Role of Carnitine Acetyl Transferase in Regulation of Nitric Oxide Signaling in Pulmonary Arterial Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
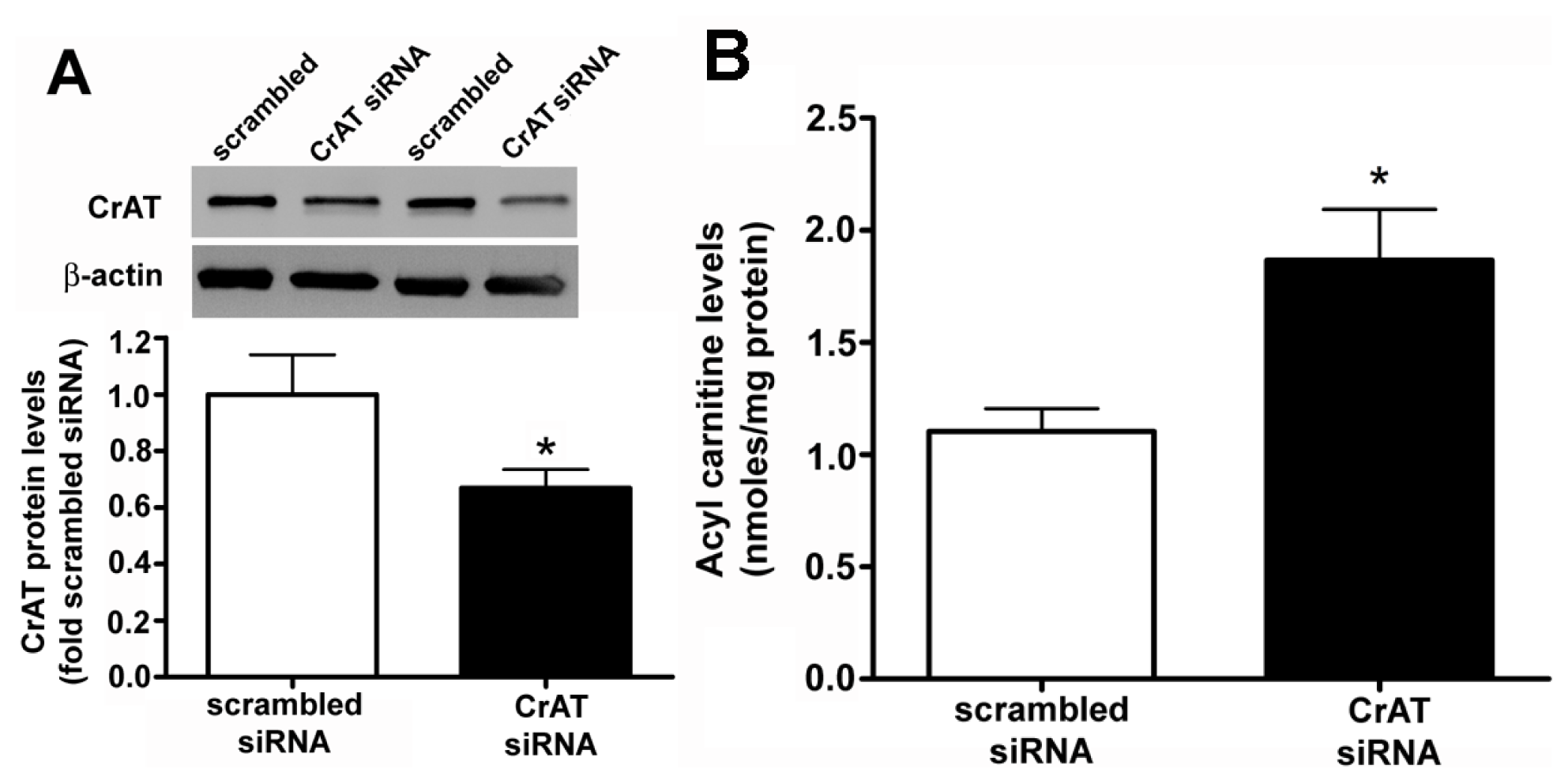
2.1. CrAT siRNA Significantly Decreases CrAT Protein Levels and Disrupts Carnitine Homeostasis in Pulmonary Arterial Endothelial Cells
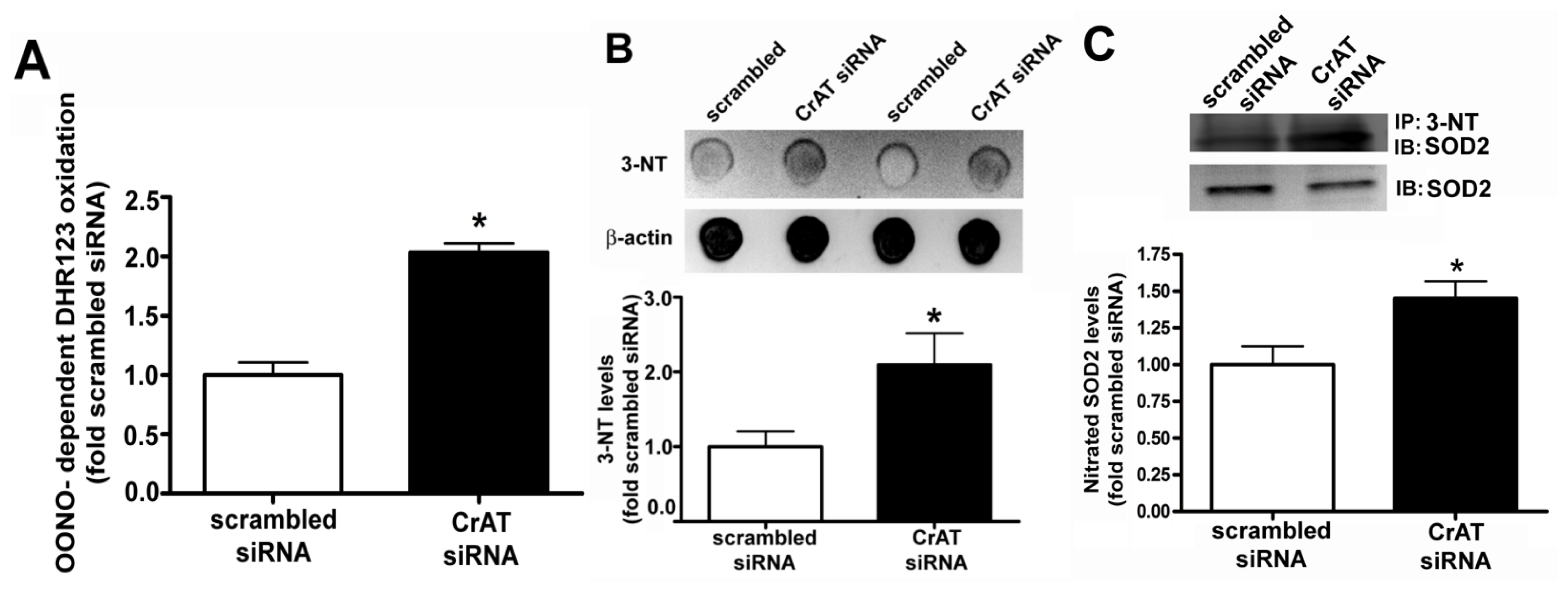
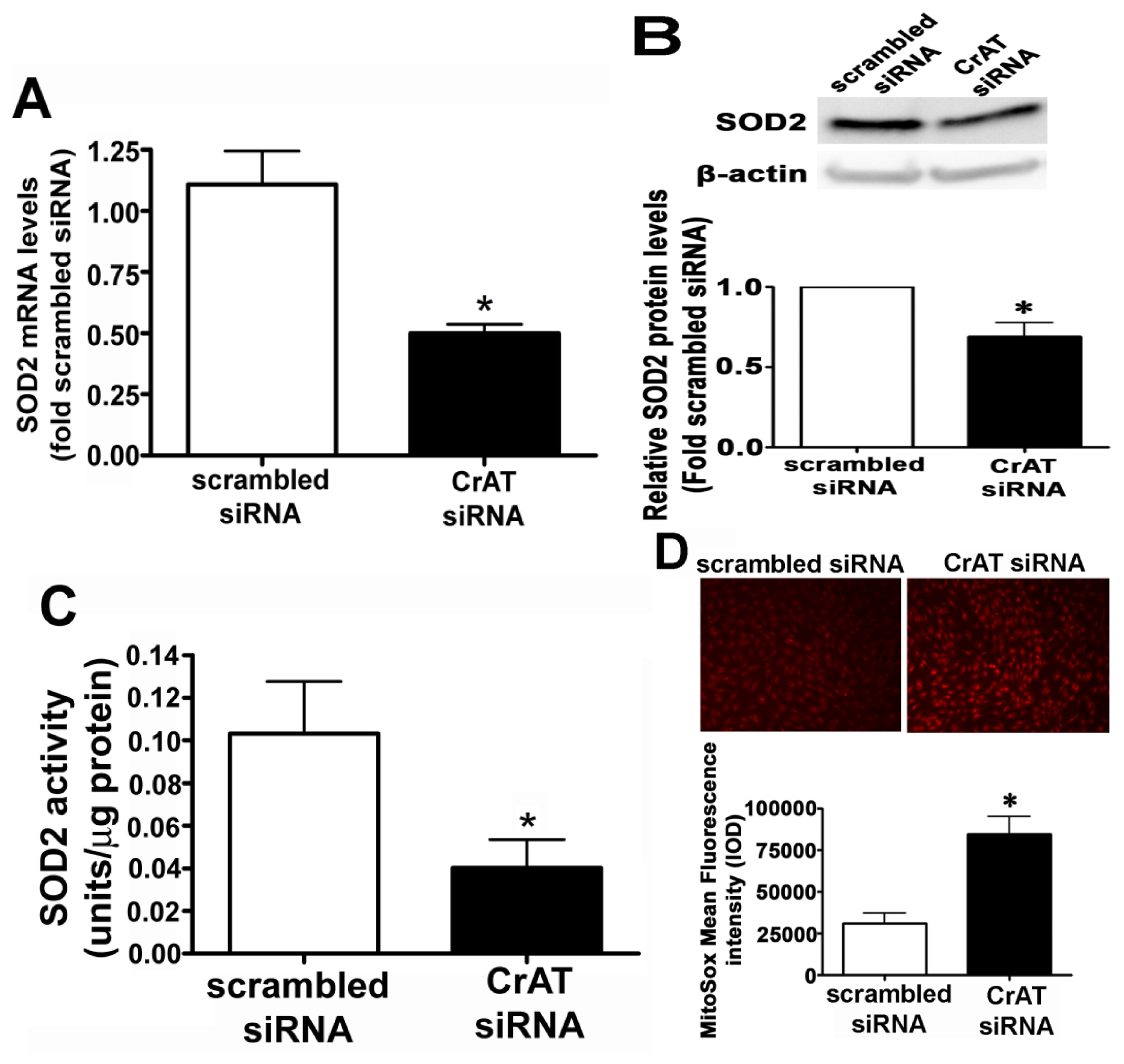
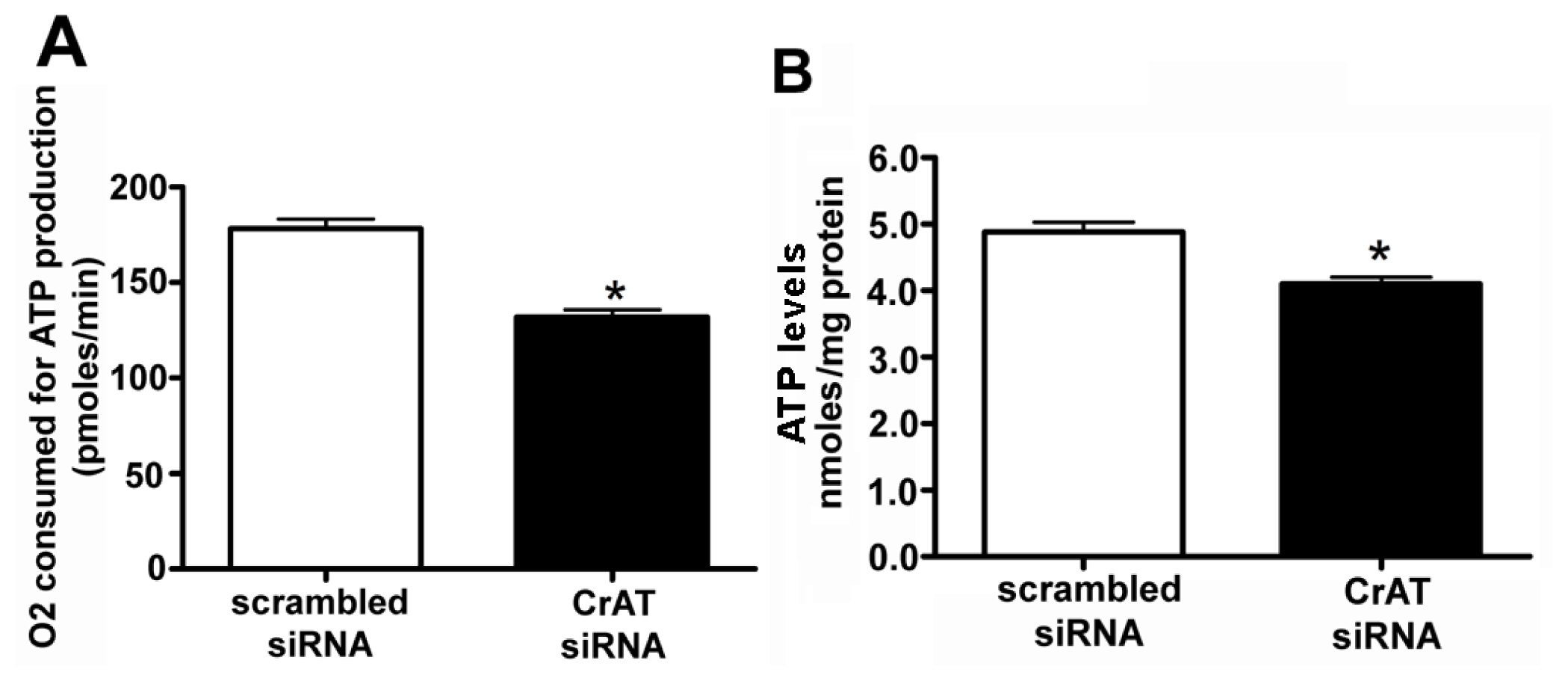
2.2. CrAT Knockdown Disrupts Superoxide Dismutase Function and Increases Mitochondrial Oxidative Stress in Pulmonary Arterial Endothelial Cells
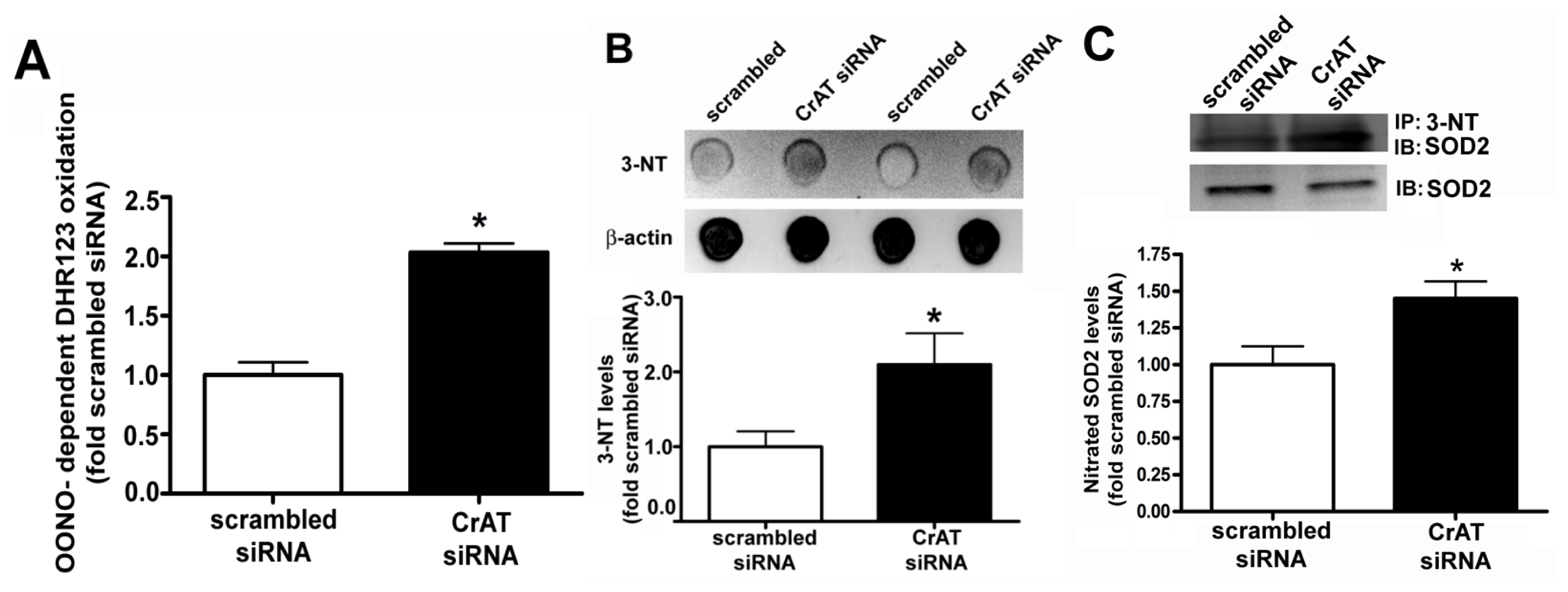
2.3. Suppressing CrAT Expression Causes Nitrative Stress in Pulmonary Arterial Endothelial Cells
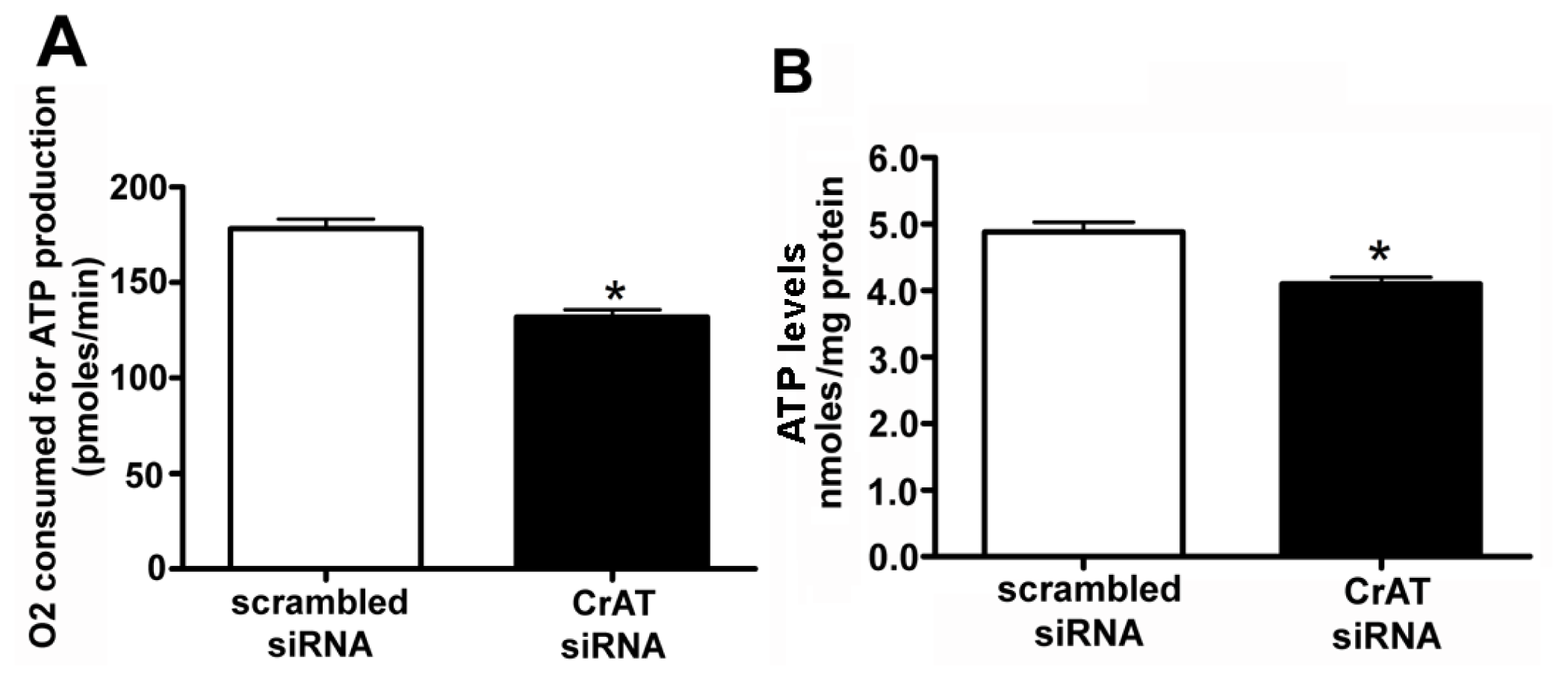
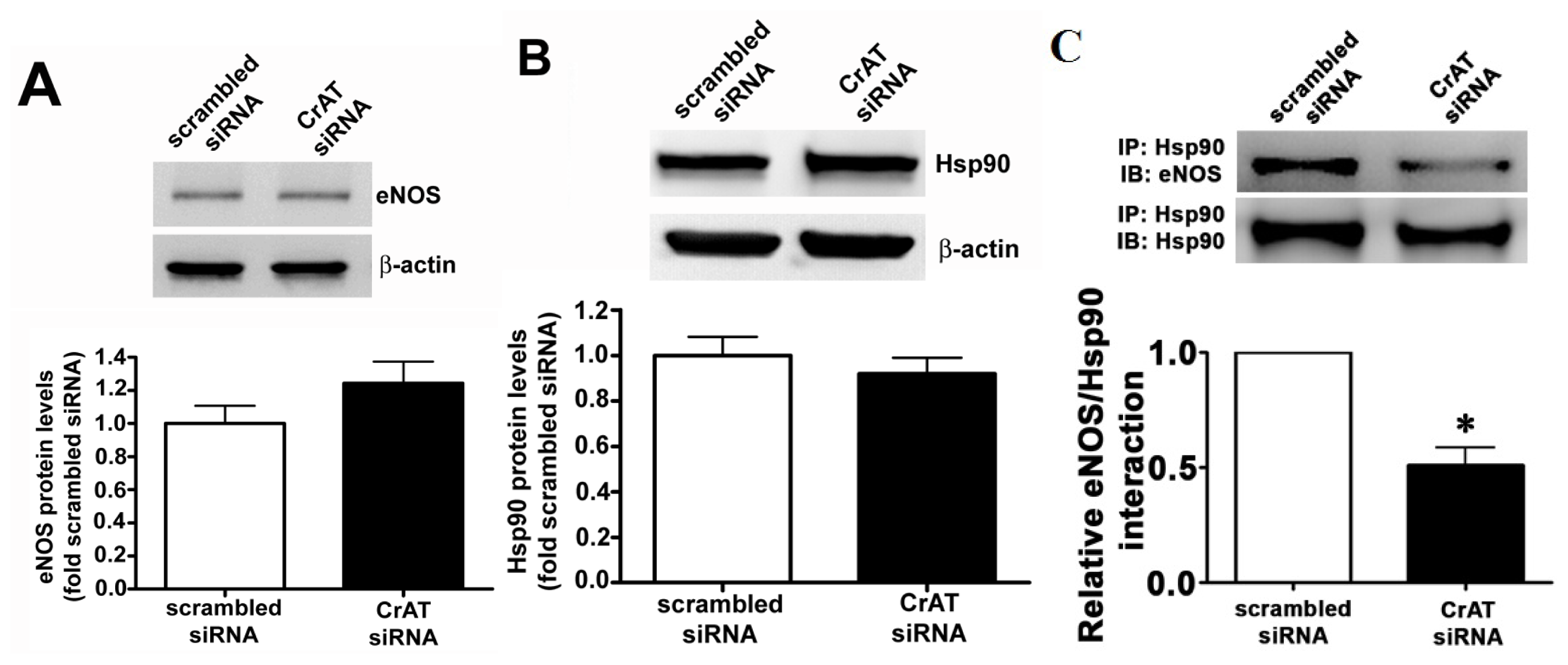
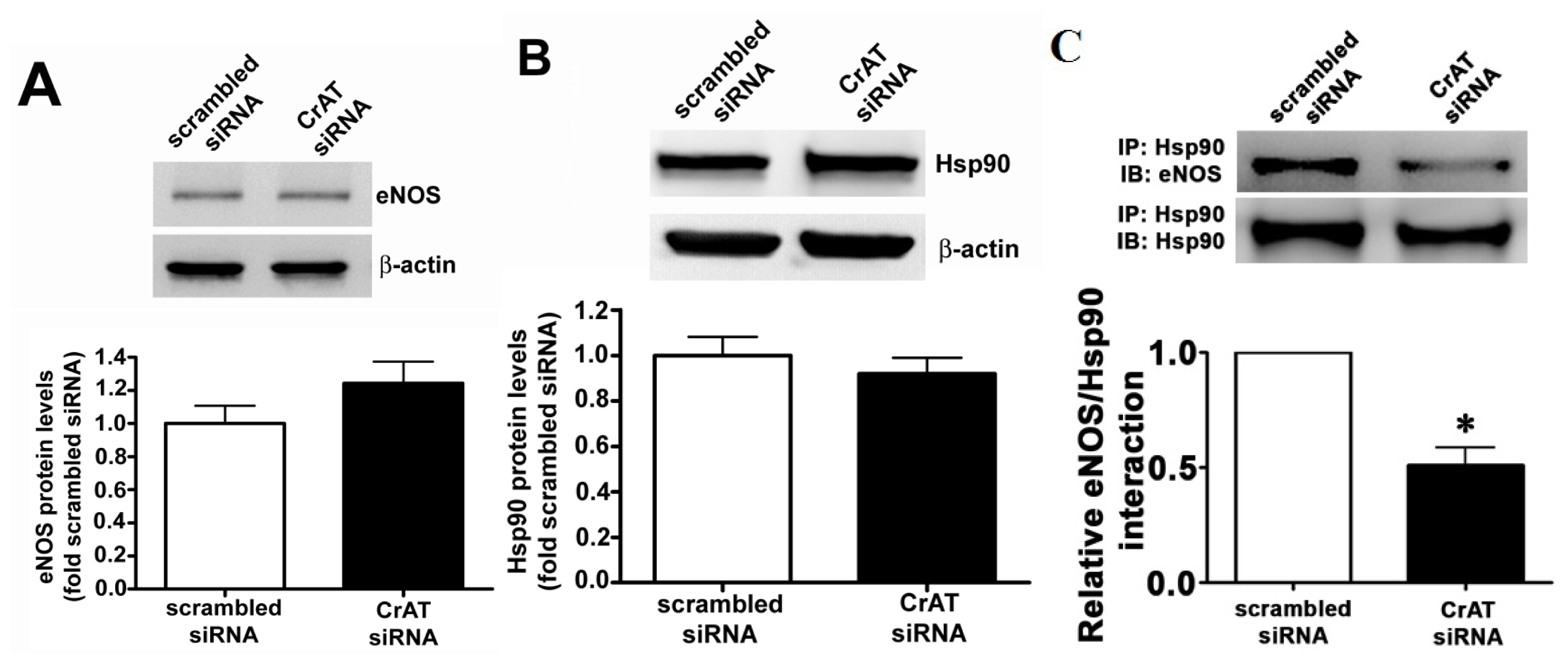
2.4. CrAT Mediated Mitochondrial Dysfunction Disrupts eNOS/Hsp90 Interactions and Leads to eNOS Uncoupling
3. Discussion
4. Methods
4.1. Culture of Pulmonary Arterial Endothelial Cells
4.2. Targeted Silencing of Carnitine Acetyl Transferase by Small-Interfering RNA
4.3. Sample Purification and Measurement of Carnitine Metabolites
4.4. Measurement of Peroxynitrite Levels
4.5. Measurement of 3-NT Levels
4.6. Real Time Quantitative (q) RT-PCR for mRNA Levels
4.7. Western Blot Analyses
4.8. Measurement of SOD2 Activity
4.9. Immunoprecipitation Analysis
4.10. Determination of Mitochondrial Superoxide
4.11. Mitochondrial Bioenergetic Analysis in Transfected Cells
4.12. Determination of Cellular ATP Levels
4.13. Shear Stress
4.14. Measurement of NOS-Derived Superoxide Levels in PAEC
4.15. Determination of Cellular NOx Levels
4.16. Statistical Analysis
5. Conclusion
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Hanley, F.L.; Heinemann, M.K.; Jonas, R.A.; Mayer, J.E., Jr; Cook, N.R.; Wessel, D.L.; Castaneda, A.R. Repair of truncus arteriosus in the neonate. J. Thorac. Cardiovasc. Surg. 1993, 105, 1047–1056. [Google Scholar]
- Black, S.M.; Fineman, J.R. Oxidative and nitrosative stress in pediatric pulmonary hypertension: Roles of endothelin-1 and nitric oxide. Vasc. Pharmacol 2006, 45, 308–316. [Google Scholar]
- Celermajer, D.S.; Cullen, S.; Deanfield, J.E. Impairment of endothelium-dependent pulmonary artery relaxation in children with congenital heart disease and abnormal pulmonary hemodynamics. Circulation 1993, 87, 440–446. [Google Scholar]
- Lakshminrusimha, S.; Wiseman, D.; Black, S.M.; Russell, J.A.; Gugino, S.F.; Oishi, P.; Steinhorn, R.H.; Fineman, J.R. The role of nitric oxide synthase-derived reactive oxygen species in the altered relaxation of pulmonary arteries from lambs with increased pulmonary blood flow. Am. J. Physiol. Heart Circ. Physiol 2007, 293, H1491–H1497. [Google Scholar]
- Zhang, D.X.; Gutterman, D.D. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am. J. Physiol. Heart Circ. Physiol 2007, 292, H2023–H2031. [Google Scholar]
- Puddu, P.; Puddu, G.M.; Cravero, E.; de Pascalis, S.; Muscari, A. The putative role of mitochondrial dysfunction in hypertension. Clin. Exp. Hypertens 2007, 29, 427–434. [Google Scholar]
- Genova, M.L.; Pich, M.M.; Bernacchia, A.; Bianchi, C.; Biondi, A.; Bovina, C.; Falasca, A.I.; Formiggini, G.; Castelli, G.P.; Lenaz, G. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann. N. Y. Acad. Sci 2004, 1011, 86–100. [Google Scholar]
- Mancuso, C.; Scapagini, G.; Curro, D.; Giuffrida Stella, A.M.; de Marco, C.; Butterfield, D.A.; Calabrese, V. Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front. Biosci 2007, 12, 1107–1123. [Google Scholar]
- Puddu, P.; Puddu, G.M.; Cravero, E.; de Pascalis, S.; Muscari, A. The emerging role of cardiovascular risk factor-induced mitochondrial dysfunction in atherogenesis. J. Biomed. Sci 2009, 16, 112. [Google Scholar]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 2010, 1802, 80–91. [Google Scholar]
- Calo, L.A.; Pagnin, E.; Davis, P.A.; Semplicini, A.; Nicolai, R.; Calvani, M.; Pessina, A.C. Antioxidant effect of l-carnitine and its short chain esters: Relevance for the protection from oxidative stress related cardiovascular damage. Int. J. Cardiol 2006, 107, 54–60. [Google Scholar]
- Rodriguez-Iturbe, B.; Zhan, C.D.; Quiroz, Y.; Sindhu, R.K.; Vaziri, N.D. Antioxidant-rich diet relieves hypertension and reduces renal immune infiltration in spontaneously hypertensive rats. Hypertension 2003, 41, 341–346. [Google Scholar]
- Alvarez de Sotomayor, M.; Bueno, R.; Perez-Guerrero, C.; Herrera, M.D. Effect of l-carnitine and propionyl-l-carnitine on endothelial function of small mesenteric arteries from SHR. J. Vasc. Res 2007, 44, 354–364. [Google Scholar]
- Reddy, V.M.; Meyrick, B.; Wong, J.; Khoor, A.; Liddicoat, J.R.; Hanley, F.L.; Fineman, J.R. In utero placement of aortopulmonary shunts. A model of postnatal pulmonary hypertension with increased pulmonary blood flow in lambs. Circulation 1995, 92, 606–613. [Google Scholar]
- Black, S.; Fineman, J.; Johengen, M.; Bristow, J.; Soifer, S. Increased pulmonary blood flow alters the molecular regulation of vascular reactivity in the lamb. Chest 1998, 114, 39S. [Google Scholar]
- Reddy, V.M.; Wong, J.; Liddicoat, J.R.; Johengen, M.; Chang, R.; Fineman, J.R. Altered endothelium-dependent responses in lambs with pulmonary hypertension and increased pulmonary blood flow. Am. J. Physiol 1996, 271, H562–H570. [Google Scholar]
- Grobe, A.C.; Wells, S.M.; Benavidez, E.; Oishi, P.; Azakie, A.; Fineman, J.R.; Black, S.M. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: Role of NADPH oxidase and endothelial NO synthase. Am. J. Physiol. Lung Cell. Mol. Physiol 2006, 290, L1069–L1077. [Google Scholar]
- Oishi, P.E.; Wiseman, D.A.; Sharma, S.; Kumar, S.; Hou, Y.; Datar, S.A.; Azakie, A.; Johengen, M.J.; Harmon, C.; Fratz, S.; et al. Progressive dysfunction of nitric oxide synthase in a lamb model of chronically increased pulmonary blood flow: A role for oxidative stress. Am. J. Physiol. Lung Cell. Mol. Physiol 2008, 295, L756–L766. [Google Scholar]
- Sharma, S.; Sud, N.; Wiseman, D.A.; Carter, A.L.; Kumar, S.; Hou, Y.; Rau, T.; Wilham, J.; Harmon, C.; Oishi, P.; et al. Altered carnitine homeostasis is associated with decreased mitochondrial function and altered nitric oxide signaling in lambs with pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol 2008, 294, L46–L56. [Google Scholar]
- Sud, N.; Sharma, S.; Wiseman, D.A.; Harmon, C.; Kumar, S.; Venema, R.C.; Fineman, J.R.; Black, S.M. Nitric oxide and superoxide generation from endothelial NOS: Modulation by HSP90. Am. J. Physiol. Lung Cell. Mol. Physiol 2007, 293, L1444–L1453. [Google Scholar]
- Binienda, Z.; Przybyla-Zawislak, B.; Virmani, A.; Schmued, L. l-carnitine and neuroprotection in the animal model of mitochondrial dysfunction. Ann. N. Y. Acad. Sci 2005, 1053, 174–182. [Google Scholar]
- Henique, C.; Mansouri, A.; Fumey, G.; Lenoir, V.; Girard, J.; Bouillaud, F.; Prip-Buus, C.; Cohen, I. Increased mitochondrial fatty acid oxidation is sufficient to protect skeletal muscle cells from palmitate-induced apoptosis. J. Biol. Chem 2010, 285, 36818–36827. [Google Scholar]
- Sayed-Ahmed, M.M.; Shouman, S.A.; Rezk, B.M.; Khalifa, M.H.; Osman, A.M.; El-Merzabani, M.M. Propionyl-l-carnitine as potential protective agent against adriamycin-induced impairment of fatty acid beta-oxidation in isolated heart mitochondria. Pharmacol. Res 2000, 41, 143–150. [Google Scholar]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Chronic fatigue syndrome and mitochondrial dysfunction. Int. J. Clin. Exp. Med 2009, 2, 1–16. [Google Scholar]
- Pande, S.V.; Blanchaer, M.C. Reversible inhibition of mitochondrial adenosine diphosphate phosphorylation by long chain acyl coenzyme A esters. J. Biol. Chem 1971, 246, 402–411. [Google Scholar]
- Ramsay, R.R.; Zammit, V.A. Carnitine acyltransferases and their influence on CoA pools in health and disease. Mol. Aspects Med 2004, 25, 475–493. [Google Scholar]
- Zammit, V.A.; Ramsay, R.R.; Bonomini, M.; Arduini, A. Carnitine, mitochondrial function and therapy. Adv. Drug Deliv. Rev 2009, 61, 1353–1362. [Google Scholar]
- Iqbal, M.; Cawthon, D.; Wideman, R.F., Jr; Bottje, W.G. Lung mitochondrial dysfunction in pulmonary hypertension syndrome I. Site-specific defects in the electron transport chain. Poult. Sci. 2001, 80, 485–495. [Google Scholar]
- Kasahara, E.; Lin, L.R.; Ho, Y.S.; Reddy, V.N. SOD2 protects against oxidation-induced apoptosis in mouse retinal pigment epithelium: Implications for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 2005, 46, 3426–3434. [Google Scholar]
- Sharma, S.; Grobe, A.C.; Wiseman, D.A.; Kumar, S.; Englaish, M.; Najwer, I.; Benavidez, E.; Oishi, P.; Azakie, A.; Fineman, J.R.; et al. Lung antioxidant enzymes are regulated by development and increased pulmonary blood flow. Am. J. Physiol. Lung Cell. Mol. Physiol 2007, 293, L960–L971. [Google Scholar]
- Fijalkowska, I.; Xu, W.; Comhair, S.A.; Janocha, A.J.; Mavrakis, L.A.; Krishnamachary, B.; Zhen, L.; Mao, T.; Richter, A.; Erzurum, S.C.; et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am. J. Pathol 2010, 176, 1130–1138. [Google Scholar]
- Fessel, J.P.; Hamid, R.; Wittmann, B.M.; Robinson, L.J.; Blackwell, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; Hemnes, A.R.; West, J.D. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm. Circ 2012, 2, 201–213. [Google Scholar]
- Surmeli, N.B.; Litterman, N.K.; Miller, A.F.; Groves, J.T. Peroxynitrite Mediates Active Site Tyrosine Nitration in Manganese Superoxide Dismutase. Evidence of a Role for the Carbonate Radical Anion. J. Am. Chem. Soc 2010, 132, 17174–17185. [Google Scholar]
- Sener, G.; Paskaloglu, K.; Satiroglu, H.; Alican, I.; Kacmaz, A.; Sakarcan, A. l-carnitine ameliorates oxidative damage due to chronic renal failure in rats. J. Cardiovasc. Pharmacol 2004, 43, 698–705. [Google Scholar]
- Arafa, H.M.; Sayed-Ahmed, M.M. Protective role of carnitine esters against alcohol-induced gastric lesions in rats. Pharmacol. Res 2003, 48, 285–290. [Google Scholar]
- Aydogdu, N.; Atmaca, G.; Yalcin, O.; Taskiran, R.; Tastekin, E.; Kaymak, K. Protective effects of l-carnitine on myoglobinuric acute renal failure in rats. Clin. Exp. Pharmacol. Physiol 2006, 33, 119–124. [Google Scholar]
- Chavez, M.D.; Lakshmanan, N.; Kavdia, M. Impact of superoxide dismutase on nitric oxide and peroxynitrite levels in the microcirculation—A computational model. Conf. Proc. IEEE Eng. Med. Biol. Soc 2007, 2007, 1022–1026. [Google Scholar]
- Quijano, C.; Hernandez-Saavedra, D.; Castro, L.; McCord, J.M.; Freeman, B.A.; Radi, R. Reaction of peroxynitrite with Mn-superoxide dismutase. Role of the metal center in decomposition kinetics and nitration. J. Biol. Chem 2001, 276, 11631–11638. [Google Scholar]
- Konduri, G.G.; Mattei, J. Role of oxidative phosphorylation and ATP release in mediating birth-related pulmonary vasodilation in fetal lambs. Am. J. Physiol. Heart Circ. Physiol 2002, 283, H1600–H1608. [Google Scholar]
- Sessa, W.C. eNOS at a glance. J. Cell. Sci 2004, 117, 2427–2429. [Google Scholar]
- Gratton, J.P.; Fontana, J.; O’Connor, D.S.; Garcia-Cardena, G.; McCabe, T.J.; Sessa, W.C. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J. Biol. Chem 2000, 275, 22268–22272. [Google Scholar]
- Vescovo, G.; Ravara, B.; Gobbo, V.; Sandri, M.; Angelini, A.; Della Barbera, M.; Dona, M.; Peluso, G.; Calvani, M.; Mosconi, L.; et al. l-Carnitine: A potential treatment for blocking apoptosis and preventing skeletal muscle myopathy in heart failure. Am. J. Physiol. Cell. Physiol 2002, 283, C802–C810. [Google Scholar]
- Gulcin, I. Antioxidant and antiradical activities of l-carnitine. Life Sci 2006, 78, 803–811. [Google Scholar]
- Arduini, A. Carnitine and its acyl esters as secondary antioxidants? Am. Heart J 1992, 123, 1726–1727. [Google Scholar]
- Iliceto, S.; Scrutinio, D.; Bruzzi, P.; D’Ambrosio, G.; Boni, L.; Di Biase, M.; Biasco, G.; Hugenholtz, P.G.; Rizzon, P. Effects of l-carnitine administration on left ventricular remodeling after acute anterior myocardial infarction: The l-Carnitine Ecocardiografia Digitalizzata Infarto Miocardico (CEDIM) Trial. J. Am. Coll. Cardiol 1995, 26, 380–387. [Google Scholar]
- Aliev, G.; Liu, J.; Shenk, J.C.; Fischbach, K.; Pacheco, G.J.; Chen, S.G.; Obrenovich, M.E.; Ward, W.F.; Richardson, A.G.; Smith, M.A.; et al. Neuronal mitochondrial amelioration by feeding acetyl-l-carnitine and lipoic acid to aged rats. J. Cell. Mol. Med 2009, 13, 320–333. [Google Scholar]
- Abdul, H.M.; Calabrese, V.; Calvani, M.; Butterfield, D.A. Acetyl-l-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1–42-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. J. Neurosci. Res 2006, 84, 398–408. [Google Scholar]
- Miguel-Carrasco, J.L.; Mate, A.; Monserrat, M.T.; Arias, J.L.; Aramburu, O.; Vazquez, C.M. The role of inflammatory markers in the cardioprotective effect of l-carnitine in L-NAME-induced hypertension. Am. J. Hypertens 2008, 21, 1231–1237. [Google Scholar]
- Pauly, D.F.; Pepine, C.J. The role of carnitine in myocardial dysfunction. Am. J. Kidney Dis 2003, 41, S35–S43. [Google Scholar]
- Tarantini, G.; Scrutinio, D.; Bruzzi, P.; Boni, L.; Rizzon, P.; Iliceto, S. Metabolic treatment with l-carnitine in acute anterior ST segment elevation myocardial infarction. A randomized controlled trial. Cardiology 2006, 106, 215–223. [Google Scholar]
- Herrera, M.D.; Bueno, R.; de Sotomayor, M.A.; Perez-Guerrero, C.; Vazquez, C.M.; Marhuenda, E. Endothelium-dependent vasorelaxation induced by l-carnitine in isolated aorta from normotensive and hypertensive rats. J. Pharm. Pharmacol 2002, 54, 1423–1427. [Google Scholar]
- Bueno, R.; Alvarez de Sotomayor, M.; Perez-Guerrero, C.; Gomez-Amores, L.; Vazquez, C.M.; Herrera, M.D. l-carnitine and propionyl-L-carnitine improve endothelial dysfunction in spontaneously hypertensive rats: Different participation of NO and COX-products. Life Sci 2005, 77, 2082–2097. [Google Scholar]
- Wedgwood, S.; Black, S.M. Endothelin-1 decreases endothelial NOS expression and activity through ETA receptor-mediated generation of hydrogen peroxide. Am. J. Physiol. Lung Cell. Mol. Physiol 2005, 288, L480–L487. [Google Scholar]
- Song, P.; Wu, Y.; Xu, J.; Xie, Z.; Dong, Y.; Zhang, M.; Zou, M.H. Reactive nitrogen species induced by hyperglycemia suppresses Akt signaling and triggers apoptosis by upregulating phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) in an LKB1-dependent manner. Circulation 2007, 116, 1585–1595. [Google Scholar]
- Black, S.M.; Bekker, J.M.; Johengen, M.J.; Parry, A.J.; Soifer, S.J.; Fineman, J.R. Altered regulation of the ET-1 cascade in lambs with increased pulmonary blood flow and pulmonary hypertension. Pediatr. Res 2000, 47, 97–106. [Google Scholar]
- McMullan, D.M.; Bekker, J.M.; Johengen, M.J.; Hendricks-Munoz, K.; Gerrets, R.; Black, S.M.; Fineman, J.R. Inhaled nitric oxide-induced rebound pulmonary hypertension: Role for endothelin-1. Am. J. Physiol. Heart Circ. Physiol 2001, 280, H777–H785. [Google Scholar]
- Wedgwood, S.; McMullan, D.M.; Bekker, J.M.; Fineman, J.R.; Black, S.M. Role for endothelin-1-induced superoxide and peroxynitrite production in rebound pulmonary hypertension associated with inhaled nitric oxide therapy. Circ. Res 2001, 89, 357–364. [Google Scholar]
- Sud, N.; Wells, S.M.; Sharma, S.; Wiseman, D.A.; Wilham, J.; Black, S.M. Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterial endothelial cells: Role of mitochondrial dysfunction. Am. J. Physiol. Cell. Physiol 2008, 294, C1407–C1418. [Google Scholar]
- Kumar, S.; Sud, N.; Fonseca, F.V.; Hou, Y.; Black, S.M. Shear stress stimulates nitric oxide signaling in pulmonary arterial endothelial cells via a reduction in catalase activity: Role of protein kinase C delta. Am. J. Physiol. Lung Cell. Mol. Physiol 2010, 298, L105–L116. [Google Scholar]
- Laurindo, F.R.; Pedro Mde, A.; Barbeiro, H.V.; Pileggi, F.; Carvalho, M.H.; Augusto, O.; da Luz, P.L. Vascular free radical release ex vivo and in vivo evidence for a flow-dependent endothelial mechanism. Circ. Res 1994, 74, 700–709. [Google Scholar]
- Lakshminrusimha, S.; Wiseman, D.; Black, S.M.; Russell, J.A.; Gugino, S.F.; Oishi, P.; Steinhorn, R.H.; Fineman, J.R. The role of nitric oxide synthase-derived reactive oxygen species in the altered relaxation of pulmonary arteries from lambs with increased pulmonary blood flow. Am. J. Physiol. Heart Circ. Physiol 2007, 293, H1491–H1497. [Google Scholar]
- Black, S.M.; Heidersbach, R.S.; McMullan, D.M.; Bekker, J.M.; Johengen, M.J.; Fineman, J.R. Inhaled nitric oxide inhibits NOS activity in lambs: Potential mechanism for rebound pulmonary hypertension. Am. J. Physiol 1999, 277, H1849–H1856. [Google Scholar]
- McMullan, D.M.; Bekker, J.M.; Parry, A.J.; Johengen, M.J.; Kon, A.; Heidersbach, R.S.; Black, S.M.; Fineman, J.R. Alterations in endogenous nitric oxide production after cardiopulmonary bypass in lambs with normal and increased pulmonary blood flow. Circulation 2000, 102, III172–178. [Google Scholar]

© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sharma, S.; Sun, X.; Agarwal, S.; Rafikov, R.; Dasarathy, S.; Kumar, S.; Black, S.M. Role of Carnitine Acetyl Transferase in Regulation of Nitric Oxide Signaling in Pulmonary Arterial Endothelial Cells. Int. J. Mol. Sci. 2013, 14, 255-272. https://doi.org/10.3390/ijms14010255
Sharma S, Sun X, Agarwal S, Rafikov R, Dasarathy S, Kumar S, Black SM. Role of Carnitine Acetyl Transferase in Regulation of Nitric Oxide Signaling in Pulmonary Arterial Endothelial Cells. International Journal of Molecular Sciences. 2013; 14(1):255-272. https://doi.org/10.3390/ijms14010255
Chicago/Turabian StyleSharma, Shruti, Xutong Sun, Saurabh Agarwal, Ruslan Rafikov, Sridevi Dasarathy, Sanjiv Kumar, and Stephen M. Black. 2013. "Role of Carnitine Acetyl Transferase in Regulation of Nitric Oxide Signaling in Pulmonary Arterial Endothelial Cells" International Journal of Molecular Sciences 14, no. 1: 255-272. https://doi.org/10.3390/ijms14010255