Nitric Oxide Synthesis Is Increased in Cybrid Cells with m.3243A>G Mutation

Abstract
:1. Introduction
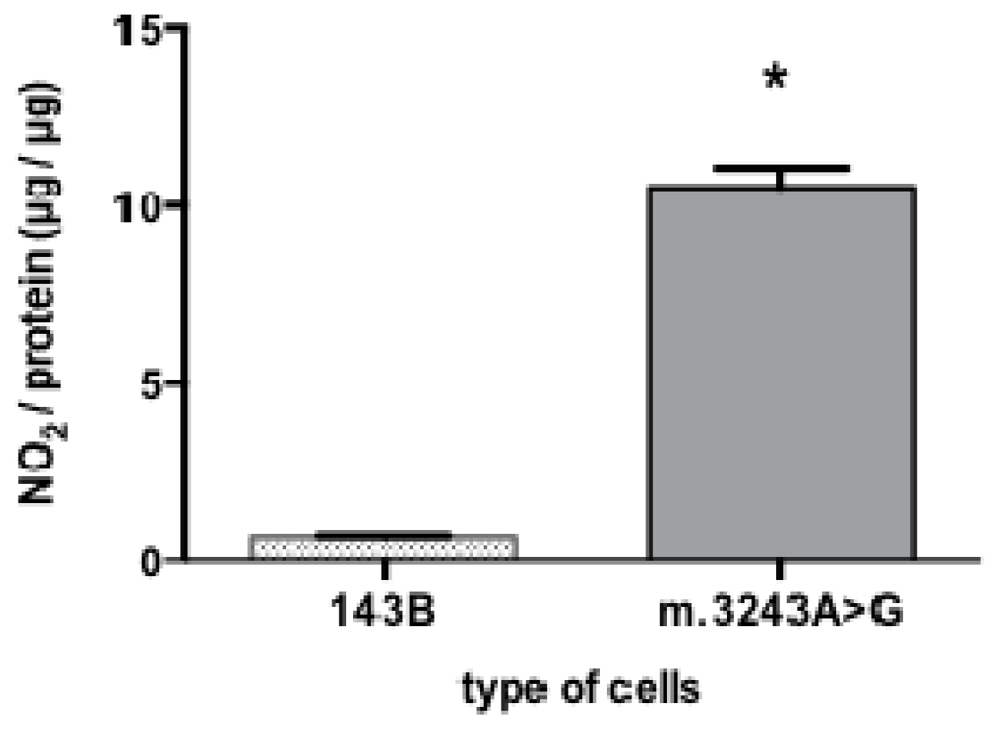
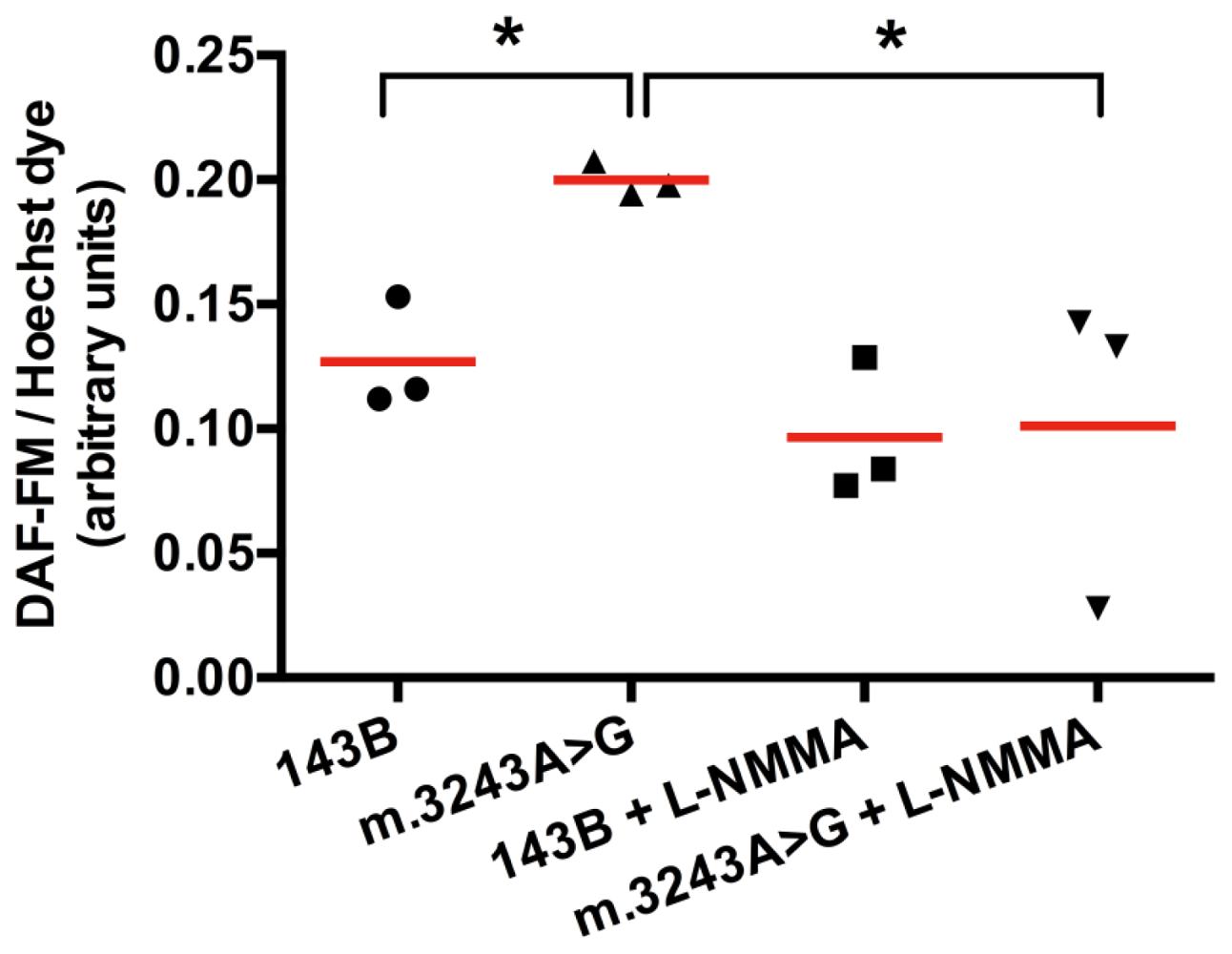
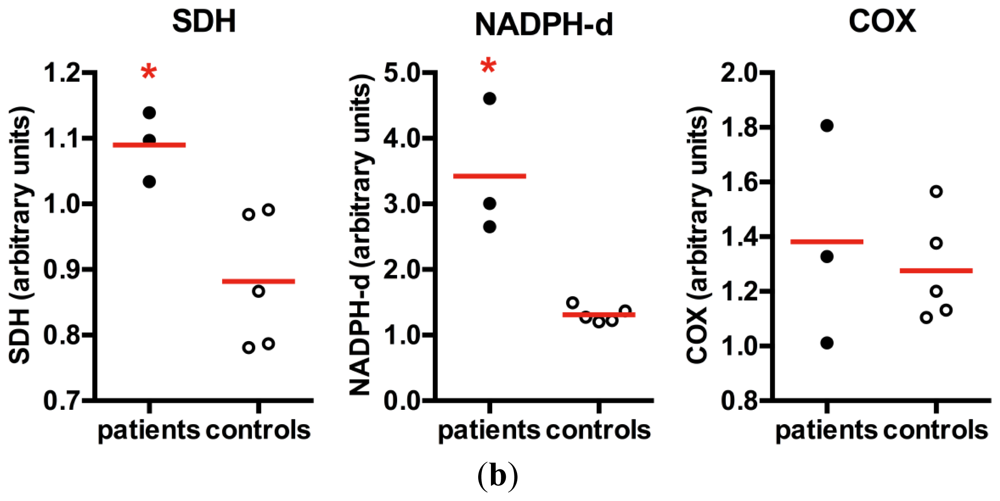
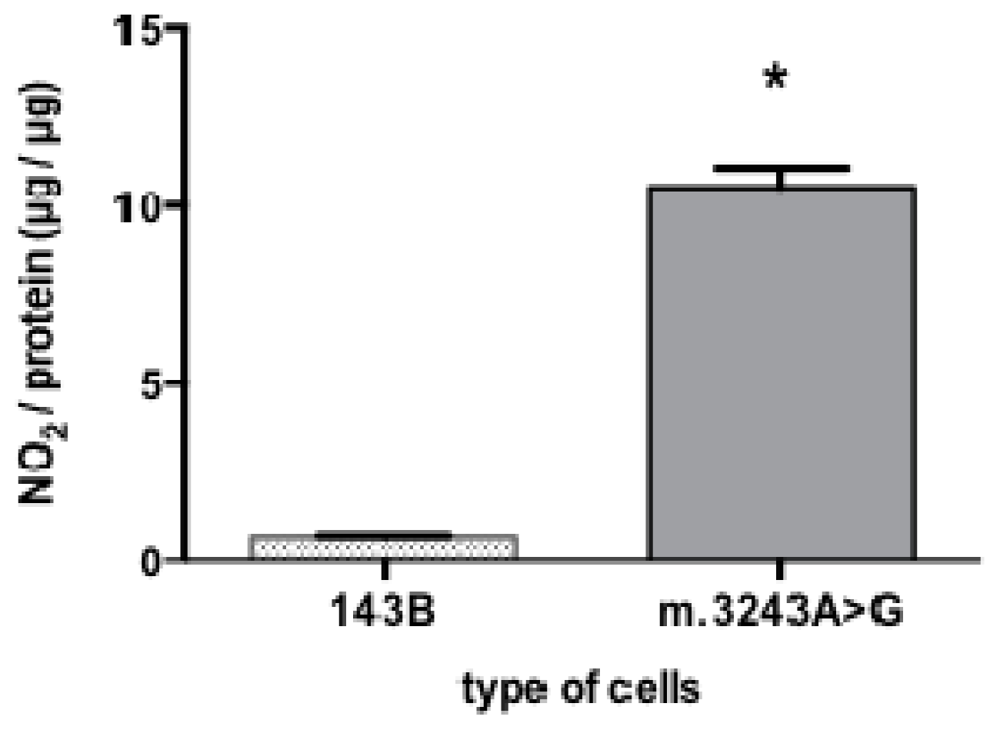
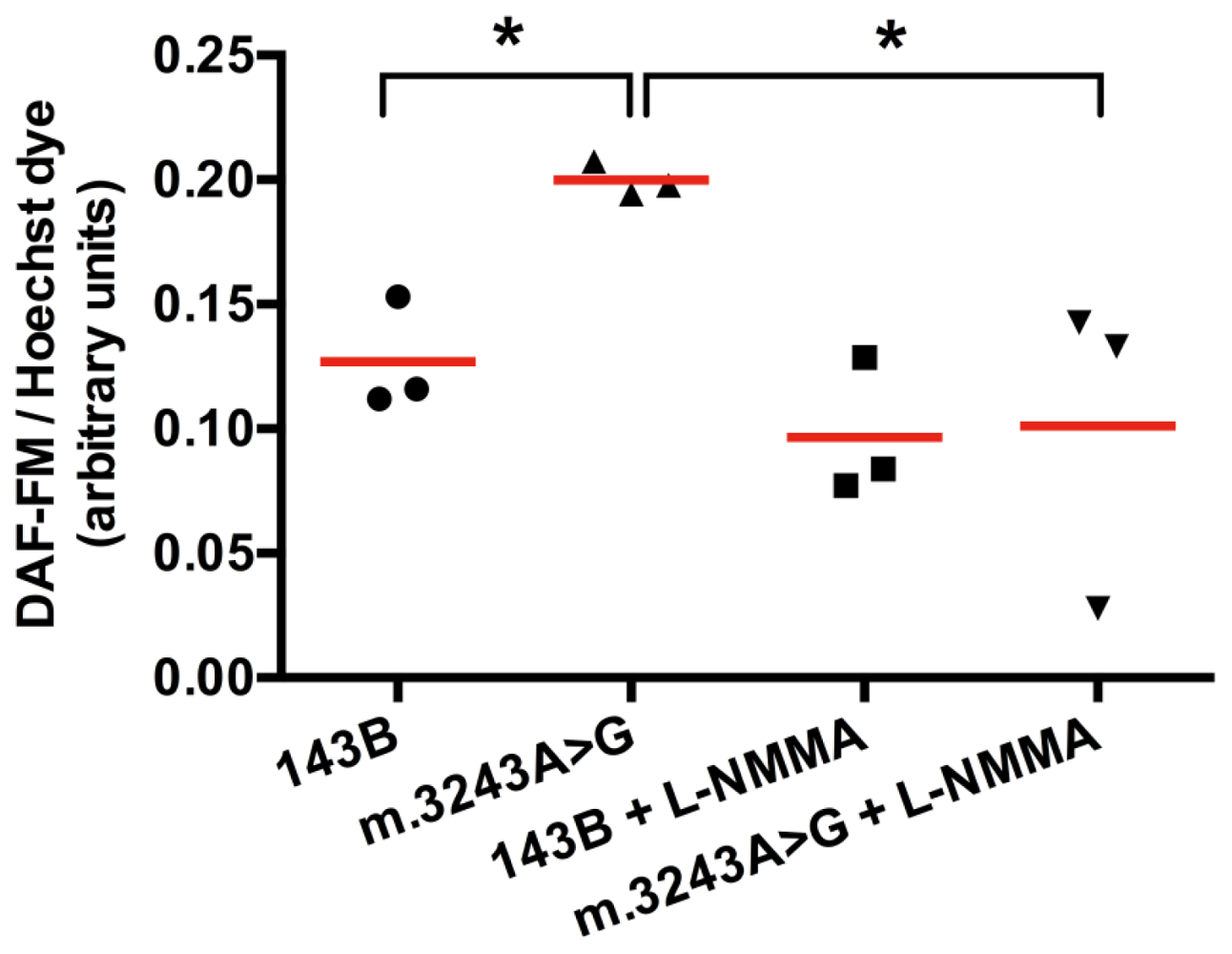
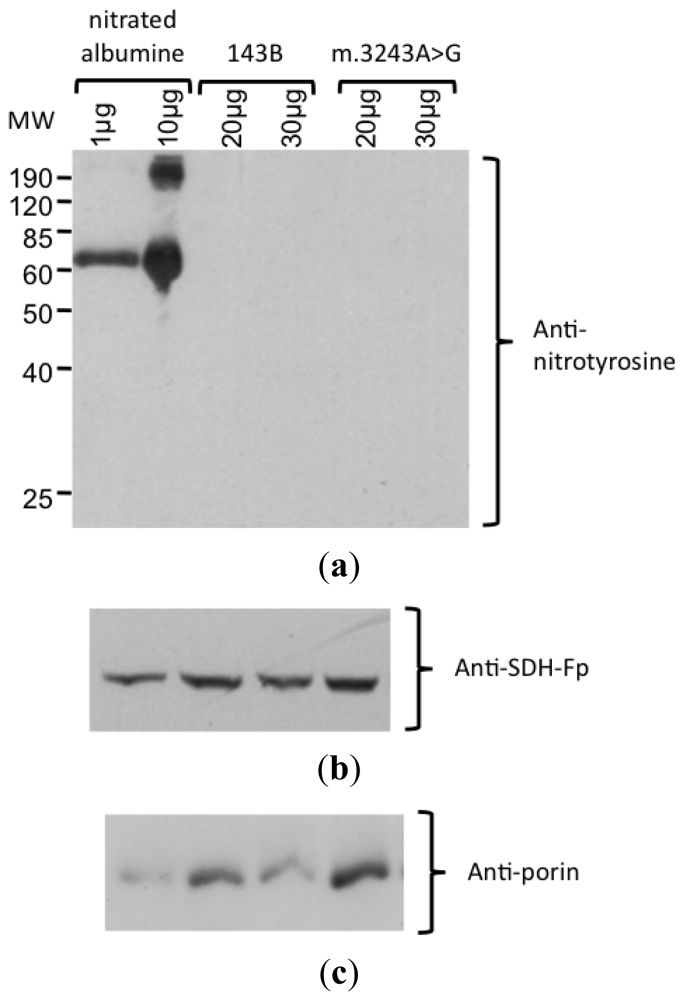
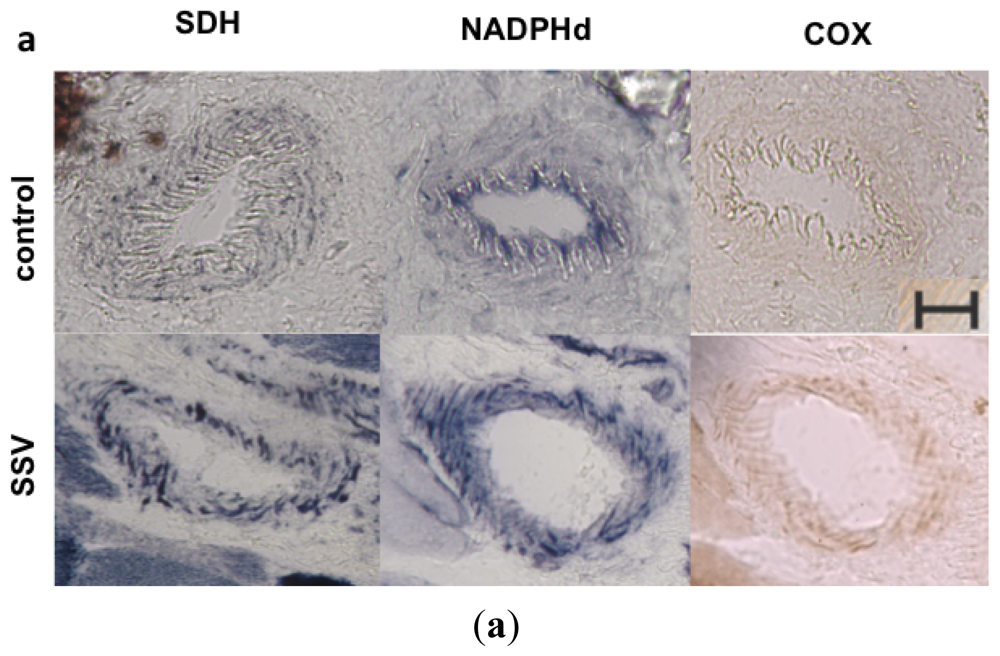
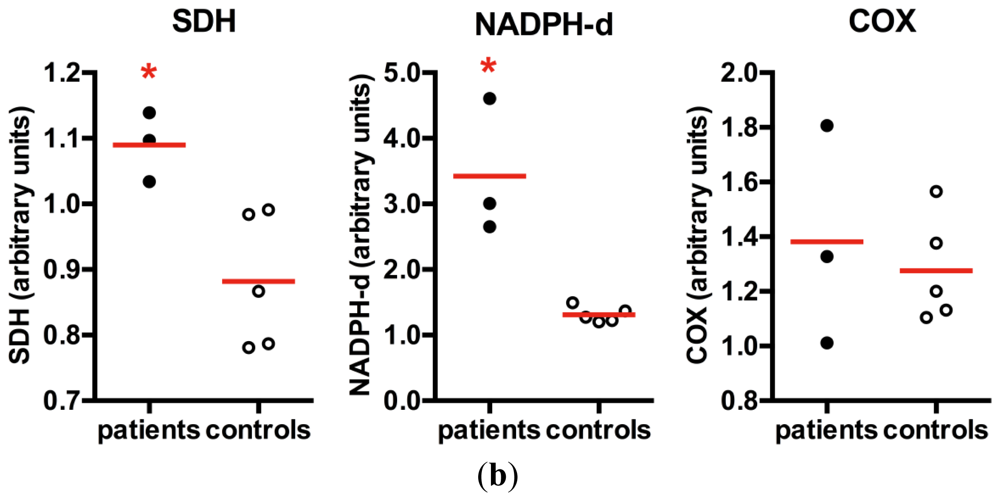
2. Results and Discussion
3. Experimental Section
3.1. Cell Culture
3.2. Cell lines Characterization
3.2.1. Genotyping
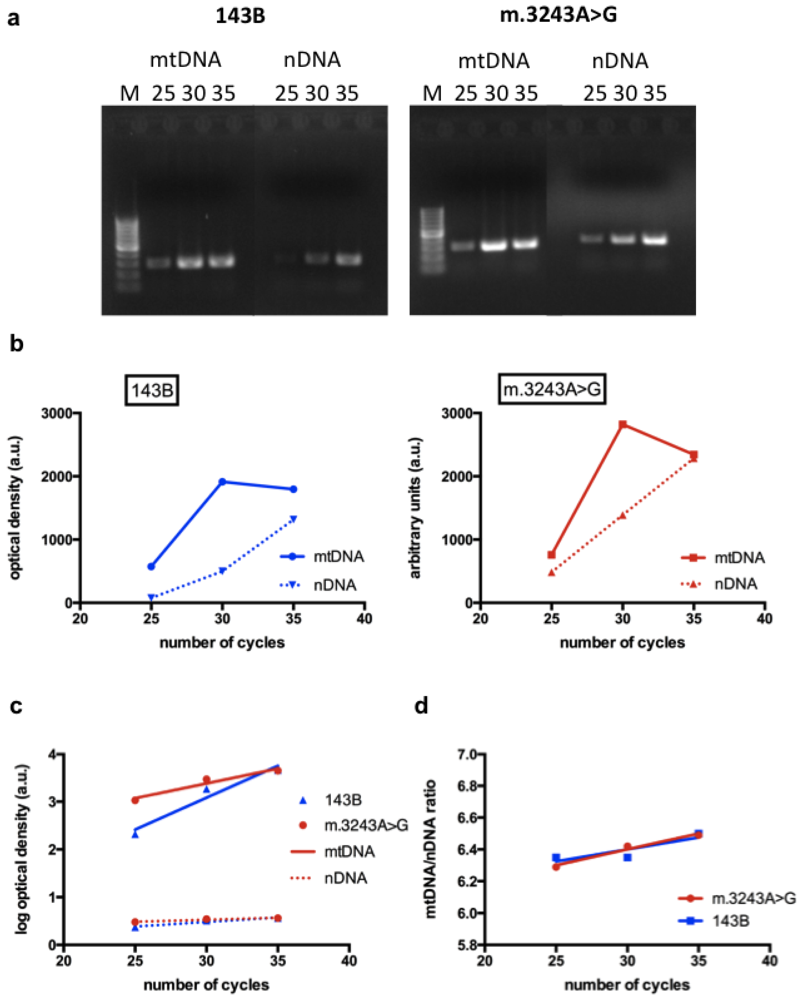
3.2.2. Evaluation of mtDNA Content by Semi-Quantitative PCR
3.2.3. Biochemical Enzyme Assays
3.2.3.1. Isolation of Mitochondria
3.2.3.2. Complex I (NADH ubiquinone oxido-reductase, EC 1.6.5.3)
3.2.3.3. Complex II (Succinate Decylubiquinone DCPIP Reductase, EC 1.10.2.2)
3.2.3.4. Complex II + III (Succinate Cytochrome c Reductase, EC 1.3.5.1 + EC 1.10.2.2)
3.2.3.5. Complex IV (Cytochrome c Oxidase, EC 1.9.3.1)
3.2.3.6. Citrate Synthase
3.3. Quantification of Nitrite
3.4. Quantitative Analysis of Intracellular NO
3.5. Detection of Nitrate Protein by Western Blotting
3.6. Muscle Histochemistry
3.7. Quantification of NOS Activity in Muscle Vessels
3.8. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-14-00394-s001.pdfAcknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci 2005, 26, 190–195. [Google Scholar]
- Nathan, C.; Xie, Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar]
- Brown, G.C. Nitric oxide and neuronal death. Nitric Oxide 2010, 23, 153–165. [Google Scholar]
- Zeviani, M.; di Donato, S. Mitochondrial disorders. Brain 2004, 127, 2153–2172. [Google Scholar]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar]
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. A neurological perspective on mitochondrial disease. Lancet Neurol 2010, 9, 829–840. [Google Scholar]
- Goto, Y.; Nonaka, I.; Horai, S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar]
- Sproule, D.M.; Kaufmann, P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: Basic concepts, clinical phenotype, and therapeutic management of melas syndrome. Ann. N. Y. Acad. Sci 2008, 1142, 133–158. [Google Scholar]
- Koga, Y.; Akita, Y.; Junko, N.; Yatsuga, S.; Povalko, N.; Fukiyama, R.; Ishii, M.; Matsuishi, T. Endothelial dysfunction in melas improved by l-arginine supplementation. Neurology 2006, 66, 1766–1769. [Google Scholar]
- Koga, Y.; Ishibashi, M.; Ueki, I.; Yatsuga, S.; Fukiyama, R.; Akita, Y.; Matsuishi, T. Effects of l-arginine on the acute phase of strokes in three patients with melas. Neurology 2002, 58, 827–828. [Google Scholar]
- Koga, Y.; Akita, Y.; Nishioka, J.; Yatsuga, S.; Povalko, N.; Tanabe, Y.; Fujimoto, S.; Matsuishi, T. l-arginine improves the symptoms of strokelike episodes in melas. Neurology 2005, 64, 710–712. [Google Scholar]
- Koga, Y.; Akita, Y.; Nishioka, J.; Yatsuga, S.; Povalko, N.; Katayama, K.; Matsuishi, T. MELAS and l-arginine therapy. Mitochondrion 2007, 7, 133–139. [Google Scholar]
- King, M.P.; Koga, Y.; Davidson, M.; Schon, E.A. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA(Leu(UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol. Cell. Biol 1992, 12, 480–490. [Google Scholar]
- Guevara, I.; Iwanejko, J.; Dembinska-Kiec, A.; Pankiewicz, J.; Wanat, A.; Anna, P.; Golabek, I.; Bartus, S.; Malczewska-Malec, M.; Szczudlik, A. Determination of nitrite/nitrate in human biological material by the simple Griess reaction. Clin. Chim. Acta 1998, 274, 177–188. [Google Scholar]
- Yoneda, M.; Ikawa, M.; Arakawa, K.; Kudo, T.; Kimura, H.; Fujibayashi, Y.; Okazawa, H. In vivo functional brain imaging and a therapeutic trial of l-arginine in melas patients. Biochim. Biophys. Acta 2012, 1820, 615–618. [Google Scholar]
- Kubota, M.; Sakakihara, Y.; Mori, M.; Yamagata, T.; Momoi-Yoshida, M. Beneficial effect of l-arginine for stroke-like episode in MELAS. Brain Dev. 2004, 26, 481–483, Discussion 480. [Google Scholar]
- El-Hattab, A.W.; Hsu, J.W.; Emrick, L.T.; Wong, L.J.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Restoration of impaired nitric oxide production in melas syndrome with citrulline and arginine supplementation. Mol. Genet. Metab 2012, 105, 607–614. [Google Scholar]
- Koga, Y.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T. MELAS and l-arginine therapy: Pathophysiology of stroke-like episodes. Ann. N. Y. Acad. Sci 2010, 1201, 104–110. [Google Scholar]
- El-Hattab, A.W.; Emrick, L.T.; Craigen, W.J.; Scaglia, F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol. Genet. Metab 2012, 107, 247–252. [Google Scholar]
- Naini, A.; Kaufmann, P.; Shanske, S.; Engelstad, K.; de Vivo, D.C.; Schon, E.A. Hypocitrullinemia in patients with melas: An insight into the “melas paradox”. J. Neurol. Sci. 2005, 229–230, 187–193. [Google Scholar]
- Frandsen, U.; Lopez-Figueroa, M.; Hellsten, Y. Localization of nitric oxide synthase in human skeletal muscle. Biochem. Biophys. Res. Commun 1996, 227, 88–93. [Google Scholar]
- Planitzer, G.; Baum, O.; Gossrau, R. Skeletal muscle fibres show NADPH diaphorase activity associated with mitochondria, the sarcoplasmic reticulum and the NOS-1-containing sarcolemma. Histochem. J 2000, 32, 303–312. [Google Scholar]
- Tengan, C.H.; Kiyomoto, B.H.; Godinho, R.O.; Gamba, J.; Neves, A.C.; Schmidt, B.; Oliveira, A.S.; Gabbai, A.A. The role of nitric oxide in muscle fibers with oxidative phosphorylation defects. Biochem. Biophys. Res. Commun 2007, 359, 771–777. [Google Scholar]
- Vattemi, G.; Mechref, Y.; Marini, M.; Tonin, P.; Minuz, P.; Grigoli, L.; Guglielmi, V.; Klouckova, I.; Chiamulera, C.; Meneguzzi, A.; et al. Increased protein nitration in mitochondrial diseases: Evidence for vessel wall involvement. Mol. Cell. Proteomics 2011, 10, M110.002964. [Google Scholar]
- Coman, D.; Yaplito-Lee, J.; Boneh, A. New indications and controversies in arginine therapy. Clin. Nutr 2008, 27, 489–496. [Google Scholar]
- Arakawa, K.; Kudo, T.; Ikawa, M.; Morikawa, N.; Kawai, Y.; Sahashi, K.; Lee, J.D.; Kuriyama, M.; Miyamori, I.; Okazawa, H.; et al. Abnormal myocardial energy-production state in mitochondrial cardiomyopathy and acute response to l-arginine infusion. C-11 acetate kinetics revealed by positron emission tomography. Circ. J 2010, 74, 2702–2711. [Google Scholar]
- Desquiret-Dumas, V.; Gueguen, N.; Barth, M.; Chevrollier, A.; Hancock, S.; Wallace, D.C.; Amati-Bonneau, P.; Henrion, D.; Bonneau, D.; Reynier, P.; et al. Metabolically induced heteroplasmy shifting and l-arginine treatment reduce the energetic defect in a neuronal-like model of melas. Biochim. Biophys. Acta 2012, 1822, 1019–1029. [Google Scholar]
- Gerard, J.M.; Luisiri, A. A fatal overdose of arginine hydrochloride. J. Toxicol. Clin. Toxicol 1997, 35, 621–625. [Google Scholar]
- Abraham, M.B.; van der Westhuyzen, J.; Khanna, V. Arginine extravasation leading to skin necrosis. J. Paediatr. Child Health 2012, 48, E96–E97. [Google Scholar]
- Nisoli, E.; Carruba, M.O. Nitric oxide and mitochondrial biogenesis. J. Cell. Sci 2006, 119, 2855–2862. [Google Scholar]
- Stamler, J.S.; Meissner, G. Physiology of nitric oxide in skeletal muscle. Physiol. Rev 2001, 81, 209–237. [Google Scholar]
- Boveris, A.; Costa, L.E.; Poderoso, J.J.; Carreras, M.C.; Cadenas, E. Regulation of mitochondrial respiration by oxygen and nitric oxide. Ann. N. Y. Acad. Sci 2000, 899, 121–135. [Google Scholar]
- Oliveira, K.K.; Kiyomoto, B.H.; Tengan, C.H. Complex I spectrophotometric assay in cultured cells: Detailed analysis of key factors. Anal. Biochem. 2012. [Google Scholar] [CrossRef]
- Medja, F.; Allouche, S.; Frachon, P.; Jardel, C.; Malgat, M.; Mousson de Camaret, B.; Slama, A.; Lunardi, J.; Mazat, J.P.; Lombes, A. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009, 9, 331–339. [Google Scholar]
- James, A.M.; Wei, Y.H.; Pang, C.Y.; Murphy, M.P. Altered mitochondrial function in fibroblasts containing MELAS or MERRF mitochondrial DNA mutations. Biochem. J 1996, 318, 401–407. [Google Scholar]
- Barrientos, A.; Fontanesi, F.; Diaz, F. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. 2009. [Google Scholar] [CrossRef]
- De Wit, L.E.; Sluiter, W. Chapter 9 reliable assay for measuring complex I activity in human blood lymphocytes and skin fibroblasts. Meth. Enzymology 2009, 456, 169–181. [Google Scholar]
- Barrientos, A. In vivo and in organello assessment of oxphos activities. Methods 2002, 26, 307–316. [Google Scholar]
- Ridnour, L.A.; Sim, J.E.; Hayward, M.A.; Wink, D.A.; Martin, S.M.; Buettner, G.R.; Spitz, D.R. A spectrophotometric method for the direct detection and quantitation of nitric oxide, nitrite, and nitrate in cell culture media. Anal. Biochem 2000, 281, 223–229. [Google Scholar]
- Miranda, K.M.; Espey, M.G.; Wink, D.A. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 2001, 5, 62–71. [Google Scholar]
- Kojima, H.; Nakatsubo, N.; Kikuchi, K.; Kawahara, S.; Kirino, Y.; Nagoshi, H.; Hirata, Y.; Nagano, T. Detection and imaging of nitric oxide with novel fluorescent indicators: Diaminofluoresceins. Anal. Chem 1998, 70, 2446–2453. [Google Scholar]
- Haqqani, A.S.; Kelly, J.F.; Birnboim, H.C. Selective nitration of histone tyrosine residues in vivo in mutatect tumors. J. Biol. Chem 2002, 277, 3614–3621. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell type | C-I (nmol/mg/min) | C-II (nmol/mg/min) | C-II+III (nmol/mg/min) | C-IV (nmol/mg/min) | CS (nmol/mg/min) |
|---|---|---|---|---|---|
| 143B | 158.11 | 109.68 | 70.68 | 78.53 | 16.54 |
| m.3243A>G | 18.48 | 112.82 | 31.41 | 29.84 | 32.35 |
| Patient | Age at biopsy | Clinical diagnosis | Muscle biopsy | m.A3243A>G (%) | Number of vessels |
|---|---|---|---|---|---|
| 1 | 45 | exercise intolerance, myopathy | RRF, COX+ | 56 | 1 |
| 2 | 19 | MELAS | RRF, COX+ | 67 | 1 |
| 3 | 4 | MELAS | RRF, COX+ | 46 | 1 |
| 4 | 31 | fatigue | normal | absent | 3 |
| 5 | 23 | fatigue | normal | absent | 1 |
| 6 | 43 | non-specific dermatitis | normal | absent | 1 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gamba, J.; Gamba, L.T.; Rodrigues, G.S.; Kiyomoto, B.H.; Moraes, C.T.; Tengan, C.H. Nitric Oxide Synthesis Is Increased in Cybrid Cells with m.3243A>G Mutation. Int. J. Mol. Sci. 2013, 14, 394-410. https://doi.org/10.3390/ijms14010394
Gamba J, Gamba LT, Rodrigues GS, Kiyomoto BH, Moraes CT, Tengan CH. Nitric Oxide Synthesis Is Increased in Cybrid Cells with m.3243A>G Mutation. International Journal of Molecular Sciences. 2013; 14(1):394-410. https://doi.org/10.3390/ijms14010394
Chicago/Turabian StyleGamba, Juliana, Luana T. Gamba, Gabriela S. Rodrigues, Beatriz H. Kiyomoto, Carlos T. Moraes, and Celia H. Tengan. 2013. "Nitric Oxide Synthesis Is Increased in Cybrid Cells with m.3243A>G Mutation" International Journal of Molecular Sciences 14, no. 1: 394-410. https://doi.org/10.3390/ijms14010394