Ovarian Cancer: In Search of Better Marker Systems Based on DNA Repair Defects


Abstract
:1. Introduction
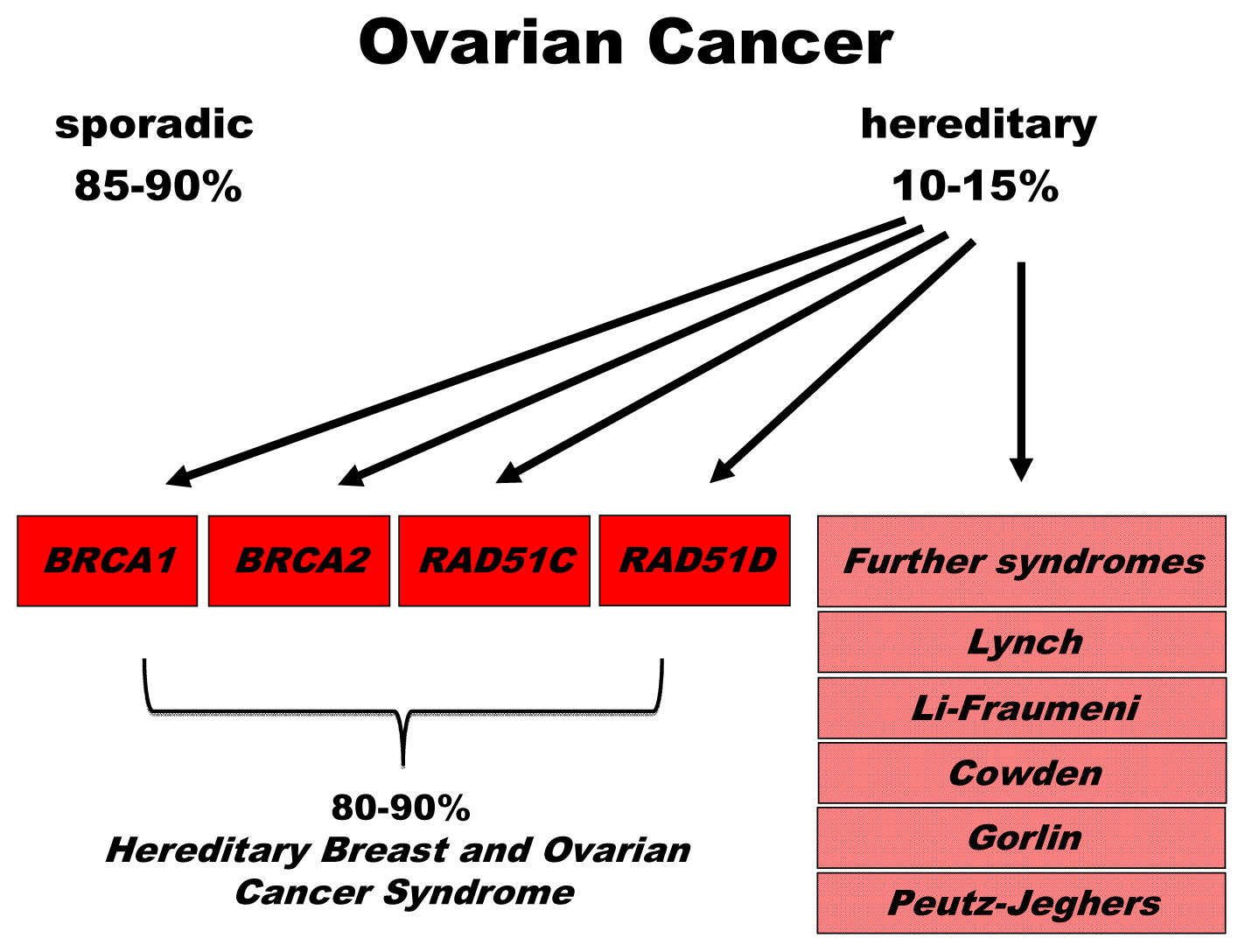
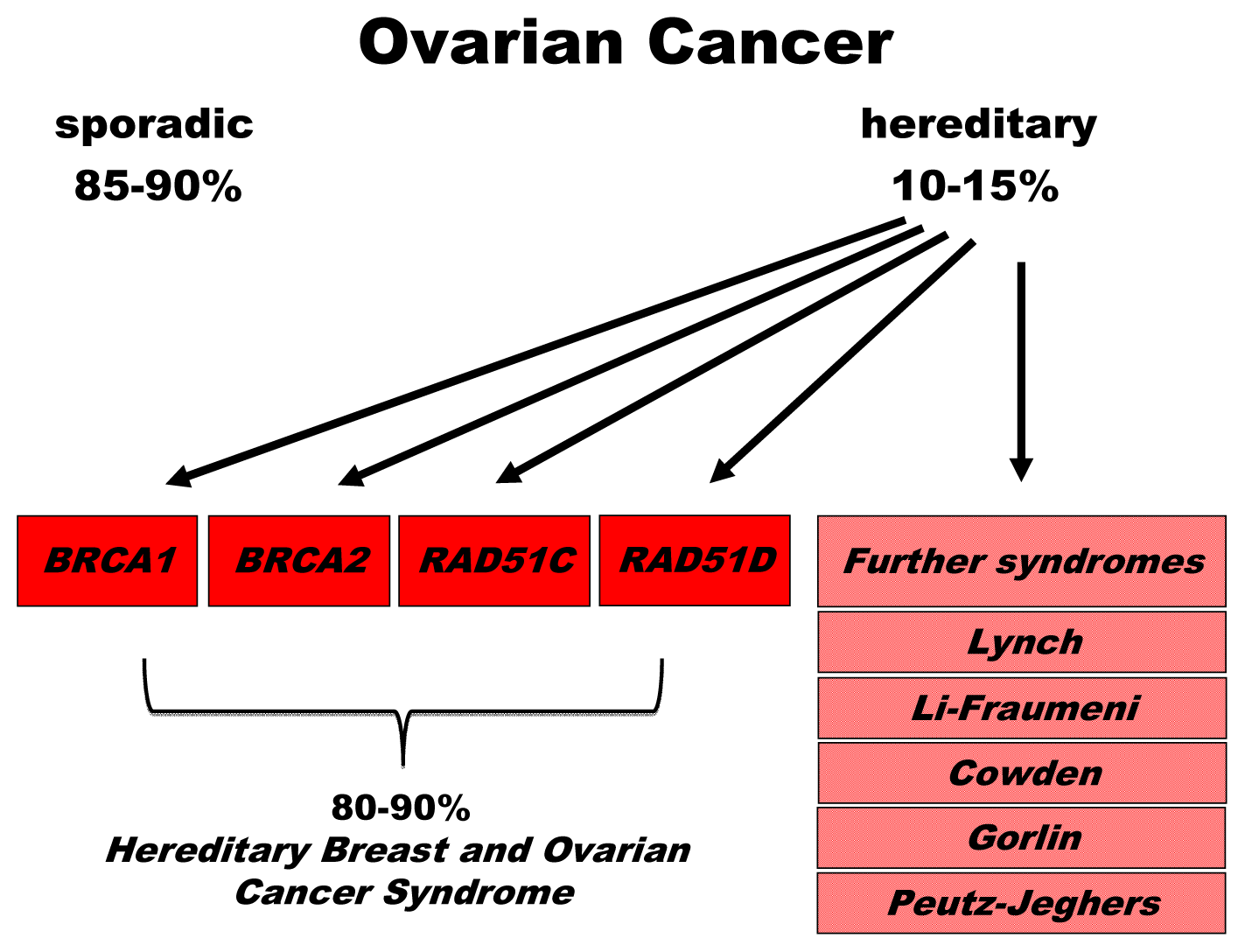
2. Genetics of Hereditary Cancer
2.1. BRCA1 and BRCA2
2.2. Susceptibility Genes with Involvement in the BRCA-Fanconi Anemia Pathway of DNA Repair
2.3. Further DNA Repair Genes and Syndromes Associated with Hereditary Ovarian Cancer
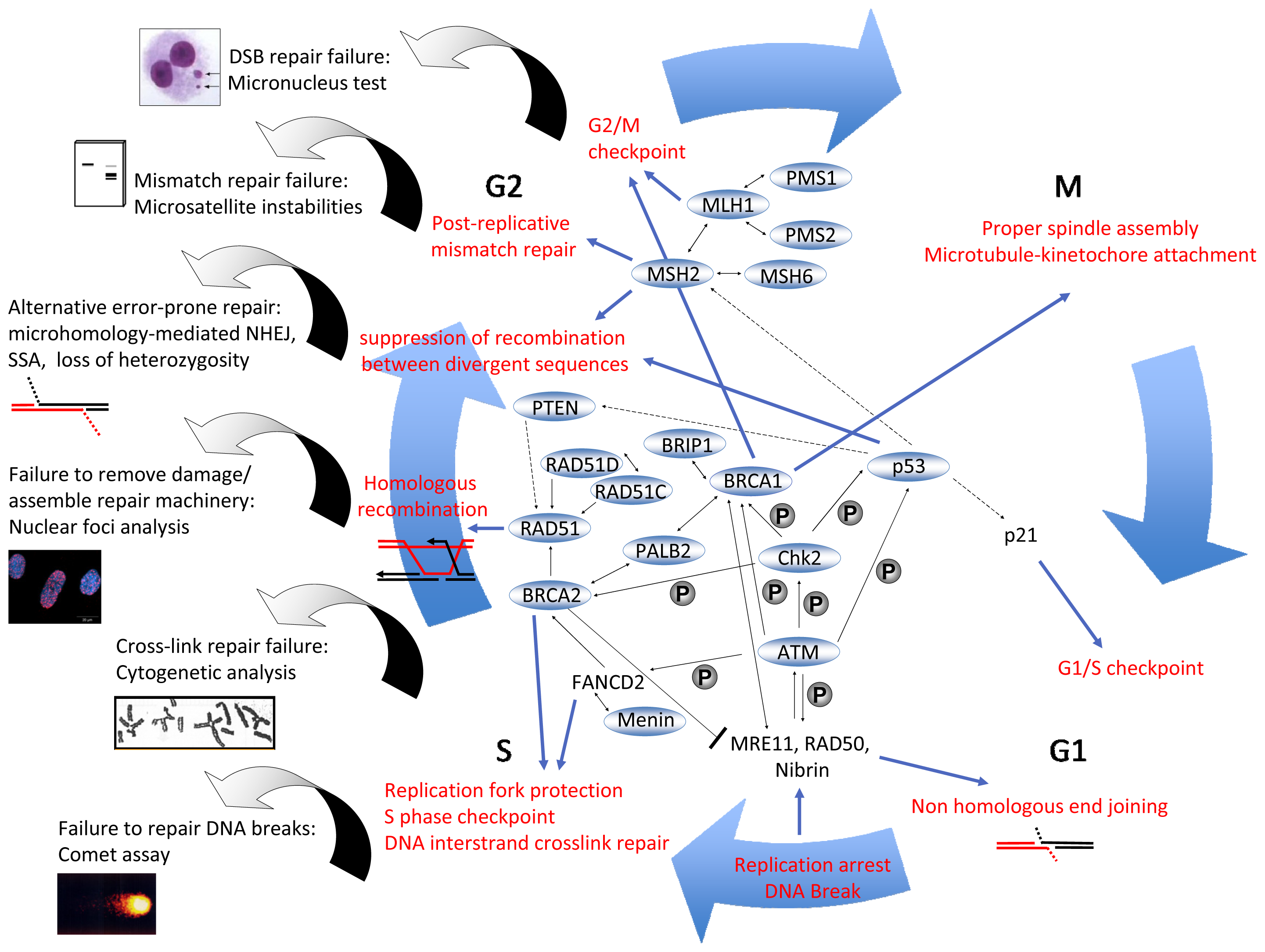
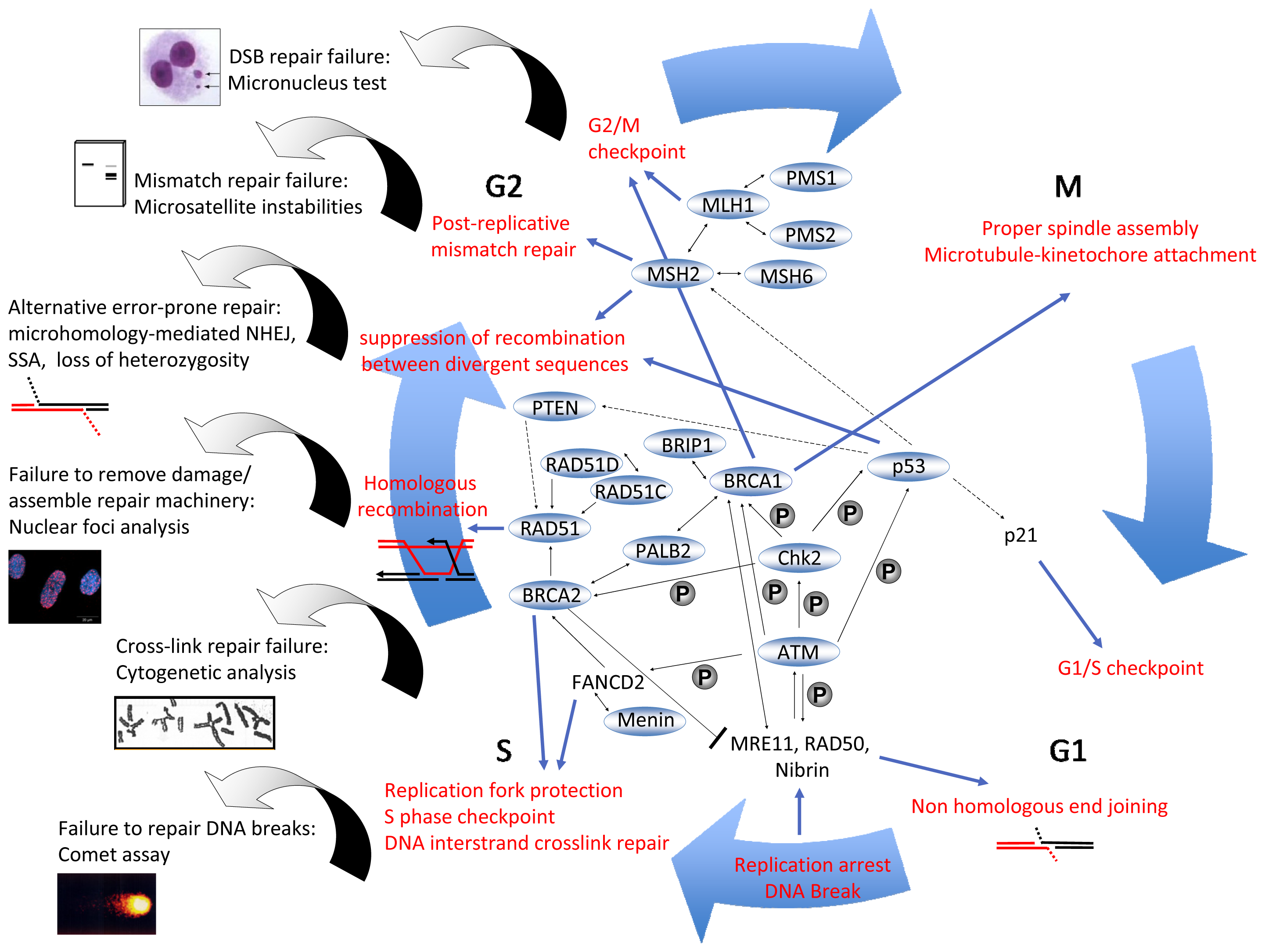
3. DNA Repair Mechanisms
4. Diagnostics: Present and Future
4.1. Pathology of Ovarian Cancer
4.2. State-of-the-Art and Recent Efforts to Identify Single Molecule or Combined Biomarkers
4.3. “Omics”-Based Approaches to Identify Potential Biomarkers
4.3.1. Protein Markers
4.3.2. Patterns of Genetic and mRNA Expression Changes
4.3.3. Epigenetic Markers
4.4. Functional Testing, a Novel Concept in Biomarker Development
4.4.1. Micronucleus Assay
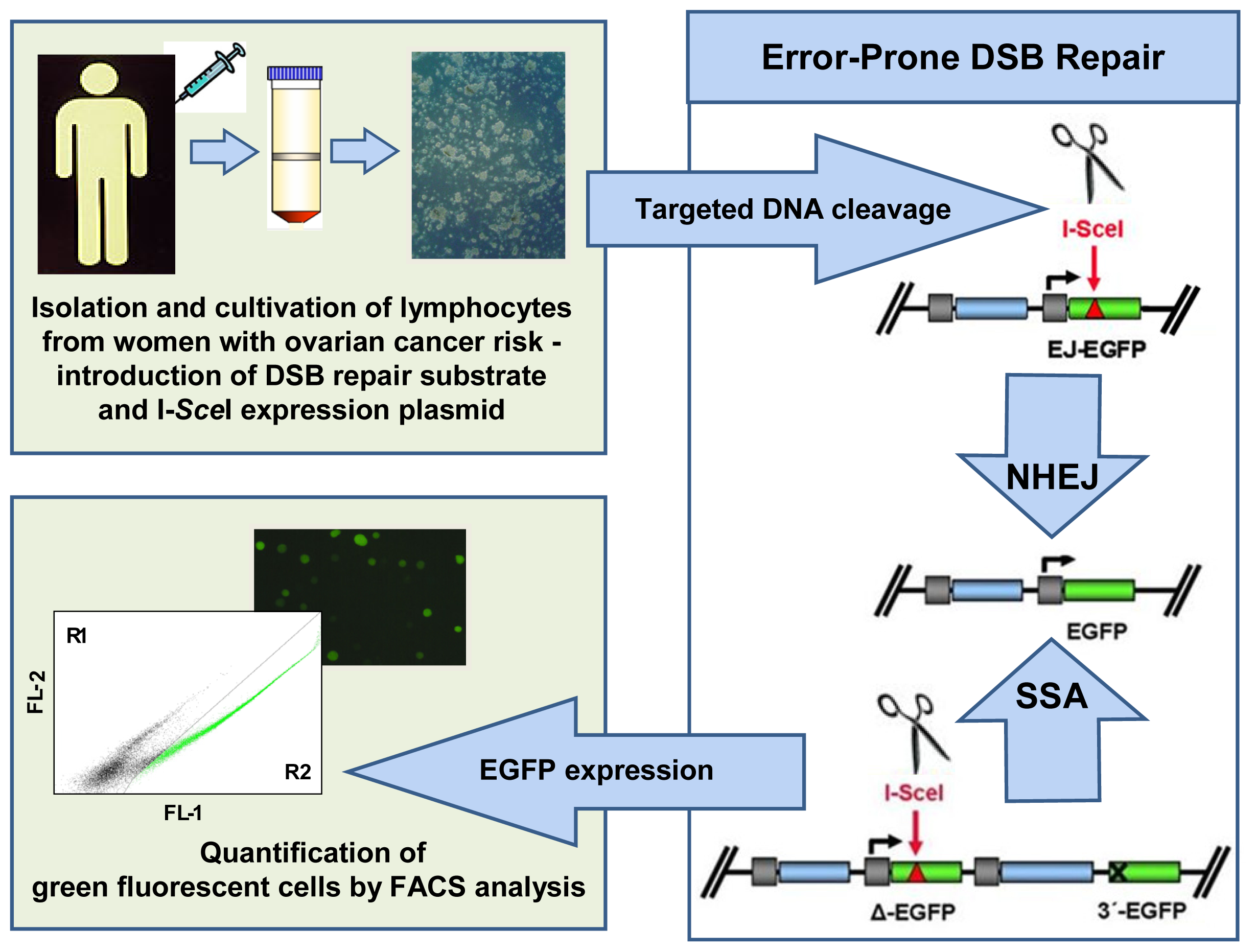
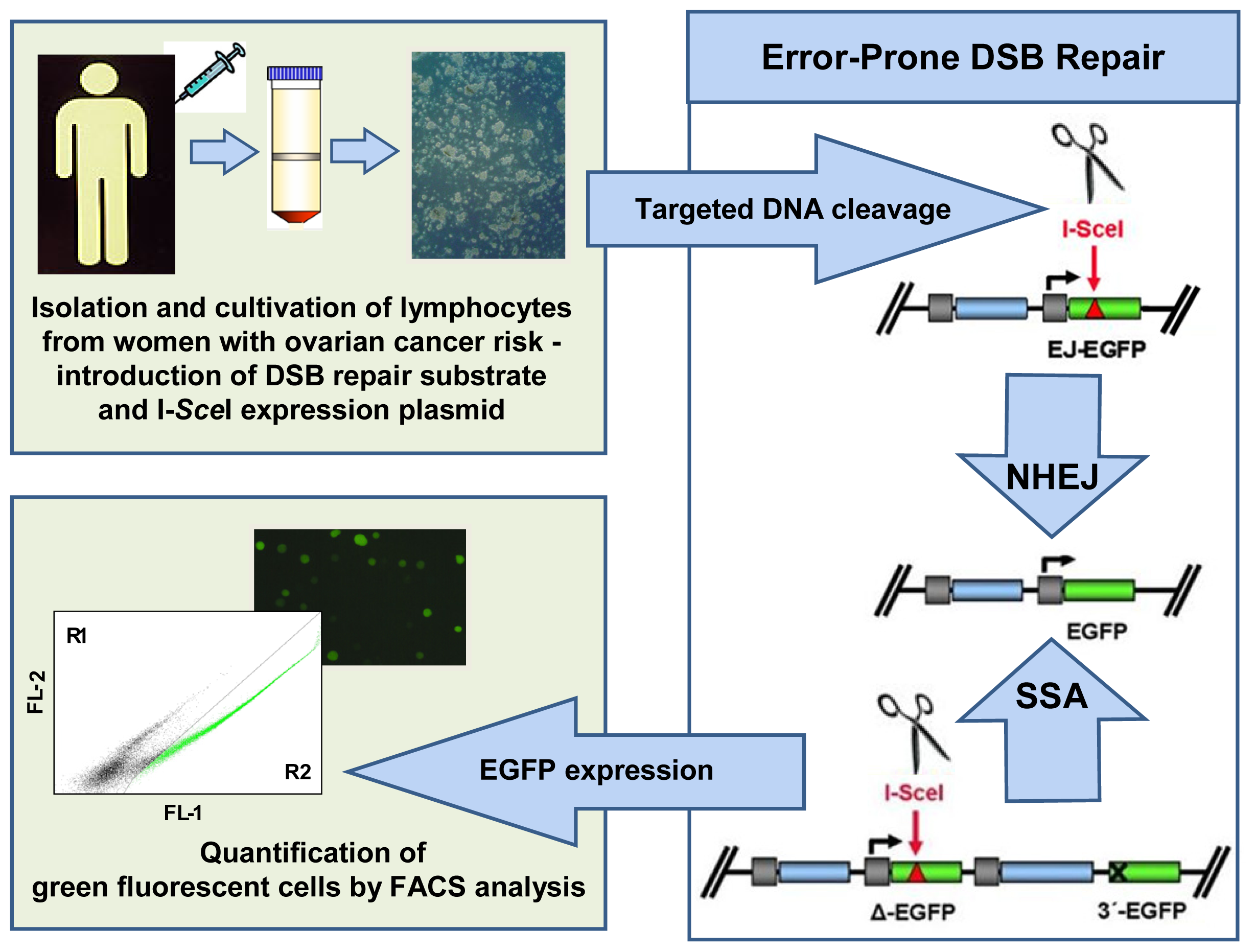
4.4.2. Pathway-Specific DSB Repair Analysis
5. Therapy: Reality and Hopes
5.1. Standard of Care and Its Refinement
5.2. The Potential of PARP Inhibitor Therapy
6. Conclusions
Acknowledgments
Abbreviations
| AT | Ataxia telangiectasia |
| ATM | Ataxia telangiectasia mutated |
| Bcl-2 | B-cell lymphoma 2 |
| BARD1 | BRCA1-associated RING domain protein 1 |
| BACH1 | BRCA1-associated C-terminal helicase 1 |
| BAP1 | BRCA1-associated protein 1 |
| BRCA1 | breast-cancer susceptibility gene 1 |
| BRCA2 | breast-cancer susceptibility gene 2 |
| BRIP1 | BRCA1 interacting protein 1 |
| CA125 | cancer antigen 125 |
| Chk2 | checkpoint kinase 2 |
| CAN | acquired somatic copy number aberration |
| CNV | copy number variation |
| CAT | computerized axial tomography |
| CtIP | C-terminal-binding protein interacting protein |
| DSB | DNA double-strand break |
| EGFP | enhanced green fluorescent protein |
| EGFR | epidermal growth factor receptor |
| FACS | fluorescence activated cell sorting |
| FA | Fanconi anemia |
| HER2 | human epidermal growth factor receptor 2 |
| HE4 | human epididymis protein 4 |
| FIGO | International Federation of Obstetricians and Gynecologists |
| LFS | Li-Fraumeni syndrome |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK/ERK kinase |
| MGMT | O6-methylguanine-DNA-methyltransferase |
| MMR | mismatch repair |
| MLPA | multiplex ligation-dependent probe amplification |
| MRE11 | meiotic recombination 11 |
| MRN-complex | protein complex consisting of MRE11, RAD50, and Nibrin |
| MLH | MutL homolog |
| MSH | MutS homolog |
| NHEJ | non-homologous end-joining |
| MRI | nuclear magnetic resonance imaging |
| NER | nucleotide excision repair |
| PALB2 | partner and localizer of BRCA2 |
| PJS | Peutz-Jeghers syndrome |
| PI3K | phosphatidylinositol 3-kinase |
| PTEN | phosphatase and tensin homolog |
| γH2AX | phospho-histone AX |
| PARP | Poly(ADP-ribose)polymerase |
| PMS | postmeiotic segregation |
| RAD51C | RAD51 homolog C |
| RAD51D | RAD51 homolog D |
| Ras | rat sarcoma |
| Raf | rapidly accelerated fibrosarcoma or rat fibrosarcoma |
| ROMA | risk of ovarian malignancy algorithm |
| STK11 | serine/threonine kinase 11 |
| SNP | single nucleotide polymorphism |
| SSA | single-strand annealing |
| TNFα | tumor necrosis factor α |
| TP53 | gene encoding human p53 |
- Conflict of InterestThe authors declare no conflict of interest.
References
- Hunn, J.; Rodriguez, G.C. Ovarian cancer: Etiology, risk factors, and epidemiology. Clin. Obstet. Gynecol 2012, 55, 3–23. [Google Scholar]
- Altekruse, S.F.; Kosary, C.L.; Krapcho, M.; Neuman, N.; Aminou, R.; Waldron, W.; Ruhl, J.; Howlader, N.; Tatalovich, Z.; Cho, H.; et al. SEER Cancer Statistics Review, 1975–2009. Available online: http://seer.cancer.gov/csr/1975_2007/ accessed on 17 July 2012.
- Stratton, J.F.; Pharoah, P.; Smith, S.K.; Easton, D.; Ponder, B.A. A systematic review and meta-analysis of family history and risk of ovarian cancer. Br. J. Obstet. Gynaecol 1998, 105, 493–499. [Google Scholar]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet 2010, 42, 410–414. [Google Scholar]
- Loveday, C.; Turnbull, C.; Ruark, E.; Xicola, R.M.; Ramsay, E.; Hughes, D.; Warren-Perry, M.; Snape, K.; Eccles, D.; et al. Breast Cancer Susceptibility Collaboration (UK). Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet 2012, 44, 475–476. [Google Scholar]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet 2003, 72, 1117–1130. [Google Scholar]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Fan, I.; Tang, J.; Li, S.; Zhang, S.; Shaw, P.A.; et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: A kin-cohort study in Ontario, Canada. J. Natl. Cancer Inst 2006, 98, 1694–706. [Google Scholar]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol 2007, 25, 1329–1233. [Google Scholar]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet 2011, 43, 879–882. [Google Scholar]
- Lynch, H.T.; Casey, M.J.; Snyder, C.L.; Bewtra, C.; Lynch, J.F.; Butts, M.; Godwin, A.K. Hereditary ovarian cancer: Molecular genetics, pathology, management, and heterogeneity. Mol. Oncol 2009, 3, 97–137. [Google Scholar]
- Pennington, K.P.; Swisher, E.M. Hereditary ovarian cancer: Beyond he usual suspects. Gynecol. Oncol 2012, 124, 347–353. [Google Scholar]
- Sourbier, C. Ovarian cancer: Emerging molecular-targeted therapies. Biologics 2012, 6, 147–154. [Google Scholar]
- Slomski, A. Screening women for ovarian cancer still does more harm than good. JAMA 2012, 307, 2474–2475. [Google Scholar]
- Longuespee, R.; Boyon, C.; Desmons, A.; Vinatier, D.; Leblanc, E.; Farre, I.; Wisztorski, M.; Ly, K.; D’Anjou, F.; Day, R.; et al. Ovarian cancer molecular pathology. Cancer Metastasis Rev 2012, 31, 713–732. [Google Scholar]
- Valentini, A.L.; Gui, B.; Miccò, M.; Mingote, M.C.; De Gaetano, A.M.; Ninivaggi, V.; Bonomo, L. Benign and suspicious ovarian masses—MR imaging criteria for characterization: Pictorial review. J. Oncol 2012, 2012, 481806. [Google Scholar]
- Rigakos, G.; Razis, E. BRCAness: Finding the achilles heel in ovarian cancer. Oncologist 2012, 17, 956–962. [Google Scholar]
- Claus, E.B.; Schwartz, P.E. Familial ovarian cancer. Update and clinical applications. Cancer 1995, 76, 1998–2003. [Google Scholar]
- Cass, I.; Baldwin, R.L.; Varkey, T.; Moslehi, R.; Narod, S.A.; Karlan, B.Y. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer 2003, 97, 2187–2195. [Google Scholar]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol 2012, 30, 2654–2663. [Google Scholar]
- Kinzler, K.W.; Vogelstein, B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997, 386, 761–763. [Google Scholar]
- Howlett, N.G.; Taniguchi, T.; Durkin, S.G.; D’Andrea, A.D.; Glover, T.W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet 2005, 14, 693–701. [Google Scholar]
- Kennedy, R.D.; D’Andrea, A.D. The Fanconi anemia/BRCA pathway: New faces in the crowd. Genes Dev 2005, 19, 2925–2940. [Google Scholar]
- Walsh, T.; King, M.C. Ten genes for inherited breast cancer. Cancer Cell 2007, 11, 103–105. [Google Scholar]
- Surtees, J.A.; Argueso, J.L.; Alani, E. Mismatch repair proteins: Key regulators of genetic recombination. Cytogenet. Genome Res 2004, 107, 146–159. [Google Scholar]
- Siehler, S.Y.; Schrauder, M.; Gerischer, U.; Cantor, S.; Marra, G.; Wiesmüller, L. Human Mlh1 monitors homologous recombination independently of mismatch repair and damage signaling. DNA Repair 2009, 8, 242–252. [Google Scholar]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol 2008, 9, 759–769. [Google Scholar]
- Gatz, S.A.; Wiesmüller, L. p53 in recombination and repair. Cell Death Differ 2006, 13, 1003–1006. [Google Scholar]
- Thompson, L.H.; Hinz, J.M. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: Mechanistic insights. Mutat. Res 2009, 668, 54–72. [Google Scholar]
- O’Brien, V.; Brown, R. Signalling cell cycle arrest and cell death through the MMR system. Carcinogenesis 2006, 27, 682–692. [Google Scholar]
- Ewald, B.; Sampath, D.; Plunkett, W. ATM and the Mre11-Rad50-Nbs1 complex respond to nucleoside analogue-induced stalled replication forks and contribute to drug resistance. Cancer Res 2008, 68, 7947–7955. [Google Scholar]
- Sato, K.; Ohta, T.; Venkitaraman, A.R. A mitotic role for the DNA damage-responsive CHK2 kinase. Nat. Cell Biol 2010, 12, 424–425. [Google Scholar]
- Hombauer, H.; Srivatsan, A.; Putnam, C.D.; Kolodner, R.D. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 2011, 334, 1713–1716. [Google Scholar]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar]
- Mukhopadhyay, A.; Elattar, A.; Cerbinskaite, A.; Wilkinson, S.J.; Drew, Y.; Kyle, S.; Los, G.; Hostomsky, Z.; Edmondson, R.J.; Curtin, N.J. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin. Cancer Res. 2010, 16, 2344–2351. [Google Scholar]
- Rothfuss, A.; Schütz, P.; Bochum, S.; Volm, T.; Eberhardt, E.; Kreienberg, R.; Vogel, W.; Speit, G. Induced micronucleus frequencies in peripheral lymphocytes as a screening test for carriers of a BRCA1 mutation in breast cancer families. Cancer Res 2000, 60, 390–394. [Google Scholar]
- Redon, C.E.; Nakamura, A.J.; Zhang, Y.W.; Ji, J.J.; Bonner, W.M.; Kinders, R.J.; Parchment, R.E.; Doroshow, J.H.; Pommier, Y. Histone γH2AX and Poly(ADP-Ribose) as clinical pharmacodynamic biomarkers. Clin. Cancer Res 2010, 16, 4532–4542. [Google Scholar]
- Keimling, M.; Deniz, M.; Varga, D.; Stahl, A.; Schrezenmeier, H.; Kreienberg, R.; Hoffmann, I.; König, J.; Wiesmüller, L. The power of DNA double-strand break (DSB) repair testing to predict breast cancer susceptibility. FASEB J 2012, 26, 2094–2104. [Google Scholar]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar]
- Somyajit, K.; Subramanya, S.; Nagaraju, G. RAD51C: A novel cancer susceptibility gene is linked to FANconi anemia and breast cancer. Carcinogenesis 2010, 12, 2031–2038. [Google Scholar]
- Bozzao, C.; Lastella, P.; Stella, A. Anticipation in lynch syndrome: Where we are where we go. Curr. Genomics 2011, 12, 451–465. [Google Scholar]
- Thorstenson, Y.R.; Roxas, A.; Kroiss, R.; Jenkins, M.A.; Yu, K.M.; Bachrich, T.; Muhr, D.; Wayne, T.L.; Chu, G.; Davis, R.W.; et al. Contributions of ATM mutations to familial breast and ovarian cancer. Cancer Res 2003, 63, 3325–3333. [Google Scholar]
- Roemer, K. Mutant p53: Gain-of-function oncoproteins and wild-type p53 inactivators. Biol. Chem 1999, 380, 879–887. [Google Scholar]
- Malkin, D. Li-Fraumeni syndrome. Genes Cancer 2011, 2, 475–484. [Google Scholar]
- McCuaig, J.M.; Armel, S.R.; Novokmet, A.; Ginsburg, O.M.; Demsky, R.; Narod, S.A.; Malkin, D. Routine TP53 testing for breast cancer under age 30: Ready for prime time? Fam. Cancer 2012, 11, 607–613. [Google Scholar]
- Bertrand, P.; Saintigny, Y.; Lopez, B.S. p53’s double life: Transactivation-independent repression of homologous recombination. Trends Genet 2004, 20, 235–243. [Google Scholar]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar]
- Jin, S.; Mao, H.; Schnepp, R.W.; Sykes, S.M.; Silva, A.C.; D’Andrea, A.D.; Hua, X. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res 2003, 63, 4204–4210. [Google Scholar]
- Marek, L.R.; Kottemann, M.C.; Glazer, P.M.; Bale, A.E. MEN1 and FANCD2 mediate distinct mechanisms of DNA crosslink repair. DNA Repair 2008, 7, 476–486. [Google Scholar]
- Gallo, A.; Agnese, S.; Esposito, I.; Galgani, M.; Avvedimento, V.E. Menin stimulates homology-directed DNA repair. FEBS Lett 2010, 584, 4531–4536. [Google Scholar]
- Lindor, N.M.; McMaster, M.L.; Lindor, C.J.; Greene, M.H. Concise handbook of familial cancer susceptibility syndromes- second edition. J. Natl. Cancer Inst. Monogr 2008, 38, 1–93. [Google Scholar]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar]
- Schrauder, M.; Wiesmüller, L. DNA Repair. In Apoptosis and Cancer Therapy, 1st ed; Debatin, K.M., Fulda, S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; Volume 2, pp. 822–846. [Google Scholar]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet 2008, 9, 619–631. [Google Scholar]
- Liu, Y.; Kadyrov, F.A.; Modrich, P. PARP-1 enhances the mismatch-dependence of 5′-directed excision in human mismatch repair in vitro. DNA Repair 2011, 10, 1145–1153. [Google Scholar]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol 2010, 17, 11–16. [Google Scholar]
- Weterings, E.; Chen, D.J. The endless tale of non-homologous end-joining. Cell Res 2008, 18, 114–124. [Google Scholar]
- O’Donovan, P.J.; Livingston, D.M. BRCA1 and BRCA2: Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 2010, 31, 961–967. [Google Scholar]
- Nussenzweig, A.; Nussenzweig, M.C. A backup DNA repair pathway moves to the forefront. Cell 2007, 131, 223–225. [Google Scholar]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 2009, 28, 2601–2615. [Google Scholar]
- Malpica, A.; Deavers, M.T.; Lu, K.; Bodurka, D.C.; Atkinson, E.N.; Gershenson, D.M.; Silva, E.G. Grading ovarian serous carcinoma using a two-tier system. Am. J. Surg. Pathol 2004, 28, 496–504. [Google Scholar]
- Shih, I.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol 2004, 164, 1511–1518. [Google Scholar]
- Cho, K.R.; Shih, I. Ovarian cancer. Annu. Rev. Pathol 2009, 4, 287–313. [Google Scholar]
- Merritt, M.A.; Cramer, D.W. Molecular pathogenesis of endometrial and ovarian cancer. Cancer Biomarkers 2010, 9, 287–305. [Google Scholar]
- Kaku, T.; Ogawa, S.; Kawano, Y.; Ohishi, Y.; Kobayashi, H.; Hirakawa, T.; Nakano, H. Histological classification of ovarian cancer. Med. Electron. Microsc 2003, 36, 9–17. [Google Scholar]
- Cai, K.Q.; Albarracin, C.; Rosen, D.; Zhong, R.; Zheng, W.; Luthra, R.; Broaddus, R.; Liu, J. Microsatellite instability and alteration of the expression of hMLH1 and hMSH2 in ovarian clear cell carcinoma. Hum. Pathol 2004, 35, 552–559. [Google Scholar]
- Naik, J.D.; Seligmann, J.; Perren, T.J. Mucinous tumours of the ovary. J. Clin. Pathol 2012, 65, 580–584. [Google Scholar]
- Wamunyokoli, F.W.; Bonome, T.; Lee, J.Y.; Feltimate, C.M.; Welch, W.R.; Radonovich, M.; Pise-Masison, C.; Brady, J.; Hao, K.; Berkowitz, R.S.; et al. Expression profiling of mucinous tumors of the ovary identifies genes of clinicopathologic importance. Clin. Cancer Res 2006, 12, 690–700. [Google Scholar]
- Koh, S.C.; Razvi, K.; Chan, Y.H.; Narasimhan, K.; Ilancheran, A.; Low, J.J.; Choolani, M. Ovarian Cancer Research Consortium of SE Asia. The association with age, human tissue kallikreins 6 and 10 and hemostatic markers for survival outcome from epithelial ovarian cancer. Arch. Gynecol. Obstet. 2011, 284, 183–190. [Google Scholar]
- Meden, H.; Fattahi-Meibodi, A. CA 125 in benign gynecological conditions. Int. J. Biol. Markers 1998, 13, 231–237. [Google Scholar]
- Kim, J.H.; Skates, S.J.; Uede, T.; Wong, K.K.; Schorge, J.O.; Feltmate, C.M.; Berkowitz, R.S.; Cramer, D.W.; Mok, S.C. Osteopontin as a potential diagnostic biomarker for ovarian cancer. JAMA 2002, 287, 1671–1679. [Google Scholar]
- Diamandis, E.P.; Scorilas, A.; Fracchioli, S.; van Gramberen, M.; de Bruijn, H.; Henrik, A.; Soosaipillai, A.; Grass, L.; Yousef, G.M.; Stenman, U.H.; et al. Human kallikrein 6 (hK6): A new potential serum biomarker for diagnosis and prognosis of ovarian carcinoma. J. Clin. Oncol 2003, 21, 1035–1043. [Google Scholar]
- Kishi, T.; Grass, L.; Soosaipillai, A.; Scorilas, A.; Harbeck, N.; Schmalfeldt, B.; Dorn, J.; Mysliwiec, M.; Schmitt, M.; Diamandis, E.P. Human kallikrein 8, a novel biomarker for ovarian carcinoma. Cancer Res 2003, 63, 2771–2774. [Google Scholar]
- Matsuzaki, H.; Kobayashi, H.; Yagyu, T.; Wakahara, K.; Kondo, T.; Kurita, N.; Sekino, H.; Inagaki, K.; Suzuki, M.; Kanayama, N.; et al. Plasma bikunin as a favorable prognostic factor in ovarian cancer. J. Clin. Oncol 2005, 23, 1463–1472. [Google Scholar]
- Nowee, M.E.; Snijders, A.M.; Rockx, D.A.; de Wit, R.M.; Kosma, V.M.; Hämäläinen, K.; Schouten, J.P.; Verheijen, R.H.; van Diest, P.J.; Albertson, D.G.; et al. DNA profiling of primary serous ovarian and fallopian tube carcinomas with array comparative genomic hybridization and multiplex ligation-dependent probe amplification. J. Pathol 2007, 213, 46–55. [Google Scholar]
- Bandiera, E.; Franceschini, R.; Specchia, C.; Bignotti, E.; Trevisiol, C.; Gion, M.; Pecorelli, S.; Santin, A.D.; Ravaggi, A. Prognostic significance of vascular endothelial growth factor serum determination in women with ovarian cancer. ISRN Obstet. Gynecol. 2012, 2012, 245756. [Google Scholar]
- Moore, R.G.; Brown, A.K.; Miller, M.C.; Skates, S.; Allard, W.J.; Verch, T.; Steinhoff, M.; Messerlian, G.; DiSilvestro, P.; Granai, C.O.; et al. The use of multiple novel tumor biomarkers for the detection of ovarian carcinoma in patients with a pelvic mass. Gynecol. Oncol 2008, 108, 402–408. [Google Scholar]
- Yurkovetsky, Z.; Skates, S.; Lomakin, A.; Nolen, B.; Pulsipher, T.; Modugno, F.; Marks, J.; Godwin, A.; Gorelik, E.; Jacobs, I.; et al. Development of a multimarker assay for early detection of ovarian cancer. J. Clin. Oncol 2010, 28, 2159–2166. [Google Scholar]
- Chan, K.K.; Chen, C.A.; Nam, J.H.; Ochiai, K.; Wilailak, S.; Choon, A.T.; Sabaratnam, S.; Hebbar, S.; Sickan, J.; Schodin, B.A.; et al. The use of HE4 in the prediction of ovarian cancer in Asian women with a pelvic mass. Gynecol. Oncol. 2012. [Google Scholar] [CrossRef]
- Escudero, J.M.; Auge, J.M.; Filella, X.; Torne, A.; Pahisa, J.; Molina, R. Comparison of serum human epididymis protein 4 with cancer antigen 125 as a tumor marker in patients with malignant and nonmalignant diseases. Clin. Chem. 2011, 57, 1534–1544. [Google Scholar]
- Freydanck, M.K.; Laubender, R.P.; Rack, B.; Schumacher, L.; Jeschke, U.; Scholz, C. Two-marker combinations for preoperative discrimination of benign and malignant ovarian masses. Anticancer Res 2012, 32, 2003–2008. [Google Scholar]
- Van Gorp, T.; Cadron, I.; Despierre, E.; Daemen, E.; Daemen, A.; Leunen, K.; Amant, F.; Timmermann, D.; de Moor, B.; Vergote, I. HE4 and CA125 as a diagnostic test in ovarian cancer: Prospective validation oft he risk of ovarian malignancy algorithm. Br. J. Cancer 2011, 104, 863–870. [Google Scholar]
- Van Gorp, T.; Veldman, J.; van Calster, B.; Cadron, I.; Leunen, K.; Amant, F.; Timmermann, D.; Vergote, I. Subjective assessment by ultrasound is superior to the risk of malignancy index (RMI) or the risk of ovarian malignancy algorithm (ROMA) in discriminating benign from malignant adnexal masses. Eur. J. Cancer 2012, 48, 1649–1656. [Google Scholar]
- Li, F.; Tie, R.; Chang, K.; Wang, F.; Deng, S.; Lu, W.; Yu, L.; Chen, M. Does risk for ovarian malignancy algorithm excel human epididymis protein 4 and ca125 in predicting epithelial ovarian cancer. A meta-analysis. BMC Cancer 2012, 19, 258. [Google Scholar]
- Trudel, D.; Tetu, B.; Gregoire, J.; Plante, M.; Renaud, M.C.; Bachvarov, D.; Douville, P.; Bairate, I. Human epididymis protein 4 (HE4) and ovarian cancer prognosis. Gynecol. Oncol. 2012. [Google Scholar] [CrossRef]
- Kong, S.Y.; Han, M.H.; Yoo, H.J.; Hwang, J.H.; Lim, M.C.; Seo, S.S.; Yoo, C.W.; Kim, J.H.; Park, S.Y.; Kang, S. Serum HE4 level is an independent prognostic factor in epithelial ovarian cancer. Ann. Surg. Oncol 2012, 19, 1707–1712. [Google Scholar]
- Georgakopoulos, P.; Mehmood, S.; Akalin, A.; Shroyer, K.R. Immunohistochemical localization of HE4 in benign, borderline, and malignant lesions of the ovary. Int. J. Gynecol. Pathol 2012, 31, 517–523. [Google Scholar]
- Novotny, Z.; Presl, J.; Kucera, R.; Topolcan, O.; Vrzalova, J.; Fuchsova, R.; Betincova, L.; Rokyta, Z. HE4 and ROMA index in Czech postmenopausal women. Anticancer Res 2012, 32, 4137–4140. [Google Scholar]
- Urban, N.; Thorpe, J.; Karlan, B.Y.; McIntosh, M.W.; Palomares, M.R.; Daly, M.B.; Paley, P.; Drescher, C.W. Interpretation of single and serial measures of HE4 and CA125 in asymptomatic women at high risk for ovarian cancer. Cancer Epidemiol. Biomarkers Prev 2012, 21, 2087–2094. [Google Scholar]
- Suh, K.S.; Park, S.W.; Castro, A.; Patel, H.; Blake, M.L.; Goy, A. Ovarian cancer biomarkers for molecular biosensors and translational medicine. Expert Rev. Mol. Diagn. 2010, 10, 1069–1083. [Google Scholar]
- Zhang, B.; Barekati, Z.; Kohler, C.; Radpour, R.; Asadollahi, R.; Holzgreve, W.; Zhong, X.Y. Proteomics and biomarkers for ovarian cancer diagnosis. Ann. Clin. Lab. Sci 2010, 40, 218–225. [Google Scholar]
- Nolen, B.M.; Lokshin, A.E. Protein biomarkers of ovarian cancer: The forest and the trees. Future Oncol 2012, 8, 55–74. [Google Scholar]
- Le Page, C.; Huntsman, D.G.; Provencher, D.M.; Mes-Masson, A.-M. Predictive and prognostic protein biomarkers in epithelial ovarian cancer: Recommendation for future studies. Cancers 2010, 2, 913–954. [Google Scholar]
- Visintin, I.; Feng, Z.; Longton, G.; Ward, D.C.; Alvero, A.B.; Lai, Y.; Tenthorey, J.; Leiser, A.; Flores-Saaib, R.; Yu, H.; et al. Diagnostic markers for early detection of ovarian cancer. Clin. Cancer Res 2008, 14, 1065–1072. [Google Scholar]
- Mcintosh, M.; Anderson, G.; Drescher, C.; Hanash, S.; Urban, N.; Brown, P.; Gambhir, S.S.; Coukos, G.; Laird, P.W.; Nelson, B.; et al. Ovarian cancer early detection claims are biased. Clin. Cancer Res 2008, 14, 7574. [Google Scholar]
- Zhang, Z.; Bast, R.C., Jr; Yu, Y.; Li, J.; Sokoll, L.J.; Rai, A.J.; Rosenzweig, J.M.; Cameron, B.; Wang, Y.Y.; Meng, X.Y.; et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 2004, 64, 5882–5890. [Google Scholar]
- Skates, S.J.; Horick, N.; Yu, Y.; Xu, F.J.; Berchuck, A.; Havrilesky, L.J.; de Bruijn, H.W.; van der Zee, A.G.; Woolas, R.P.; Jacobs, I.J.; et al. Preoperative sensitivity and specificity for early-stage ovarian cancer when combining cancer antigen CA-125II, CA 15–3, CA 72–4, and macrophage colony-stimulating factor using mixtures of multivariate normal distributions. J. Clin. Oncol 2004, 22, 4059–4066. [Google Scholar]
- Amonkar, S.D.; Bertenshaw, G.P.; Chen, T.H.; Bergstrom, K.J.; Zhao, J.; Seshaiah, P.; Yip, P.; Mansfield, B.C. Development and preliminary evaluation of a multivariate index assay for ovarian cancer. PLoS One 2009, 4, e4599. [Google Scholar]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet 2006, 7, 85–97. [Google Scholar]
- Alkan, C.; Bradley, P.C.; Eichler, E. Genome structural variation discovery and genotyping. Nat. Rev 2011, 12, 363–376. [Google Scholar]
- Stankiewicz, P.; Lupski, J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med 2010, 61, 437–455. [Google Scholar]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007, 315, 848–853. [Google Scholar]
- Albertson, D.G.; Collins, C.; McCormick, F.; Gray, J.W. Chromosome aberrations in solid tumors. Nat. Genet 2003, 34, 369–376. [Google Scholar]
- Kalb, R.; Neveling, K.; Nanda, I.; Schindler, D.; Hoehn, H. Fanconi Anemia Causes and Consequences of Genetic Instability. In Genome and Disease. Genome Dyn; Volff, J.N., Ed.; Karger: Basel, Switzerland, 2006; Volume 1, pp. 218–242. [Google Scholar]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol 2010, 11, 220–228. [Google Scholar]
- Engert, S.; Wappenschmidt, B.; Betz, B.; Kast, K.; Kutsche, M.; Hellebrand, H.; Goecke, T.O.; Kiechle, M.; Niederacher, D.; Schmutzler, R.K.; et al. MLPA screening in the BRCA1 gene from 1506 German hereditary breast cancer cases: Novel deletions, frequent involvement of exon 17, and occurrence in single early-onset cases. Hum. Mutat 2008, 29, 948–958. [Google Scholar]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.[Green Version]
- Hilton, J.L.; Geisler, J.P.; Rathe, J.A.; Hattermann-Zogg, M.A.; DeYoung, B.; Buller, R.E. Inactivation of BRCA1 and BRCA2 in ovarian cancer. J. Natl. Cancer Inst 2002, 94, 1396–1406. [Google Scholar]
- Nakayama, K.; Nakayama, N.; Kurman, R.J.; Cope, L.; Pohl, G.; Samuels, Y.; Velculescu, V.E.; Wang, T.L.; Shih, I.M. Sequence mutations and amplifications of PIK3CA and AKT2 genes in purified ovarian serous neoplasms. Cancer Biol. Ther 2006, 5, 779–785. [Google Scholar]
- Engler, D.A.; Gupta, S.; Growdon, W.B.; Drapkin, R.I.; Nitta, M.; Sergent, P.A.; Allred, S.F.; Gross, J.; Deavers, M.T.; Kuo, W.L.; et al. Genome wide DNA copy number analysis of serous type ovarian carcinomas identifies genetic markers predictive of clinical outcome. PLoS One 2012, 7, e30996. [Google Scholar]
- Gorringe, K.L.; George, J.; Anglesio, M.S.; Ramakrishna, M.; Etemadmoghadam, D.; Cowin, P.; Sridhar, A.; Williams, L.H.; Boyle, S.E.; Yanaihara, N.; et al. Copy number analysis identifies novel interactions between genomic loci in ovarian cancer. PLoS One 2010, 5, e11408. [Google Scholar]
- Schwartz, D.R.; Kardia, S.L.; Shedden, K.A.; Kuick, R.; Michailidis, G.; Taylor, J.M.; Misek, D.E.; Wu, R.; Zhai, Y.; Darrah, D.M.; et al. Gene expression in ovarian cancer reflects both morphology and biological behavior, distinguishing clear cell from other poor-prognosis ovarian carcinomas. Cancer Res 2002, 62, 4722–4729. [Google Scholar]
- Bonome, T.; Lee, J.Y.; Park, D.C.; Radonovich, M.; Pise-Masison, C.; Brady, J.; Gardner, G.J.; Hao, K.; Wong, W.H.; Barrett, J.C.; et al. Expression profiling of serous low malignant potential, low-grade, and high-grade tumors of the ovary. Cancer Res 2005, 65, 10602–10612. [Google Scholar]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res 2008, 14, 5198–5208. [Google Scholar]
- Jazaeri, A.A.; Awtrey, C.S.; Chandramouli, G.V.; Chuang, Y.E.; Khan, J.; Sotiriou, C.; Aprelikova, O.; Yee, C.J.; Zorn, K.K.; Birrer, M.J.; et al. Gene expression profiles associated with response to chemotherapy in epithelial ovarian cancers. Clin. Cancer Res 2005, 11, 6300–6310. [Google Scholar]
- Fekete, T.; Rásó, E.; Pete, I.; Tegze, B.; Liko, I.; Munkácsy, G.; Sipos, N.; Rigó, J., Jr; Györffy, B. Meta-analysis of gene expression profiles associated with histological classification and survival in 829 ovarian cancer samples. Int. J. Cancer 2012, 131, 95–105. [Google Scholar]
- Kang, J.; D’Andrea, A.D.; Kozono, D. A DNA repair pathway-focused score for prediction of outcomes in ovarian cancer treated with platinum-based chemotherapy. J. Natl. Cancer Inst 2012, 104, 670–681. [Google Scholar]
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.J.; Wilhelm-Benartzi, C.S.; Rizzo, S.; van der Zee, A.; Plumb, J.A.; Brown, R. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 2012, 31, 4567–4576. [Google Scholar]
- Balch, C.; Matei, D.E.; Huang, T.H.; Nephew, K.P. Role of epigenomics in ovarian and endometrial cancers. Epigenomics 2010, 2, 419–447. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar]
- Baldwin, R.L.; Nemeth, E.; Tran, H.; Shvartsman, H.; Cass, I.; Narod, S.; Karlan, B.Y. BRCA1 promoter region hypermethylation in ovarian carcinoma: A population-based study. Cancer Res 2000, 60, 5329–5333. [Google Scholar]
- Strathdee, G.; Appleton, K.; Illand, M.; Millan, D.W.; Sargent, J.; Paul, J.; Brown, R. Primary ovarian carcinomas display multiple methylator phenotypes involving known tumor suppressor genes. Am. J. Pathol 2001, 158, 1121–1127. [Google Scholar]
- Rathi, A.; Virmani, A.K.; Schorge, J.O.; Elias, K.J.; Maruyama, R.; Minna, J.D.; Mok, S.C.; Girard, L.; Fishman, D.A.; Gazdar, A.F. Methylation profiles of sporadic ovarian tumors and nonmalignant ovaries from high-risk women. Clin. Cancer Res 2002, 8, 3324–3331. [Google Scholar]
- Teodoridis, J.M.; Hall, J.; Marsh, S.; Kannall, H.D.; Smyth, C.; Curto, J.; Siddiqui, N.; Gabra, H.; McLeod, H.L.; Strathdee, G.; et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res 2005, 65, 8961–8967. [Google Scholar]
- De Caceres, I.; Battagli, C.; Esteller, M.; Herman, J.G.; Dulaimi, E.; Edelson, M.I.; Bergman, C.; Ehya, H.; Eisenberg, B.L.; Cairns, P. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res 2004, 64, 6476–6481. [Google Scholar]
- Wiley, A.; Katsaros, D.; Chen, H.; Rigault de la Longrais, I.A.; Beeghly, A.; Puopolo, M.; Singal, R.; Zhang, Y.; Amoako, A.; Zelterman, D.; et al. Aberrant promoter methylation of multiple genes in malignant ovarian tumors and in ovarian tumors with low malignant potential. Cancer 2006, 107, 299–308. [Google Scholar]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar]
- Maradeo, M.E.; Cairns, P. Translational application of epigenetic alterations: Ovarian cancer as a model. FEBS Lett 2011, 585, 2112–2120. [Google Scholar]
- Stefansson, O.A.; Villanueva, A.; Vidal, A.; Martí, L.; Esteller, M. BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics 2012, 7, 1225–1229. [Google Scholar]
- Ralhan, R.; Kaur, J.; Kreienberg, R.; Wiesmüller, L. Links between DNA double strand break repair and breast cancer: Accumulating evidence from both familial and nonfamilial cases. Cancer Lett 2007, 248, 1–17. [Google Scholar]
- Litman, R.; Peng, M.; Jin, Z.; Zhang, F.; Zhang, J.; Powell, S.; Andreassen, P.R.; Cantor, S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005, 8, 255–265. [Google Scholar]
- Fenech, M.; Morley, A.A. Kinetochore detection in micronuclei: An alternative method for measuring chromosome loss. Mutagenesis 1989, 4, 98–104. [Google Scholar]
- Fenech, M. The in vitro micronucleus technique. Mutat. Res 2000, 455, 81–95. [Google Scholar]
- Zunino, A.; Degan, P.; Vigo, T.; Abbondandolo, A. Hydrogen peroxide: Effects on DNA, chromosomes, cell cycle and apoptosis induction in Fanconi’s anemia cell lines. Mutagenesis 2001, 16, 283–288. [Google Scholar]
- Gutiérrez-Enríquez, S.; Hall, J. Use of the cytokinesis-block micronucleus assay to measure radiation-induced chromosome damage in lymphoblastoid cell lines. Mutat. Res 2003, 535, 1–13. [Google Scholar]
- Baeyens, A.; Thierens, H.; Claes, K.; Poppe, B.; de Ridder, L.; Vral, A. Chromosomal radiosensitivity in BRCA1 and BRCA2 mutation carriers. Int. J. Radiat. Biol 2004, 80, 745–756. [Google Scholar]
- Cardinale, F.; Bruzzi, P.; Bolognesi, C. Role of micronucleus test in predicting breast cancer susceptibility: A systematic review and meta-analysis. Br. J. Cancer 2012, 106, 780–790. [Google Scholar]
- Akyüz, N.; Boehden, G.S.; Süsse, S.; Rimek, A.; Preuss, U.; Scheidtmann, K.H.; Wiesmüller, L. DNA substrate dependence of p53-mediated regulation of double-strand break repair. Mol. Cell Biol 2002, 22, 6306–6317. [Google Scholar]
- Keimling, M.; Kaur, J.; Bagadi, S.A.; Kreienberg, R.; Wiesmüller, L.; Ralhan, R. A sensitive test for the detection of specific DSB repair defects in primary cells from breast cancer specimens. Int. J. Cancer 2008, 123, 730–736. [Google Scholar]
- Keimling, M.; Volcic, M.; Csernok, A.; Wieland, B.; Dörk, T.; Wiesmüller, L. Functional characterization connects individual patient mutations in ataxia telangiectasia mutated (ATM) with dysfunction of specific DNA double-strand break-repair signaling pathways. FASEB J 2011, 25, 3849–3860. [Google Scholar]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol 2008, 26, 3785–3790. [Google Scholar]
- Romero, I.; Bast, R.C., Jr. Minireview: Human ovarian cancer: Biology, current management, and paths to personalizing therapy. Endocrinology 2012, 153, 1593–1602. [Google Scholar]
- Heintz, A.P.; Odicino, F.; Maisonneuve, P.; Quinn, M.A.; Benedet, J.L.; Creasman, W.T.; Ngan, H.Y.; Pecorelli, S.; Beller, U. Carcinoma of the ovary. FIGO 26th annual report on the results of treatment in gynecological cancer. Int. J Gynaecol. Obstet 2006, 95, S161–S192. [Google Scholar]
- Bristow, R.E.; Tomacruz, R.S.; Armstrong, D.K.; Trimble, E.L.; Montz, FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during paltinum era: A-meta analysis. J. Clin. Oncol. 2002, 20, 1248–1259. [Google Scholar]
- Kommoss, S.; Rochon, J.; Harter, P.; Heitz, F.; Grabowski, J.P.; Ewald-Riegler, N.; Haberstroh, M.; Neunhoeffer, T.; Barinoff, J.; Gomez, R.; et al. Prognostic impact of additional extended surgical procedures in advanced-stage primary ovarian cancer. Ann. Surg. Oncol 2010, 17, 279–286. [Google Scholar]
- Harter, P.; du Bois, A. The role of surgery in ovarian cancer with special emphasis on cytoreductive surgery for recurrence. Curr. Opin. Oncol 2005, 17, 505–514. [Google Scholar]
- Rose, P.G.; Nerenstone, S.; Brady, M.F.; Clarke-Pearson, D.; Olt, G.; Rubin, S.C.; Moore, D.H.; Small, J.M. Gynecologic Oncology Group. Secondary surgical cytoreduction for advanced ovarian carcinoma. N. Eng. J. Med. 2004, 351, 2489–2497. [Google Scholar]
- Du Bois, A.; Marth, C.; Pfisterer, J.; Harter, P.; Hilpert, F.; Zeimet, A.G.; Sehouli, J. Neoadjuvant chemotherapy cannot be regarded as adequat routine therapy strategy of advanced ovarian cancer. Int. J. Gynaecol. Cancer 2012, 22, 182–185. [Google Scholar]
- International Collaborative Ovarian Neoplasm Group. Paclitaxel plus carboplatin versus standard chemotherapy with either single-agent carboplatin or cyclophosphamide, doxorubicin, and cisplatin in women with ovarian cancer: The ICON3 randomised trial. Lancet 2002, 360, 505–515.
- Du Bois, A.; Lück, H.J.; Meier, W.; Adams, H.P.; Möbus, V.; Costa, S.; Bauknecht, T.; Richter, B.; Warm, M.; Schröder, W.; et al. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J. Natl. Cancer Inst 2003, 95, 1320–1329. [Google Scholar]
- Du Bois, A.; Pfisterer, J.; Burchardi, N.; Loibl, S.; Huober, J.; Wimberger, P.; Burges, A.; Stähle, A.; Jackisch, C.; Kölbl, H.; et al. Combination therapy with pegylated liposomal doxorubicin and carboplatin in gynecologic malignancies: A prospective phase II study of the Arbeitsgemeinschaft Gynäekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and Kommission Uterus (AGO-K-Ut). Gynecol. Oncol 2007, 107, 518–525. [Google Scholar]
- Du Bois, A.; Herrstedt, J.; Hardy-Bessard, A.C.; Müller, H.H.; Harter, P.; Kristensen, G.; Joly, F.; Huober, J.; Avall-Lundqvist, E.; Weber, B.; et al. Phase III trial of carboplatin plus paclitaxel with or without gemcitabine in first-line treatment of epithelial ovarian cancer. J. Clin. Oncol 2010, 28, 4162–4169. [Google Scholar]
- Fujiwara, K.; Aotani, E.; Hamano, T.; Nagao, S.; Yoshikawa, H.; Sugiyama, T.; Kigawa, J.; Aoki, D.; Katsumata, N.; Takeuchi, M.; et al. A randomized Phase II/III trial of 3 weekly intraperitoneal versus intravenous carboplatin in combination with intravenous weekly dose-dense paclitaxel for newly diagnosed ovarian, fallopian tube and primary peritoneal cancer. Jpn. J. Clin. Oncol 2011, 41, 278–282. [Google Scholar]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. ICON7 Investigators. A phase 3 trials of bevacicumab in ovarian cancer. N. Engl. J. Med 2011, 365, 2484–2496. [Google Scholar]
- Tan, D.S.; Rothermundt, C.; Thomas, K.; Bancroft, E.; Eeles, R.; Shanley, S.; Ardern-Jones, A.; Norman, A.; Kaye, S.B.; Gore, M.E. “BRCAness” syndrome in ovarian cancer: A case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J. Clin. Oncol 2008, 26, 5530–5536. [Google Scholar]
- Huehls, A.M.; Wagner, J.M.; Huntoon, C.J.; Karnitz, L.M. Identification of DNA repair pathways that affect the survival of ovarian cancer cells treated with a PARP inhibitor in a novel drug combination. Mol. Pharmacol 2012, 82, 767–776. [Google Scholar]
- Mégnin-Chanet, F.; Bollet, M.A.; Hall, J. Targeting poly(ADP-ribose)polymerase activity for cancer therapy. Cell Mol. Life Sci 2010, 67, 3649–3662. [Google Scholar]
- Weil, M.K.; Chen, A.P. PARP inhibitor treatment in ovarian and breast cancer. Curr. Probl. Cancer 2012, 35, 7–50. [Google Scholar]
- Bryant, H.E.; Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med 2009, 361, 123–134. [Google Scholar]
- Annunziata, C.M.O.; Shaughnessy, J. Poly (ADP-ribose) polymerase as a novel therapeutic target in cancer. Clin. Cancer Res. 2010, 16, 4517–4526. [Google Scholar]
- Chionh, F.; Mitchell, G.; Lindeman, G.J.; Friedlander, M.; Scott, C.L. The role of poly adenosine diphosphate ribose polymerase inhibitors in breast and ovarian cancer: Current status and future directions. Asia Pac. J. Clin. Oncol 2011, 7, 197–211. [Google Scholar]
- Patel, A.G.; De Lorenzo, S.B.; Flatten, K.S.; Poirier, G.G.; Kaufmann, S.H. Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clin. Cancer Res 2012, 18, 1655–1662. [Google Scholar]
- Liu, X.; Shi, Y.; Maag, D.X.; Palma, J.P.; Patterson, M.J.; Ellis, P.A.; Surber, B.W.; Ready, D.B.; Soni, N.B.; Ladror, U.S.; et al. Iniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a Bona Fide PARP inhibitor. Clin. Cancer Res 2012, 18, 510–523. [Google Scholar]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol 2010, 28, 2512–2519. [Google Scholar]
- Wang, Z.C.; Birkbak, N.J.; Culhane, A.C.; Drapkin, R.; Fatima, A.; Tian, R.; Schwede, M.; Alsop, K.; Daniels, K.E.; Piao, H.; et al. Profiles of genomic instability in hgh-grade serous ovarian cancer predict treatment outcome. Clin. Cancer Res 2012, 18, 5806–5815. [Google Scholar]
- Mukhopadhyay, A.; Plummer, E.R.; Elattar, A.; Soohoo, S.; Uzir, B.; Quinn, J.E.; McCluggaga, W.G.; Maxwell, P.; Aneke, H.; Curtin, N.J.; et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: Sensitivity to PARP inhibitors, platinum, and survival. Cancer Res 2012, 15, 5675–5682. [Google Scholar]
- Nishikawa, H.; Wu, W.; Koike, A.; Kojima, R.; Gomi, H.; Fukuda, M.; Ohta, T. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res 2009, 69, 111–119. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| BRCA1 | BRCA2 | RAD51C | RAD51D | |
|---|---|---|---|---|
| Ovarian cancer | 20%–50% | 10%–20% | >9% | 10% |
| Breast cancer (female) | 50%–90% | 40%–60% | * n.s. | n.s. |
| Breast cancer (male) | 0% | 11% | 0% | ? |
| Prostate cancer | 7% | 31% | ? | ? |
| Histological subtype of ovarian cancer | Clinicopathological characteristics | Associated gene mutations or pathway defects * |
|---|---|---|
| Serous | histology: usually large; often bilateral; mixture of cystic, papillary, and solid growth; usually marked nuclear atypia | |
| low-grade: slow proliferation/progression; poor response to chemotherapy | low-grade: BRAF and KRAS mutations (Ras/Raf/MEK/MAPK pathway) | |
| high-grade: rapid proliferation/progression; poor response to chemotherapy; recurrence | high-grade: germline BRCA1/2 mutations; TP53 mutations; HER2/neu amplifications; AKT2 amplifications (PI3K pathway) | |
| Endometrioid | histology: strong resemblance to endometrial adenocarcinoma; associated with endometriosis | PTEN mutations (PI3K pathway); β-catenin mutations (WNT signaling); Microsatellite instability via loss of MMR gene expression; high-grade: TP53 mutations |
| Clear cell | histology: growth tubular, papillary, solid, or frequently mixed types; associated with endometriosis | PI3K pathway deregulation; Microsatellite instability in subset; low frequency of KRAS, BRAF, and TP53 mutations |
| Mucinous | histology: usually unilocular; ~largest of all ovarian tumors; papillary and solid forms; associated with endometriosis; poor response to chemotherapy compared with serous cancer | frequently KRAS mutations; overexpression of mucin genes (MUC2, MUC3, and MUC17) |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Varga, D.; Deniz, M.; Schwentner, L.; Wiesmüller, L. Ovarian Cancer: In Search of Better Marker Systems Based on DNA Repair Defects. Int. J. Mol. Sci. 2013, 14, 640-673. https://doi.org/10.3390/ijms14010640
Varga D, Deniz M, Schwentner L, Wiesmüller L. Ovarian Cancer: In Search of Better Marker Systems Based on DNA Repair Defects. International Journal of Molecular Sciences. 2013; 14(1):640-673. https://doi.org/10.3390/ijms14010640
Chicago/Turabian StyleVarga, Dominic, Miriam Deniz, Lukas Schwentner, and Lisa Wiesmüller. 2013. "Ovarian Cancer: In Search of Better Marker Systems Based on DNA Repair Defects" International Journal of Molecular Sciences 14, no. 1: 640-673. https://doi.org/10.3390/ijms14010640
APA StyleVarga, D., Deniz, M., Schwentner, L., & Wiesmüller, L. (2013). Ovarian Cancer: In Search of Better Marker Systems Based on DNA Repair Defects. International Journal of Molecular Sciences, 14(1), 640-673. https://doi.org/10.3390/ijms14010640