VCD Studies on Chiral Characters of Metal Complex Oligomers

Abstract
:
1. Introduction
- The VCD spectrum typically contains many well resolved vibrational bands, whereas ECD tends to show fairly broad band contour. This makes VCD assignment considerably more conclusive than ECD.
- Most of 3N-6 normal vibrations, which are all well resolved as stated above, are obtained from a single measurement and can be utilized to analyze the chiral character in the molecular vibrations of a given molecule.
- The DFT modeling of VCD spectra in the ground electronic state is more reliable than for ECD. Moreover the program is commercially available.
2. VCD Application to Chiral Mononuclear Metal Complexes
2.1. Effects of Electronic Properties of Central Metal Ions on Vibrational Energy Levels
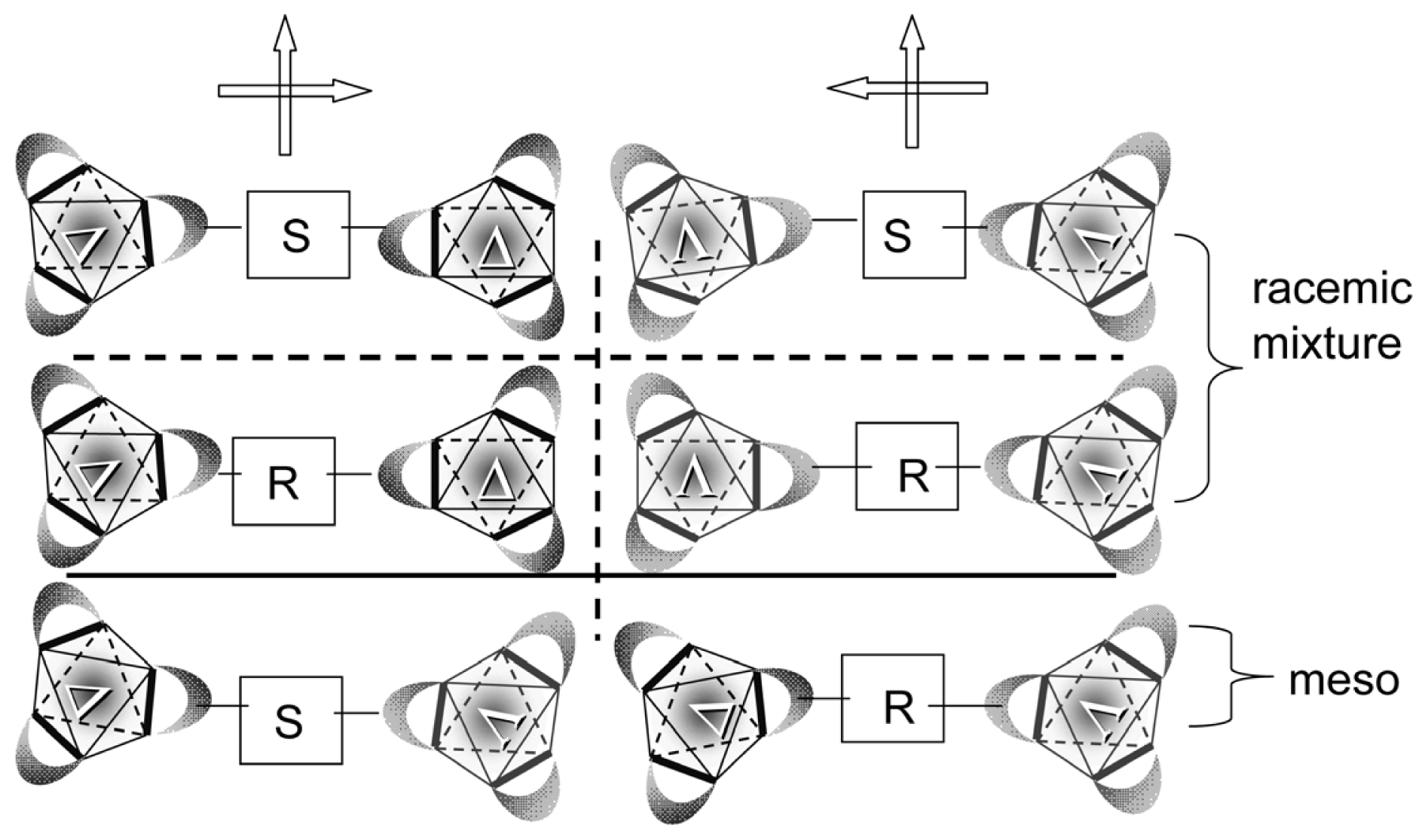
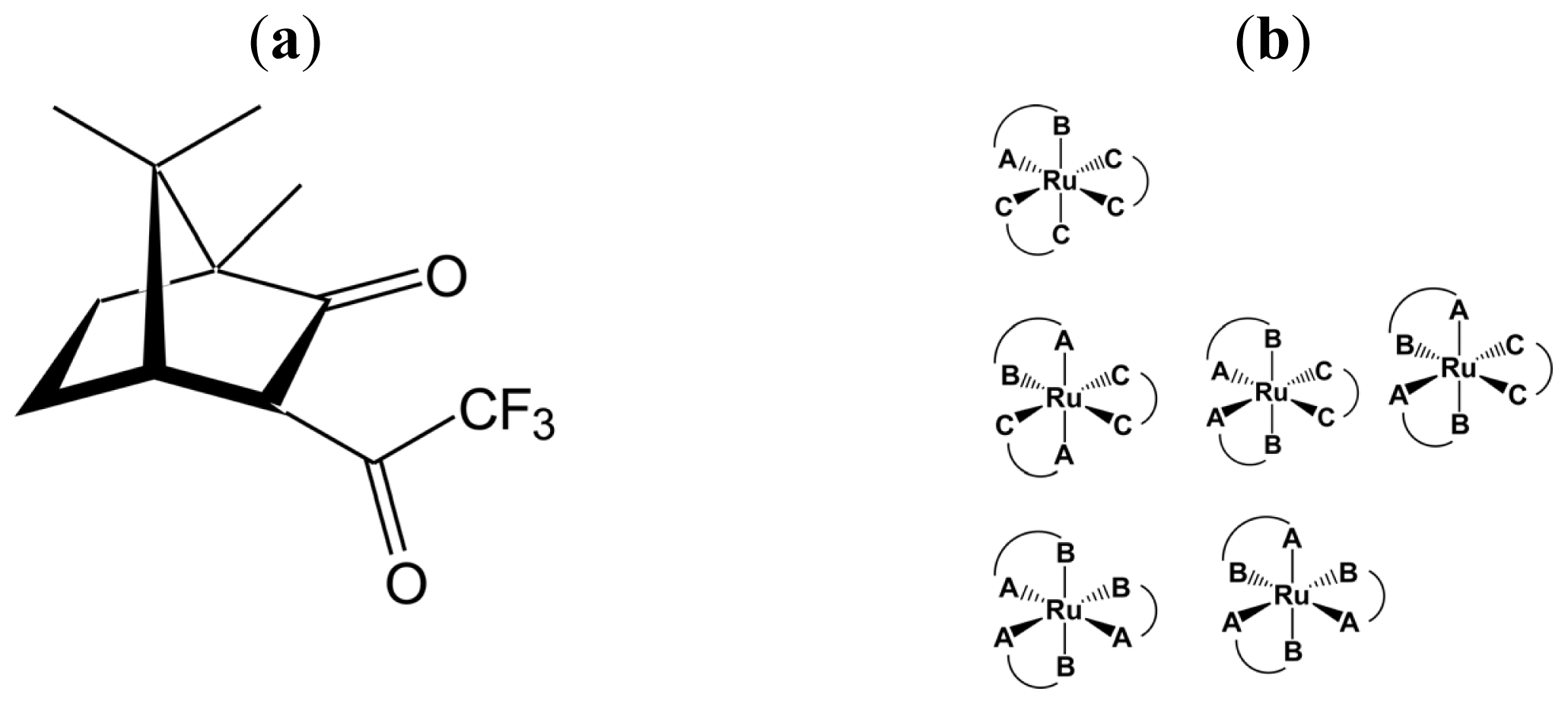
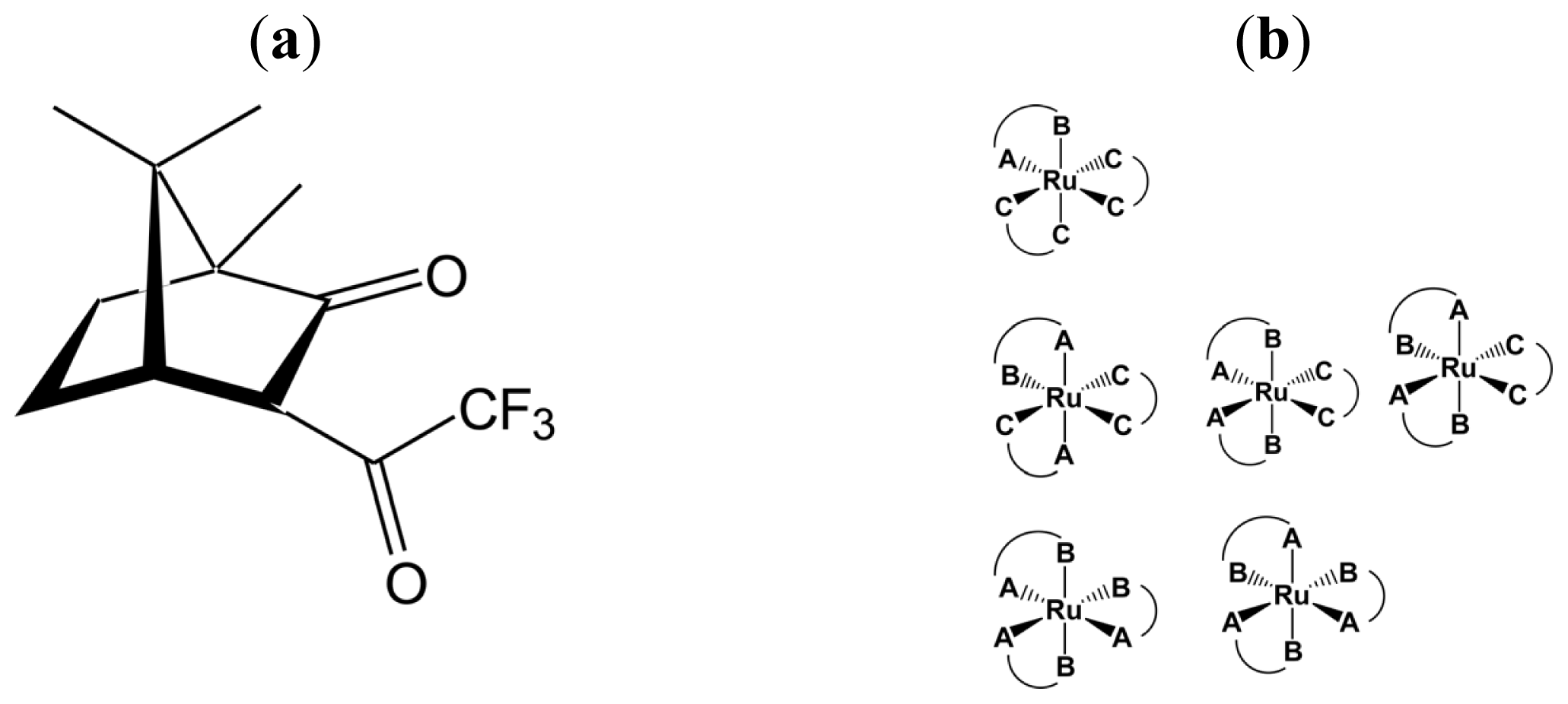
2.2. Identification of Geometrical and Diastereomeric Isomers of Octahedral Complexes
3. VCD Application to Oligomers of Chiral Metal Complexes in Solution
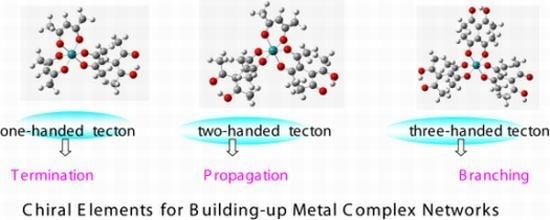
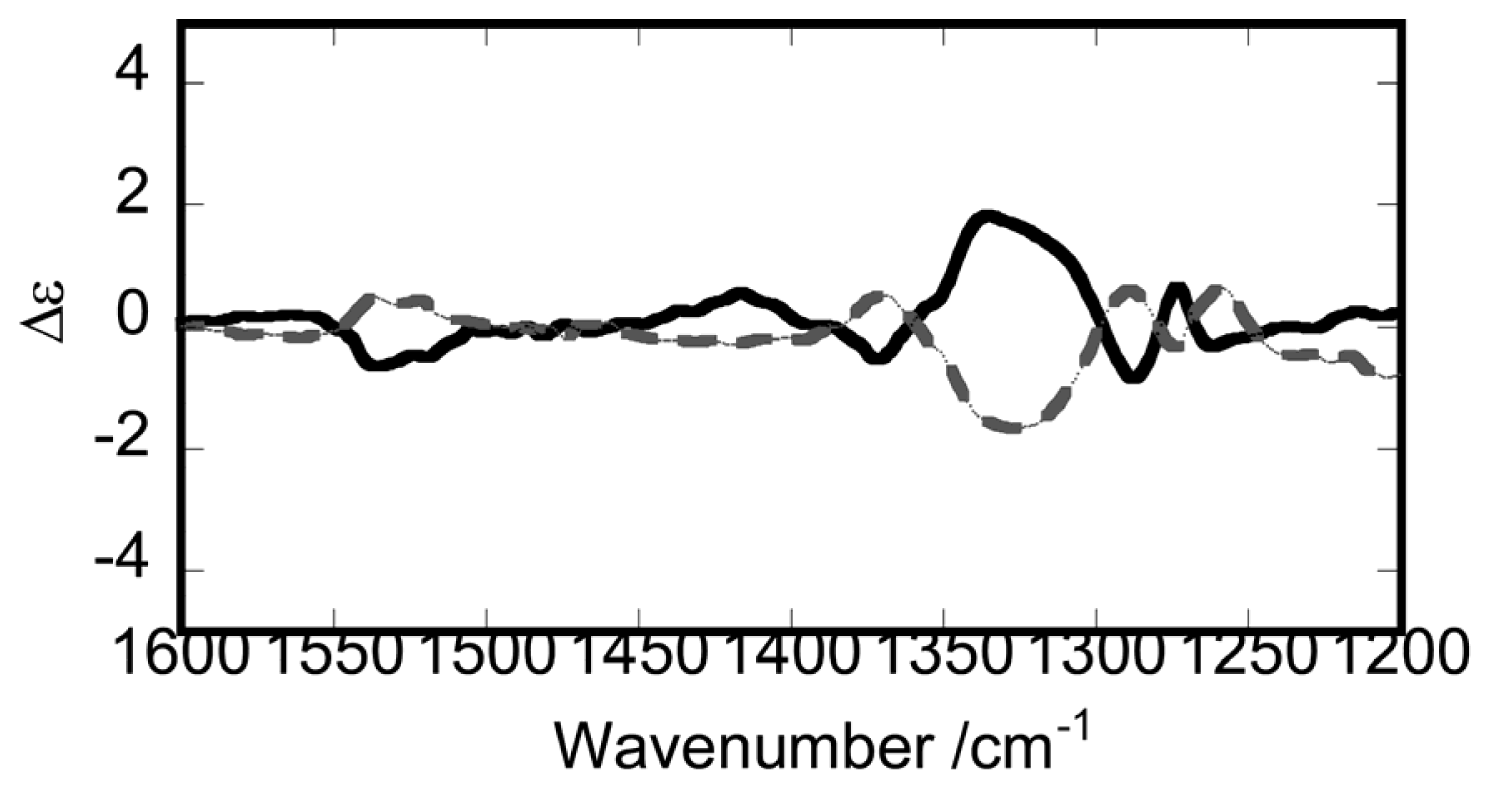
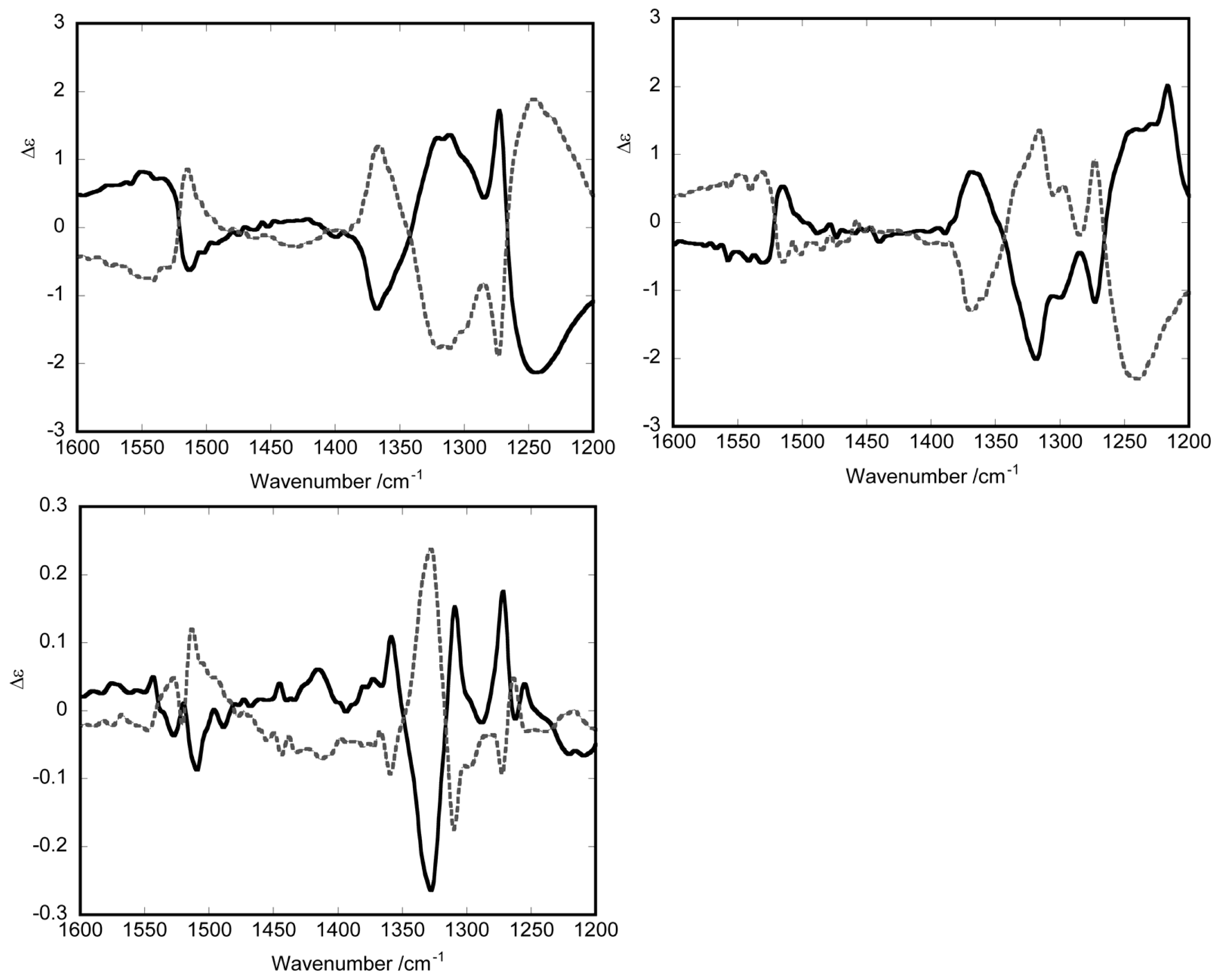
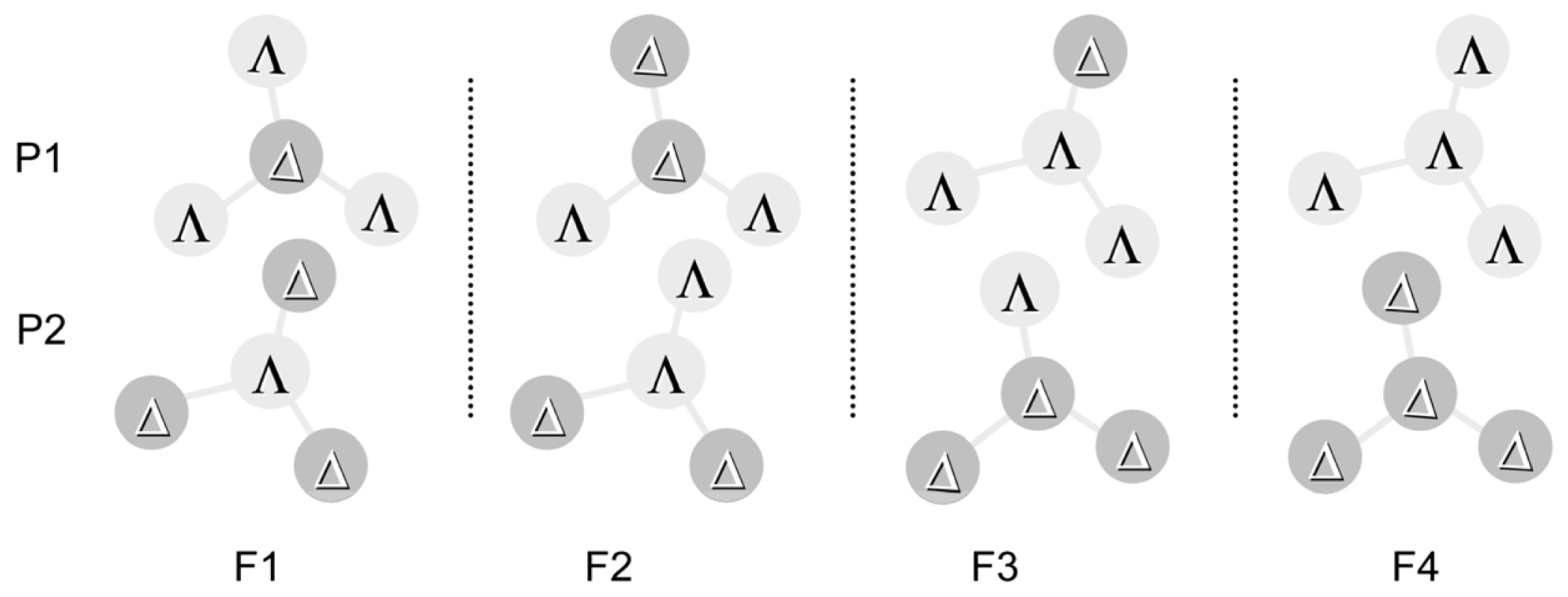
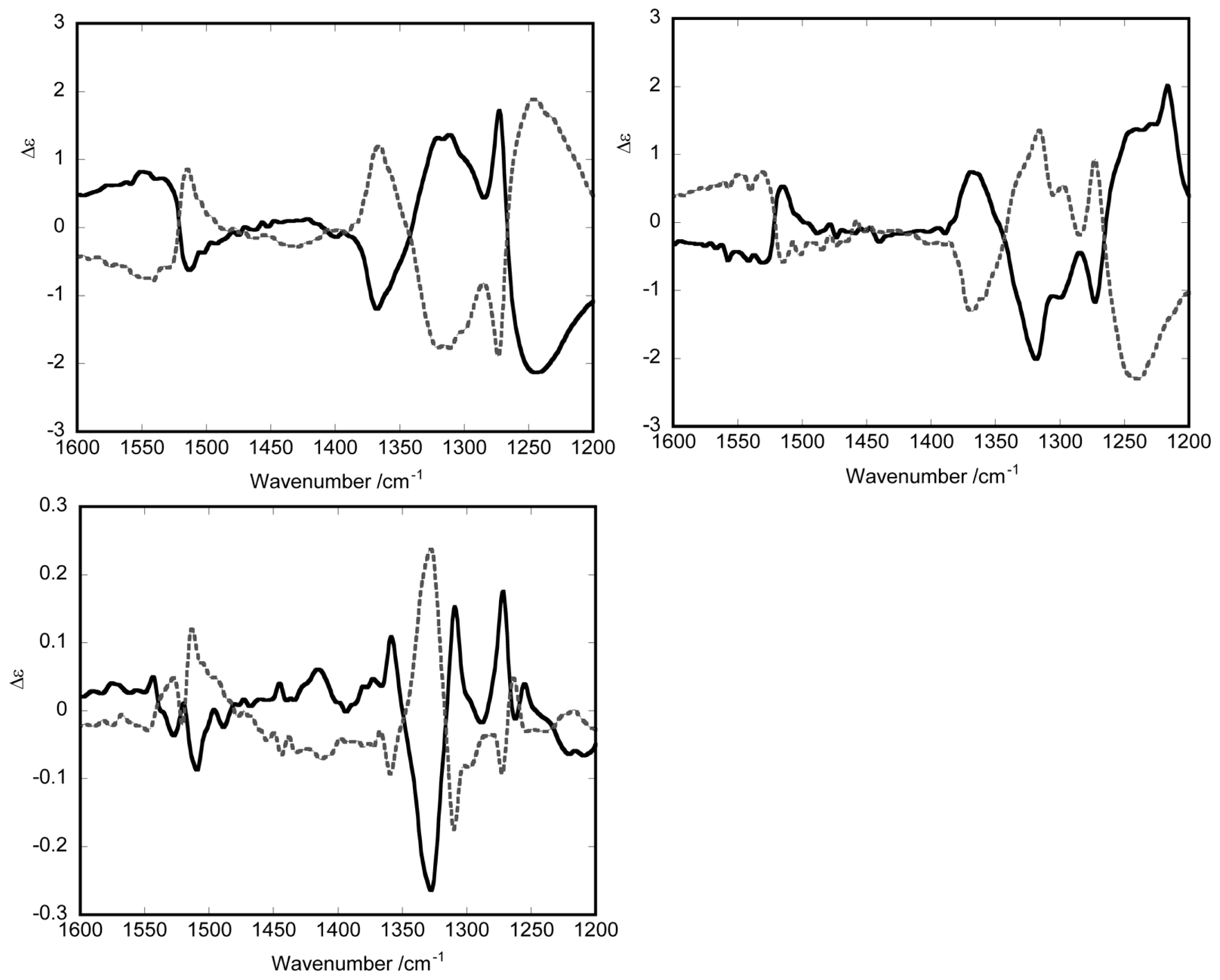
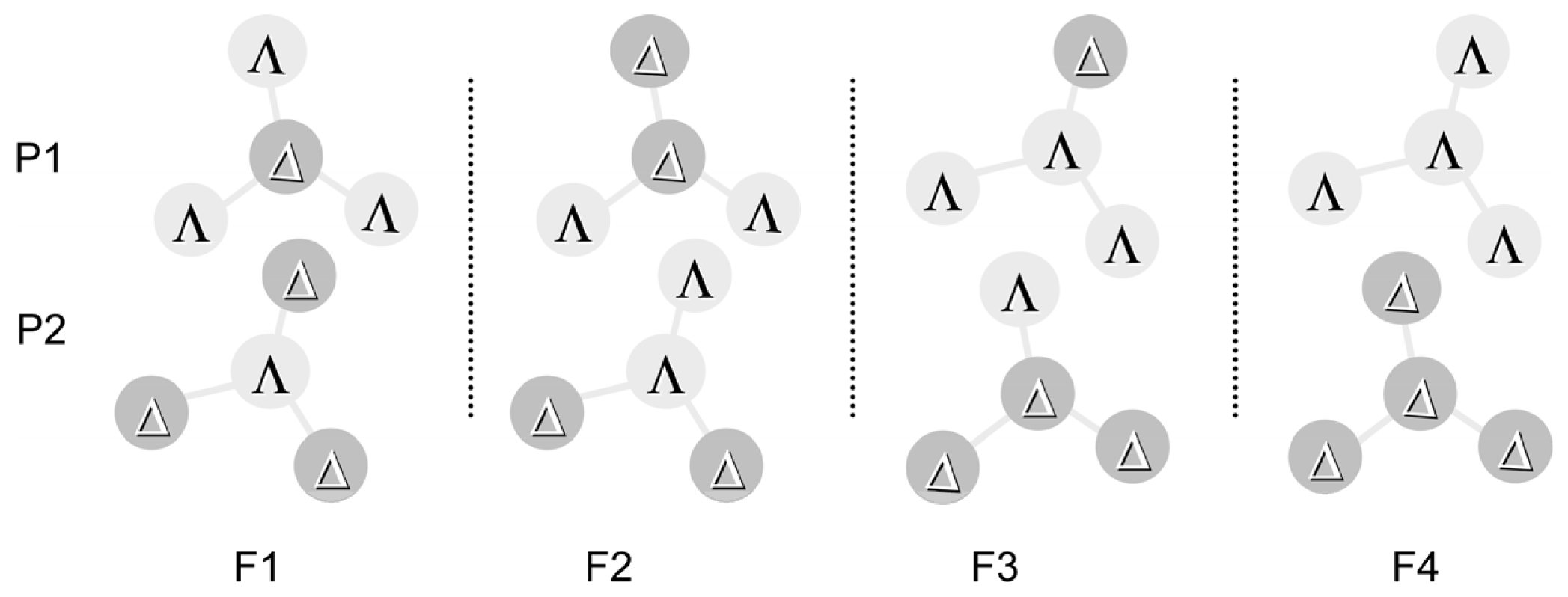
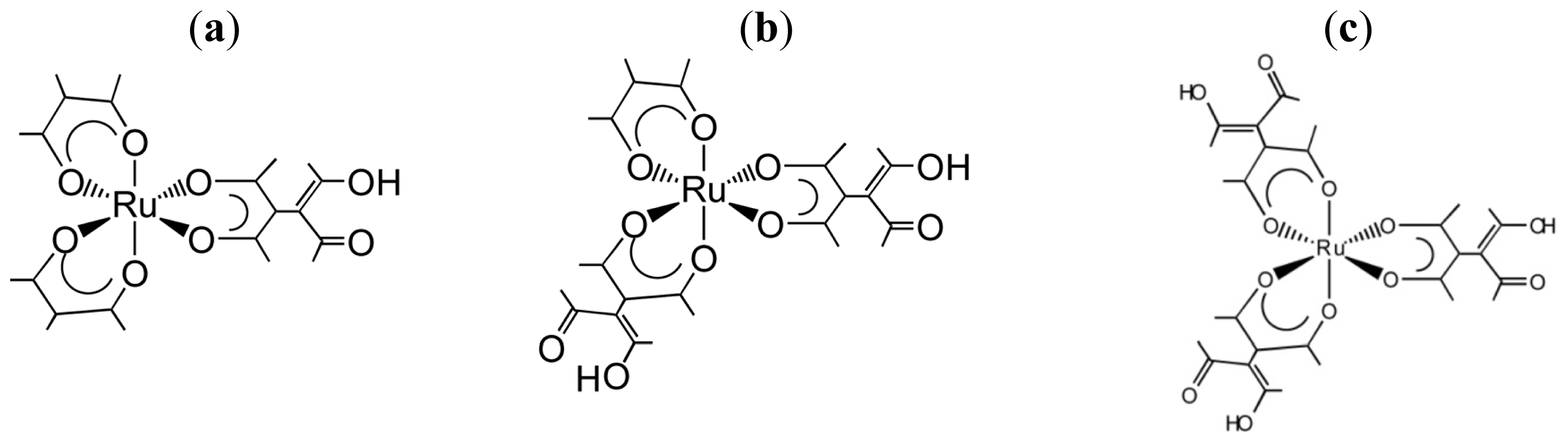
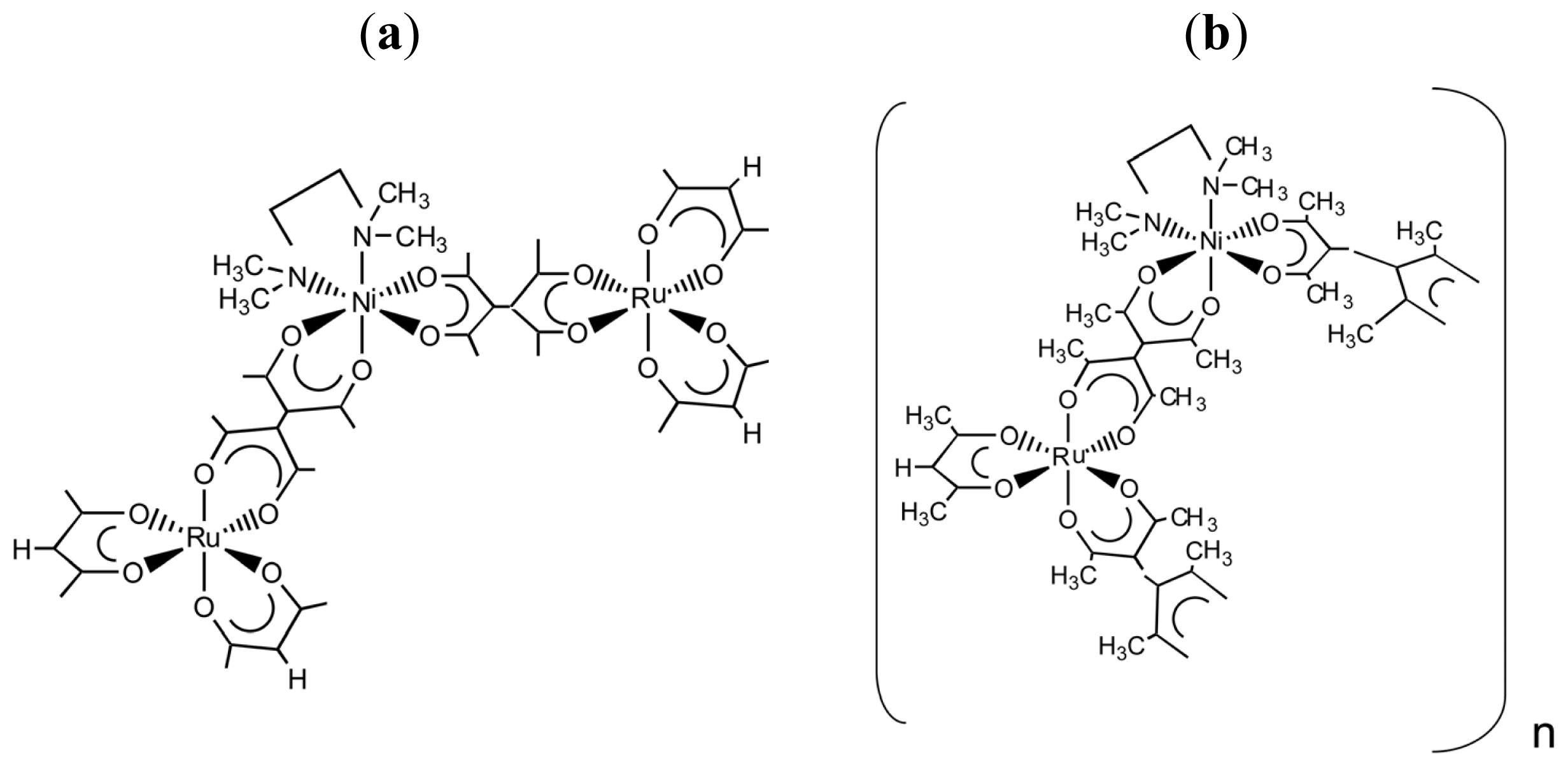
3.1. Induction of Chiral Structures in Multinuclear Complexes
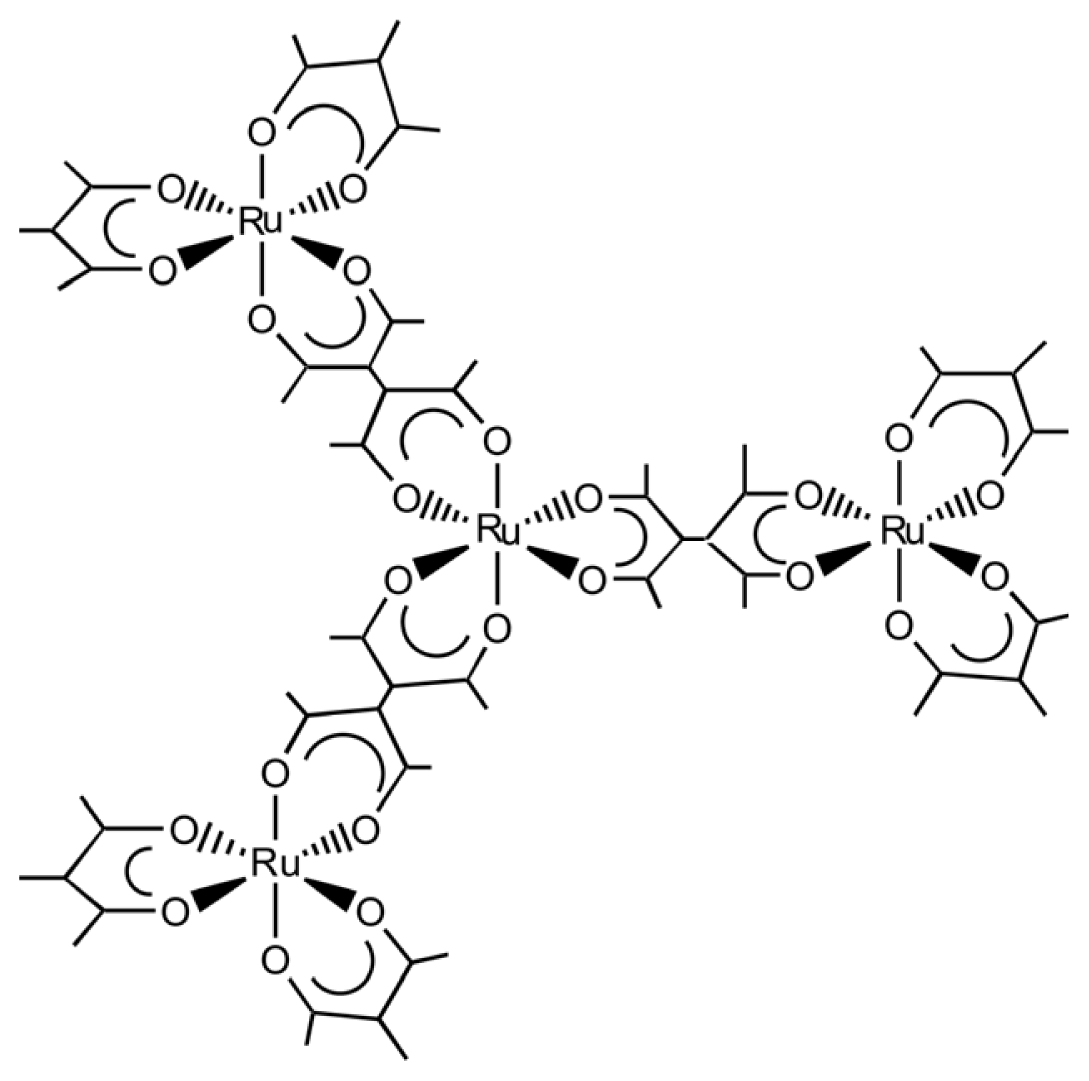
3.2. Chiral Structures of Microdomains in Multinuclear Complexes
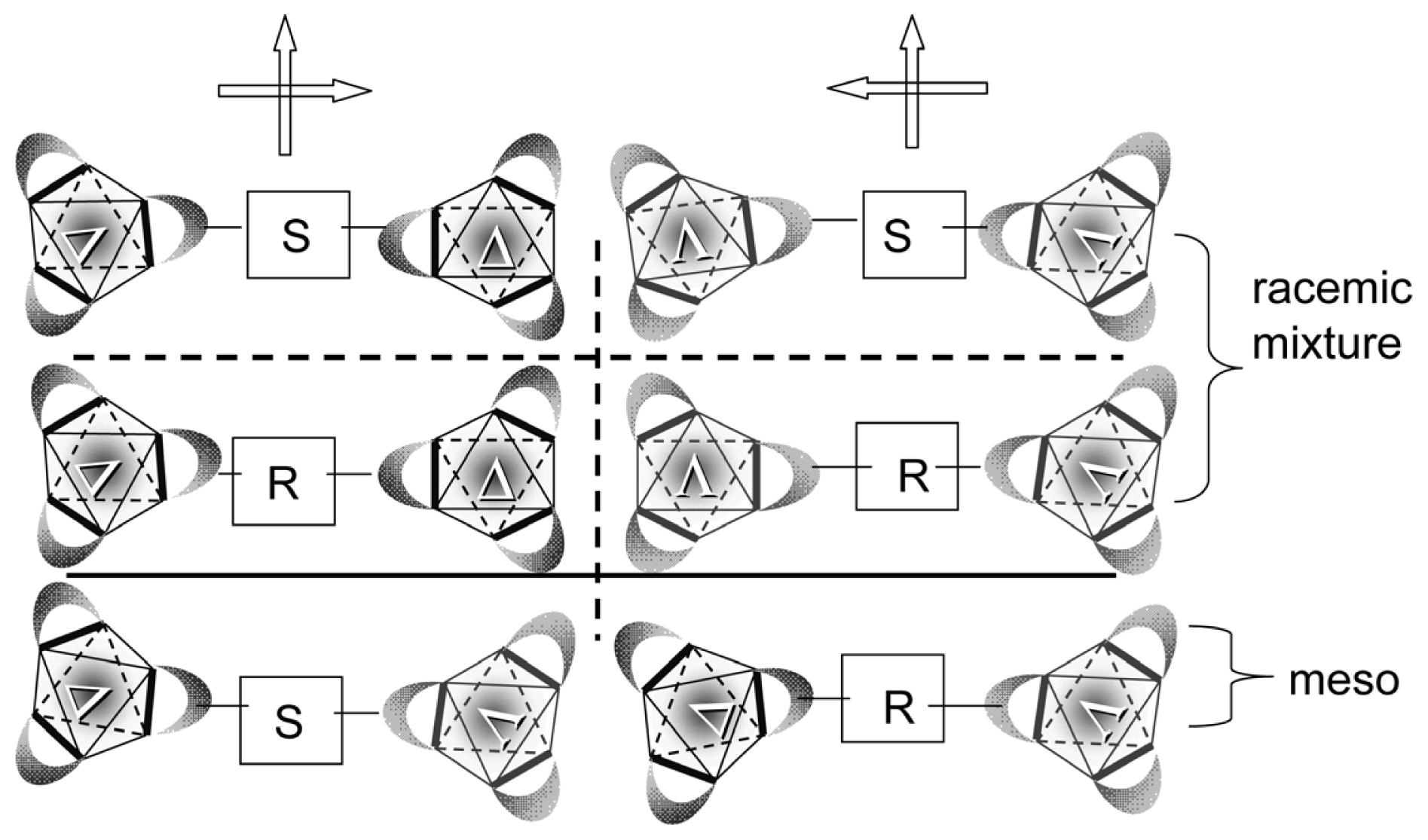
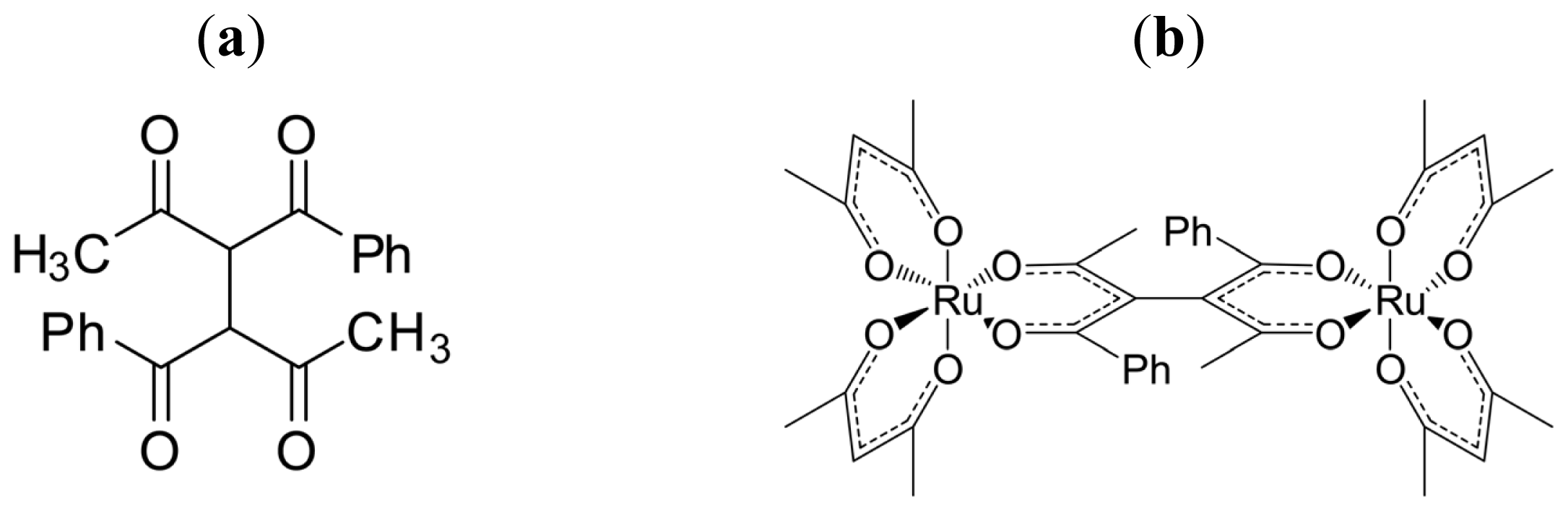
3.3. Axial Chirality in a Dimer Bridged with Non-Symmetric Bis(β-diketonato)
4. Future Development
4.1. Monitoring the Structural Change of Chiral Aggregates by Means of VCD
4.2. VCD Application to Asymmetric Reactions in Solutions
5. Summary
- Conflict of InterestThe authors declare no conflict of interest.
References
- Amouri, H.; Gruselle, M. Chirality in Transion Metal Chemsitry; Wiley: West Sussex, UK, 2008; pp. 121–232. [Google Scholar]
- Riehl, J.P.; Kaizaki, S. Absolute Chiral Structures of Inorganic Compounds. In Physical Inorganic Chemistry; Bakac, A., Ed.; Wiley: Hoboken, NJ, USA, 2010; pp. 143–197. [Google Scholar]
- Kaizaki, S. Applications of Electronic Circular Dichroism to Inorganic Sterochemsitry. In Comprehensive Chiroptical Spectroscopy; Berova, N., Polavarapu, P.L., Nakanishi, K., Woody, R.W., Eds.; Wiley: Hoboken, NJ, USA, 2012; Volume 2, pp. 451–471. [Google Scholar]
- Canary, J.W.; Dai, Z. Dynamic Stereochemistry and Chiroptical Spectroscopy of Metallo-Organic Compounds. In Comprehensive Chiroptical Spectroscopy; Berova, N., Polavarapu, P.L., Nakanishi, K., Woody, R.W., Eds.; Wiley: Hoboken, NJ, USA, 2012; Volume 2, pp. 251–287. [Google Scholar]
- Nafie, L.A. Infrared Vibrational Optical Activity: Measurement and Instrumentation. In Comprehensive Chiroptical Spectroscopy; Berova, N., Polavarapu, P.L., Nakanishi, K., Woody, R.W., Eds.; Wiley: Hoboken, NJ, USA, 2012; Volume 1, pp. 115–146. [Google Scholar]
- Knof, U.; von Zelewsky, A. Predetermined Chirality at Metal Centers. Angew. Chem. Int. Ed 1999, 38, 302–322. [Google Scholar]
- Swiegers, G.F.; Malefetse, T.J. New Self-Assembled Structural Motifs in Coordination Chemistry. Chem. Rev 2000, 100, 3483–3537. [Google Scholar]
- Hembury, G.A.; Borovkov, V.V.; Inoue, Y. Chirality-Sensing Supramolecular Systems. Chem. Rev 2008, 108, 1–69. [Google Scholar]
- Gupta, K.C.; Sutar, A.K. Catalytic Activities of Schiff Base Transition Metal Complexes. Coord. Chem. Rev 2008, 252, 1420–1450. [Google Scholar]
- Meggers, E. Chiral Auxiliaries as Emerging Tools for the Asymmetric Synthesis of Octahedral Metal Complexes. Chem. Eur. J 2010, 16, 752–758. [Google Scholar]
- Cook, T.R.; Zheng, Y.-R.; Stang, P.J. Metal-Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal-Organic Materials. Chem. Rev. 2012. [Google Scholar] [CrossRef]
- Sato, H.; Yamagishi, A. Application of the Isomerism of Octahedral Metal Complexes as a Chiral Source in Photochemistry. J. Photochem. Photobiol. C: Photochem. Rev 2007, 8, 67–84. [Google Scholar]
- Sato, H.; Yamagishi, A. Application of Δ- and Λ-isomerism of Octahedral Metal Complexes for Inducing Chiral Nematic Phases. Int. J. Mol. Sci 2009, 10, 4559–4574. [Google Scholar]
- Sato, H.; Taniguchi, T.; Monde, K.; Nishimura, S.-I.; Yamagishi, A. Dramatic Effects of d-Electron Configurations on Vibrational Circular Dichroism Spectra of Tris(acetylacetonato)metal(III). Chem. Lett 2006, 35, 364–365. [Google Scholar]
- Sato, H.; Taniguchi, T.; Nakahashi, A.; Monde, K.; Yamagishi, A. Effects of Central Metals Ions on Vibrational Circular Dichroism Spectra of Tris-(β-diketonato)Metal(III) Complexes. Inorg. Chem 2007, 46, 6755–6766. [Google Scholar]
- Sato, H.; Mori, Y.; Fukuda, Y.; Yamagishi, A. Syntheses and Vibrational Circular Dichroism Spectra of the Complete Series of [Ru((-)- or (+)-tfac)n(acac)3-n] (n = 0 ~ 3, tfac = 3-trifluoroacetylcamphorato and acac = acetylacetonato). Inorg. Chem 2009, 48, 4354–4361. [Google Scholar]
- Sato, H.; Dai Shirotani, D.; Yamanari, K.; Kaizaki, S. Vibrational Circular Dichroism of Δ-SAPR- 8- tetrakis ((+)-heptafluorobutyrylcamphorato) lanthanide(III) Complexes with an Encapsulated Alkali Metal Ion. Inorg. Chem 2010, 49, 356–358. [Google Scholar]
- Sato, H.; Uno, H.; Nakano, H. Identification of Geometrical Isomers Using Vibrational Circular Dichroism Spectroscopy: A Series of Mixed-Ligand Complexes of Diamagnetic Co(III) Ions. Dalton Trans 2011, 40, 1332–1337. [Google Scholar]
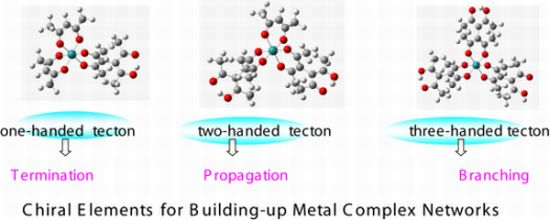
- Sato, H.; Nakao, A.; Yamagishi, A. Formation of Chiral Heterometallic Oligomers by Combination of Inert Ru(III) Tectons and Labile Ni(II) Connectors. New J. Chem 2011, 35, 1823–1828. [Google Scholar]
- Mori, H.; Yamagishi, A.; Sato, H. Theoretical Study on Vibrational Circular Dichroism Spectra of Tris(acetylacetonato)metal(III) Complexes: Anharmonic Effects and Low-lying Excited States. J. Chem. Phys 2011, 135, 84506–84512. [Google Scholar]
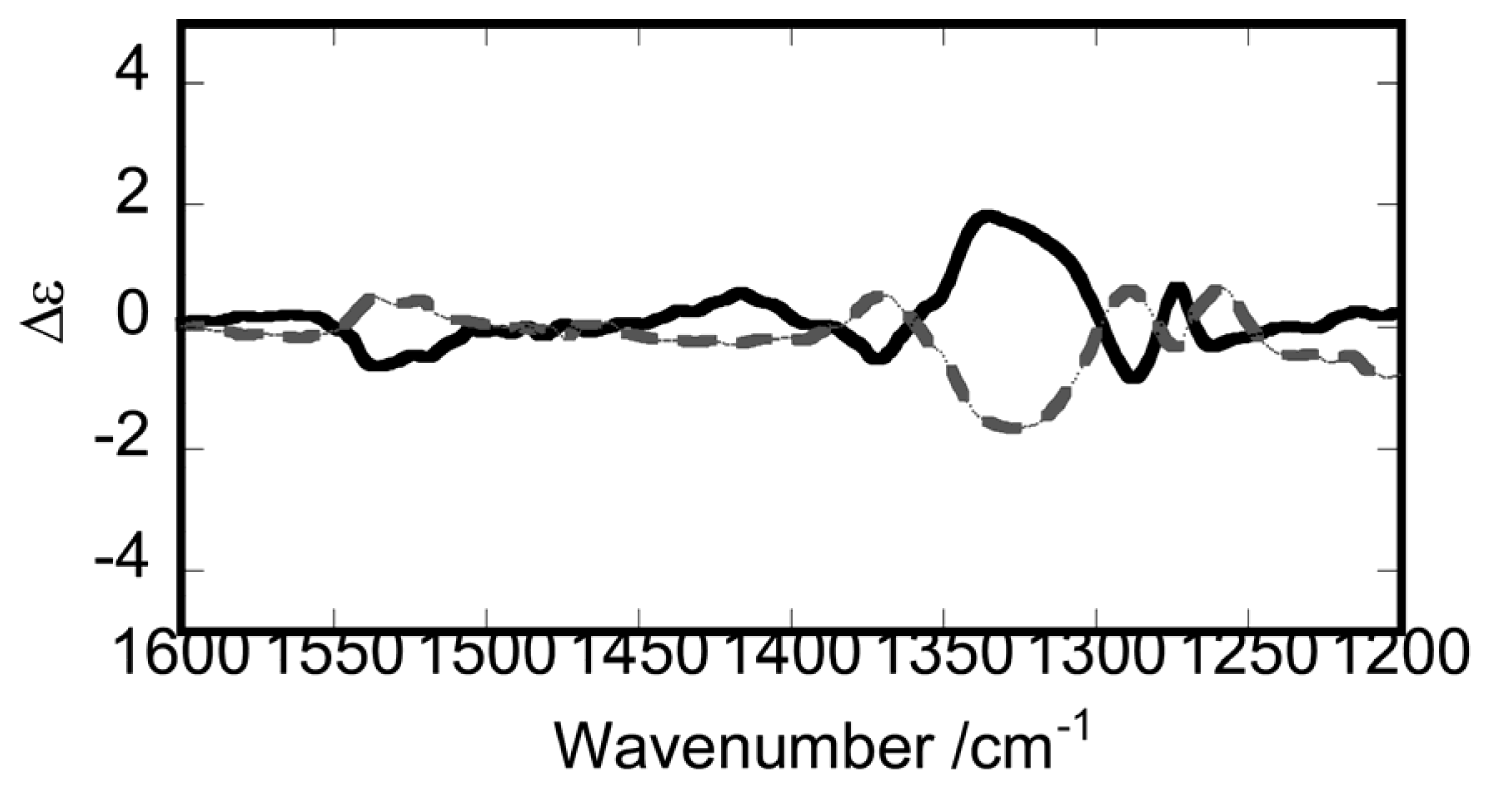
- Sato, H.; Takase, R.; Mori, Y.; Yamagishi, A. Can a meso-type dinuclear complex be chiral?: Dinuclear β-diketonato Ru(III) complexes. Dalton Trans 2012, 41, 747–751. [Google Scholar]
- Sato, H.; Sato, F.; Taniguchi, M.; Yamagishi, A. Chirality Effects on Core-periphery Connection in a Star-burst Type Tetranuclear Ru(III) Complex: Application of Vibrational Circular Dichroism Spectroscopy. Dalton Trans 2012, 41, 1709–1712. [Google Scholar]
- Shirotani, D.; Sato, H.; Yamanari, K.; Kaizaki, S. Chiroptical Spectra of Tetrakis (+)-3- heptafluorobutylrylcamphorate Ln(III) Complexes with an Encapsulated Alkali Metal Ion: Electronic Circular Dichroism in the 4f-4f Transitions of a Series of the Cs-Ln Complexes. Dalton Trans 2012, 41, 10557–10567. [Google Scholar]
- Sato, H.; Mori, Y.; Kitazawa, T.; Yamagishi, A. Inversion of Axial Chirality in Coordinated Bis-β-diketonato Ligands. Dalton Trans 2013, 42, 232–237. [Google Scholar]
- Taniguchi, T.; Monde, K.; Nishimura, S.-I.; Yoshida, J.; Sato, H.; Yamagishi, A. Rewinding of Helical Systems by Use of Cr(III) Complex as a Photoresponsive Chiral Dopant. Mol. Cryst. Liq. Cryst 2006, 460, 107–116. [Google Scholar]
- Stephen, P.J.; Lowe, M.A. Vibrational Circular Dichroism. Ann. Rev. Phys. Chem 1985, 36, 213–241. [Google Scholar]
- He, Y.; Cao, X.; Nafie, L.A.; Freedman, T.B. Ab Initio VCD Calculation of a Transition-Metal Containing Molecule and a New Intensity Enhancement Mechanism for VCD. J. Am. Chem. Soc 2001, 123, 11320–11321. [Google Scholar]
- Freedman, T.B.; Cao, X.; Dukor, R.K.; Nafie, L.A. Absolute Configuration Determination of Chiral Molecules in the Solution State using Vibrational Circular Dichroism. Chirality 2003, 15, 743–758. [Google Scholar]
- Bieri, M.; Gautier, C.; Bürgi, T. Probing Chiral Interfaces by Infrared Spectroscopic Methods. Phys. Chem. Chem. Phys 2007, 9, 671–685. [Google Scholar]
- Johannessen, C.; Thulstrup, P.W. Vibrational Circular Dichroism Spectroscopy of a Spin-Triplet Bis-(biuretato) Cobaltate(III) Coordination Compound with Low-Lying Electronic Transitions. Dalton Trans 2007, 36, 1028–1033. [Google Scholar]
- Amstrong, W.; Cotton, F.A.; Petrovic, A.G.; Polavarapu, P.L.; Warnke, M.M. Resolution of Enantiomers in Solution and Determination of the Chirality of Extended Metal Atom Chains. Inorg. Chem 2007, 46, 1535–1537. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Pan, J.-J. The Determination of the Absolute Configurations of Chiral Molecules using Vibrational Circular Dichroism (VCD) Spectroscopy. Chirality 2008, 20, 643–663. [Google Scholar]
- Polavarapu, P.L. Why is It Important to Simultaneously Use More Than One Chiroptical Spectroscopic Method for Determining the Structures of Chiral Molecules? Chirality 2008, 20, 664–672. [Google Scholar]
- Mukhopadhyay, P.; Wipf, P.; Beratan, D.N. Optical Signatures of Molecular Dissymmetry: Combining Theory with Experiments To Address Stereochemical Puzzles. Acc. Chem. Res 2009, 42, 809–819. [Google Scholar]
- Nicu, V.P.; Austschbach, J.; Baereends, E.J. Enhancement of IR and VCD Intensities due to Charge Transfer. Phys. Chem. Chem. Phys 2009, 11, 1526–1538. [Google Scholar]
- Nic, V.P.; Baerends, E.J. Robust Normal Modes in Vibrational Circular Dichroism Spectra. Phys. Chem. Chem. Phys 2009, 11, 6107–6118. [Google Scholar]
- Han, Z.-B.; Ji, J.-W.; An, H.-Y.; Zhang, W.; Zhang, G.-X.; Yang, L.-G. Two 3D Chiral Coordination Polymers with 4-connected 66 Topological Net: Synthesis, Structure and Magnetic Properties. Dalton Trans 2009, 38, 9807–9811. [Google Scholar]
- Merten, C.; Amkreutz, M.; Hartwig, A. Determining the Structure of α-phenylethyl Isocyanide in Chloroform by VCD Spectroscopy and DFT Calculations—Simple Case or Challenge? Phys. Chem. Chem. Phys 2010, 12, 11635–11641. [Google Scholar]
- Wu, T.; Li, C.-H.; Li, Y.-Z.; Zhang, Z.-G.; You, Z.-Z. Synthesis, Structure and Chiroptical Study of Chiral Macrocyclic Imine nickel(II) Coordination Compounds Derived from Camphor. Dalton Trans 2010, 39, 3227–3232. [Google Scholar]
- Soioshonok, V.A.; Ono, T.; Ueki, H.; Vanthuyne, N.; Balaban, T.S.; Bürch, J.; Fliegi, H.; Klopper, W.; Naubron, J.-V.; Bui, T.T.T.; et al. Ridge-Tile-like Chiral Topology: Synthesis, Resolution, and Complete Chiroptical Characterization of Enantiomers of Edge-Sharing Binuclear Square Planar Complexes of Ni(II) Bearing Achiral Ligands. J. Am. Chem. Soc 2010, 132, 10477–10483. [Google Scholar]
- Sadlej, J.; Dobrowolski, J.C.; Joanna, E.; Rode, J.E. VCD Spectroscopy as a Novel Probe for Chirality Transfer in Molecular. Chem. Soc. Rev 2010, 39, 1478–1488. [Google Scholar]
- Szilvágyi, G.; Majer, Z.; Vass, E.; Hollósi, M. Conformational Studies on Chiral Rhodium complexes by ECD and VCD spectroscopy. Chirality 2011, 23, 294–299. [Google Scholar]
- Chamayou, A.-C.; Lüdeke, S.; Brecht, V.; Freedman, T.B.; Nafie, L.A.; Janiak, C. Chirality and Diastereoselection of Δ/Λ-configured Tetrahedral Zinc Complexes through Enantiopure Schiff Base Complexes: Combined Vibrational Circular Dichroism, Density Functional Theory, 1H NMR, and X-ray Structural Studies. Inorg. Chem 2011, 50, 11363–11374. [Google Scholar]
- Zhang, K.; Cao, K.-S.; Wang, G.; Peng, Y.-X.; Chu, Z.-L.; Huang, W. Reversible Color Changes of Chiral Double Salts of d-(+)-camphoric Diammonium and Transition-Metal Sulfates via Gain and Loss of Crystalline and Coordination Water Molecules. Inorg. Chim. Acta 2011, 378, 224–230. [Google Scholar]
- Yang, G.; Xu, Y. Vibrational Circular Dichroism Spectroscopy of Chiral Molecules. Top. Curr. Chem 2011, 298, 189–236. [Google Scholar]
- Merten, C.; Hiller, K.; Xu, Y. Effects of Electron Configuration and Coordination Number on the Vibrational Circular Dichroism Spectra of Metal Complexes of Trans-1,2-diaminocyclohexane. Phys. Chem. Chem. Phys 2012, 14, 12884–12891. [Google Scholar]
- Dezhahang, Z.; Merten, C.; Poopari, M.R.; Xu, Y. Vibrational Circular Dichroism Spectroscopy of Two Chiral Binaphthyl Diphosphine Ligands and their Palladium Complexes in Solution. Dalton Trans 2012, 41, 10817–10824. [Google Scholar]
- Wu, T.; Zhang, X.-P.; Li, C.-H.; Bouř, P.; Li, Y.-Z.; You, X.-Z. Vibrational and Electronic Circular Dichroism Monitoring of Copper(II) Coordination with a Chiral Ligand. Chirality 2012, 24, 451–458. [Google Scholar]
- Luber, S.; Reiher, M. Prediction of Raman Optical Activity Spectra of Chiral 3-Acetylcamphorato-Cobalt Complexes. Chem. Phys. Chem 2010, 11, 1876–1887. [Google Scholar]
- Monde, K.; Miura, N.; Hashimoto, M.; Taniguchi, T.; Inabe, T. Conformational Analysis of Chiral Helical Perfluoroalkyl Chains by VCD. J. Am. Chem. Soc 2006, 128, 6000–6001. [Google Scholar]
- Sato, H.; Yajima, T.; Yamagishi, A. Molecular Origin for Helical Winding of Fibrils Formed by Perfluorinated Gelators. Chem. Commun 2011, 47, 3736–3738. [Google Scholar]
- Sato, H.; Furuno, Y.; Fukuda, Y.; Okamoto, K.; Yamagishi, A. Crystallization Induced Chiral Locking of a Central Labile Core in a Star-shaped Tetra-nuclear Complex. Dalton Trans 2008, 37, 1283–1285. [Google Scholar]
- Fujimoto, N.; Mori, Y.; Yamagishi, A.; Sato, H. Molecular Recognition of Star-burst Tetranuclear Ru(III) Complexes on a Chirally Modified Clay Surface. Chem. Commun 2010, 46, 5473–5475. [Google Scholar]
- Saito, M.; Sato, H.; Mori, Y.; Fukuda, Y. Structure and Enantio-Differentiating Behaviors of Nickel(II) Complexes with Chiral Schiff Base Ligands Derived from 1,1′-Binaphthyl-2,2′-diamine. Bull. Chem. Soc. Jpn 2009, 82, 1266–1273. [Google Scholar]
- Che, C.-M.; Huang, J.-S. Metal Complexes of Chiral Binaphthyl Schiff-base Ligands and their Application in Stereoselective Organic Transformations. Coord. Chem. Rev 2003, 242, 97–113. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sato, H.; Yamagishi, A. VCD Studies on Chiral Characters of Metal Complex Oligomers. Int. J. Mol. Sci. 2013, 14, 964-978. https://doi.org/10.3390/ijms14010964
Sato H, Yamagishi A. VCD Studies on Chiral Characters of Metal Complex Oligomers. International Journal of Molecular Sciences. 2013; 14(1):964-978. https://doi.org/10.3390/ijms14010964
Chicago/Turabian StyleSato, Hisako, and Akihiko Yamagishi. 2013. "VCD Studies on Chiral Characters of Metal Complex Oligomers" International Journal of Molecular Sciences 14, no. 1: 964-978. https://doi.org/10.3390/ijms14010964