H.C. Metabolic response to low-level toxicant exposure in a novel renal tubule epithelial cell system. Mol. Biosyst 2011, 7, 247–257. [Google Scholar]
- Jennings, P.; Weiland, C.; Limonciel, A.; Bloch, K.M.; Radford, R.; Aschauer, L.; McMorrow, T.; Wilmes, A.; Pfaller, W.; Ahr, H.J.; et al. Transcriptomic alterations induced by Ochratoxin A in rat and human renal proximal tubular in vitro models and comparison to a rat in vivo model. Arch. Toxicol 2011. [Google Scholar] [CrossRef]
- Ohmori, K.; Sasaki, K.; Asada, S.; Tanaka, N.; Umeda, M. An assay method for the prediction of tumor promoting potential of chemicals by the use of Bhas 42 cells. Mutat. Res 2004, 557, 191–202. [Google Scholar]
- Tsuchiya, T.; Umeda, M.; Tanaka, N.; Sakai, A.; Nishiyama, H.; Yoshimura, I.; Ajimi, S.; Asada, S.; Asakura, M.; Baba, H.; et al. Application of the improved BALB/c 3T3 cell transformation assay to the examination of the initiating and promoting activities of chemicals: The second interlaboratory collaborative study by the non-genotoxic carcinogen study group of Japan. Altern. Lab. Anim 2010, 38, 11–27. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Radford, R.; Frain, H.; Ryan, M.P.; Slattery, C.; McMorrow, T. Mechanisms of Chemical Carcinogenesis in the Kidneys. Int. J. Mol. Sci. 2013, 14, 19416-19433. https://doi.org/10.3390/ijms141019416
Radford R, Frain H, Ryan MP, Slattery C, McMorrow T. Mechanisms of Chemical Carcinogenesis in the Kidneys. International Journal of Molecular Sciences. 2013; 14(10):19416-19433. https://doi.org/10.3390/ijms141019416
Chicago/Turabian StyleRadford, Robert, Helena Frain, Michael P. Ryan, Craig Slattery, and Tara McMorrow. 2013. "Mechanisms of Chemical Carcinogenesis in the Kidneys" International Journal of Molecular Sciences 14, no. 10: 19416-19433. https://doi.org/10.3390/ijms141019416
APA StyleRadford, R., Frain, H., Ryan, M. P., Slattery, C., & McMorrow, T. (2013). Mechanisms of Chemical Carcinogenesis in the Kidneys. International Journal of Molecular Sciences, 14(10), 19416-19433. https://doi.org/10.3390/ijms141019416