Crystal Structure of Dimeric Flavodoxin from Desulfovibrio gigas Suggests a Potential Binding Region for the Electron-Transferring Partner

Abstract
:1. Introduction
- With the donation of electrons from the pyruvate, Fld can transport electrons from the phosphoroclastic system to the outer membrane or to the sulfate-reducing system [23]. The other organic source, aldehyde, which is reduced by aldehyde oxidoreductase (AOR), can also produce electrons. The transfer of electrons between AOR and Fld has also been reported [24].
- Through the donation of electrons from molecular dihydrogen, Fld carries the electrons from the outer membrane to the sulfate-reducing system [25]. Although some studies showed that cytochrome c3 and Fld might form a protein complex [26], the exact pathway of the electron transfer between the periplasm and the sulfate-reducing system remains unclear. Among the SRB Desulfovibrio sp., the structures of Flds from Desulfovibrio vulgaris (D. vulgaris) and Desulfovibrio desulfuricans (D. desulfuricans) have been determined [27,28]. The effects of some mutated residues, such as G61V and D95E, interacting with the FMN in three redox states were also studied in D. vulgaris Fld [29,30].
2. Results and Discussion
2.1. Crystal Characterization and X-ray Diffraction
2.2. The Crystal Structure of Fld
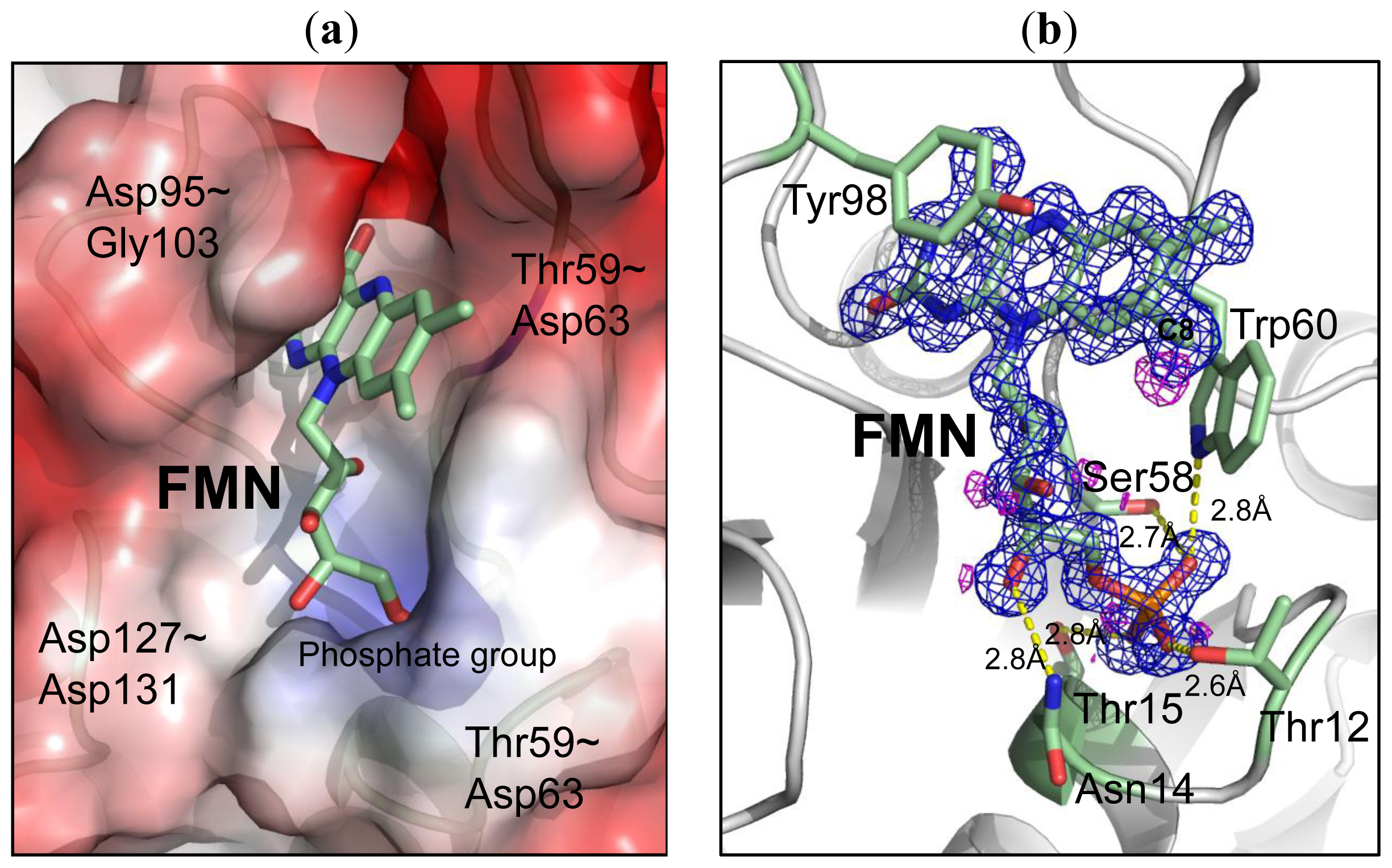
2.3. Environment of FMN
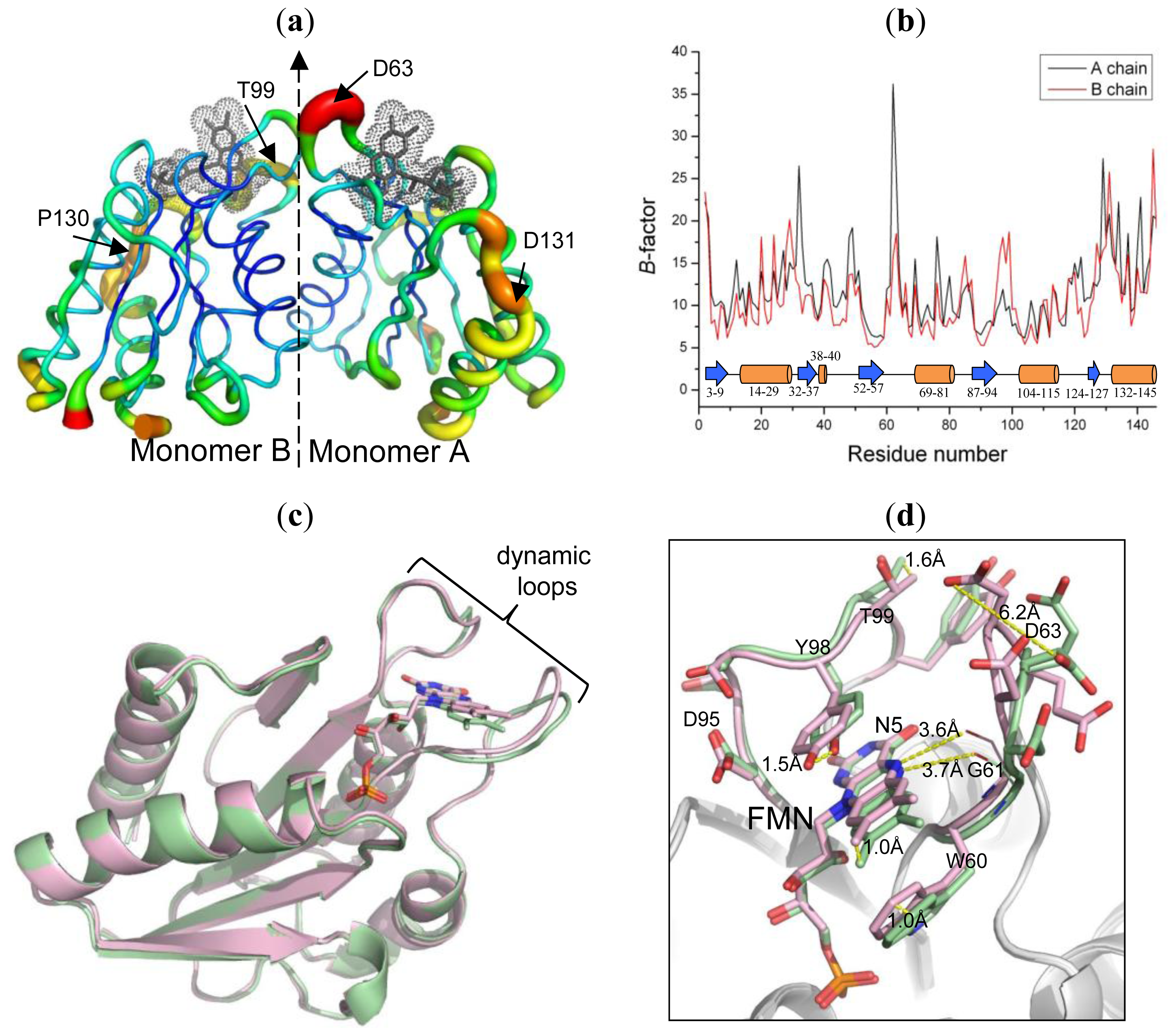
2.4. The Dynamic Characteristics in Fld
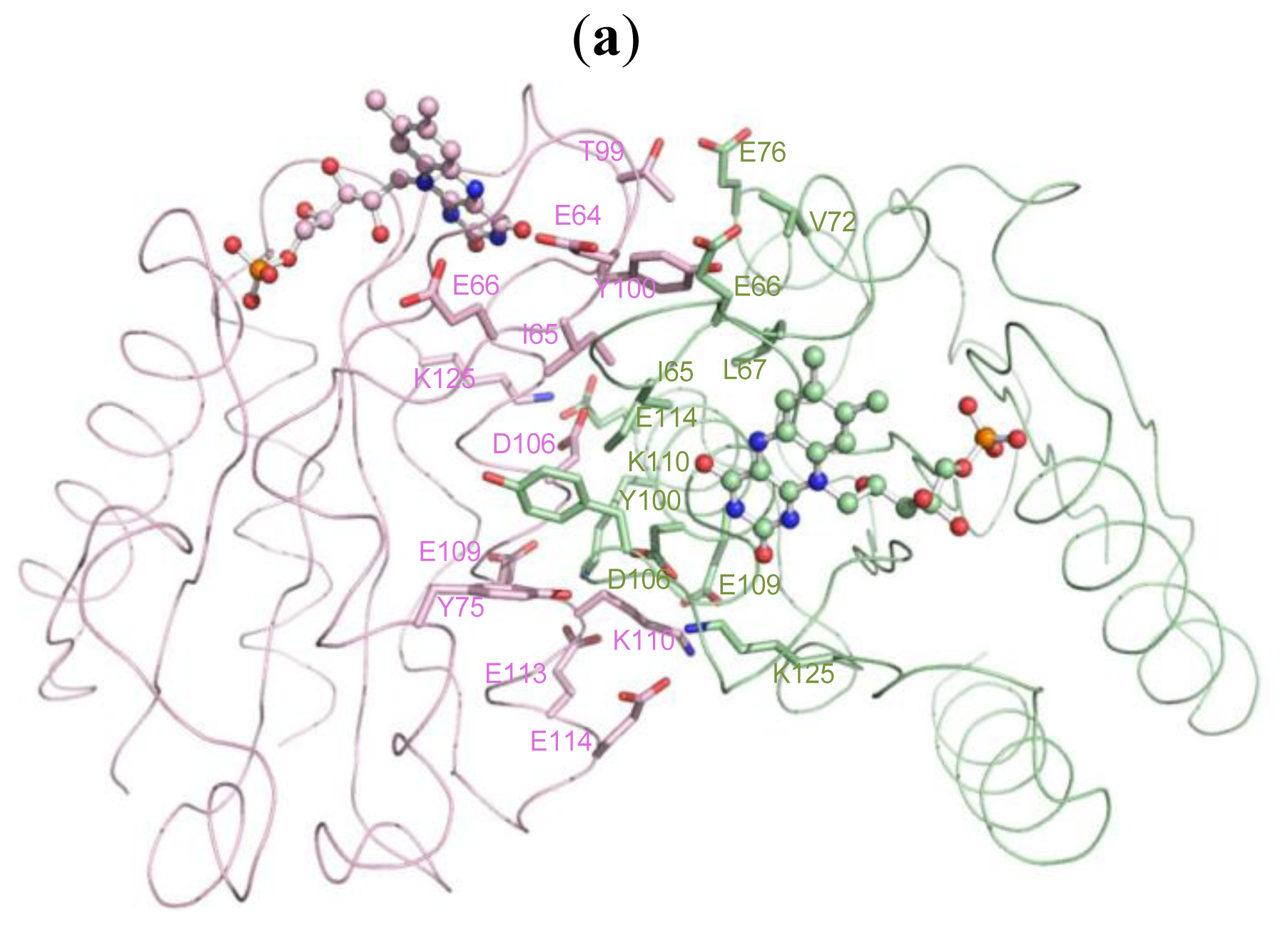
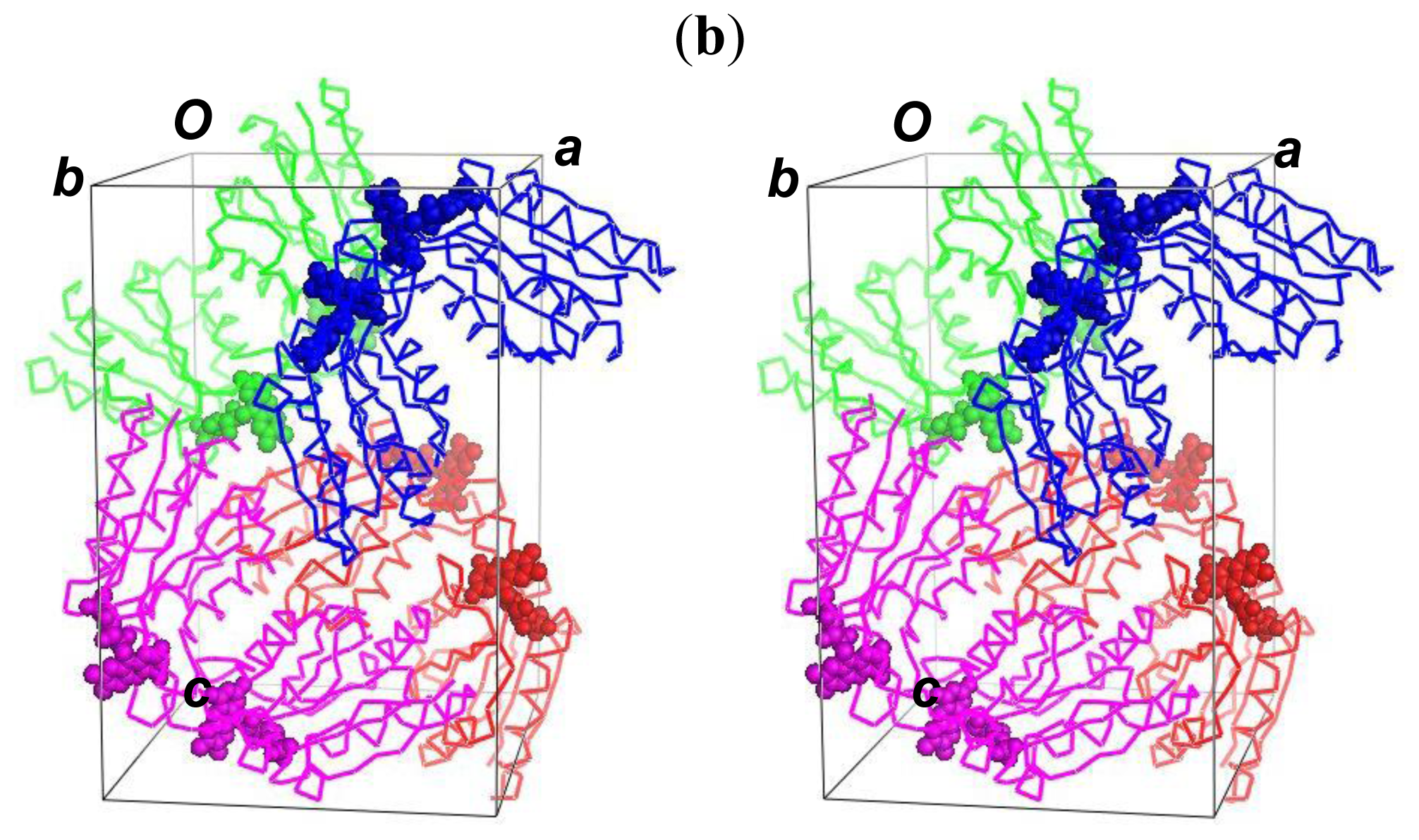
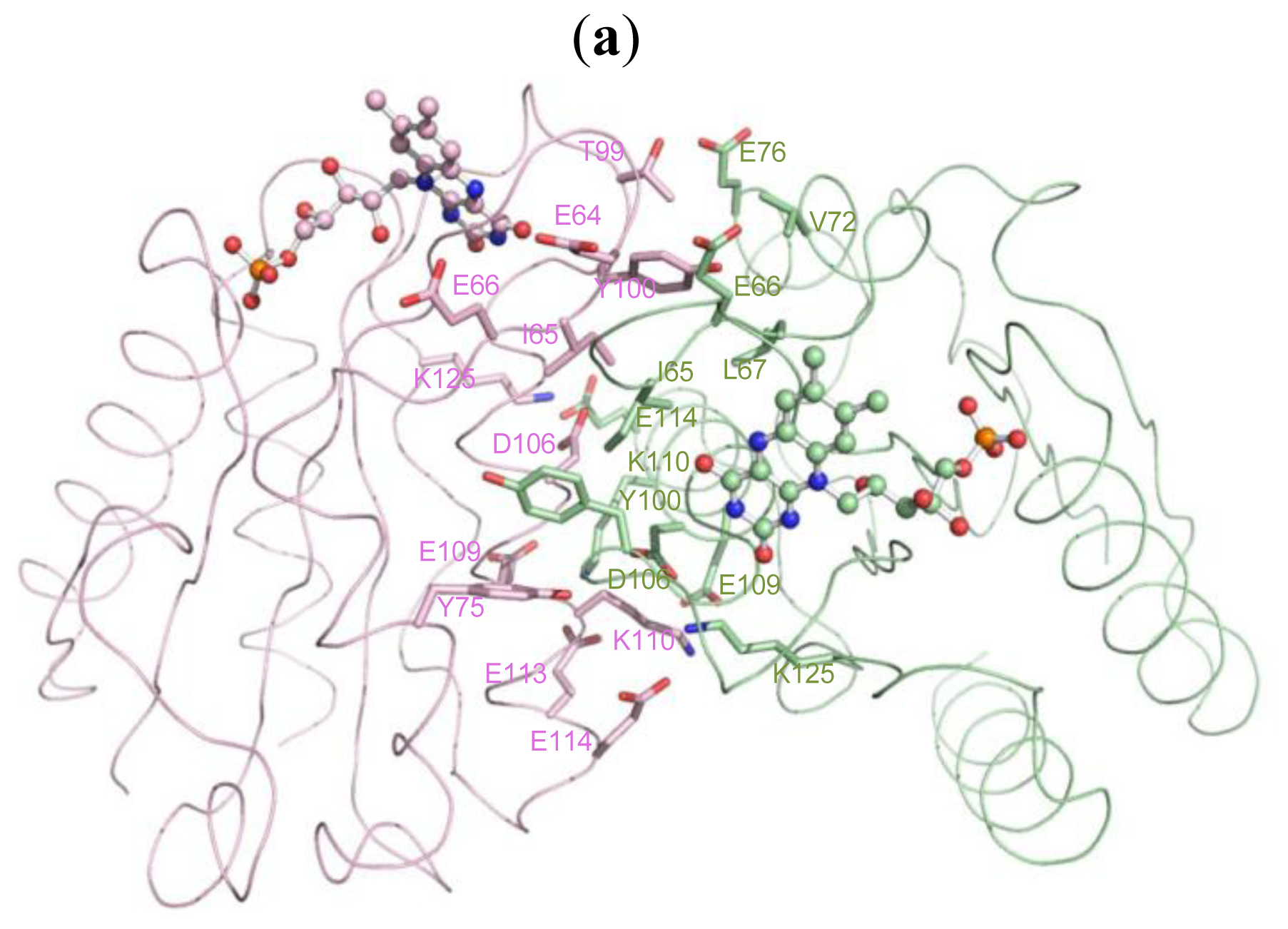
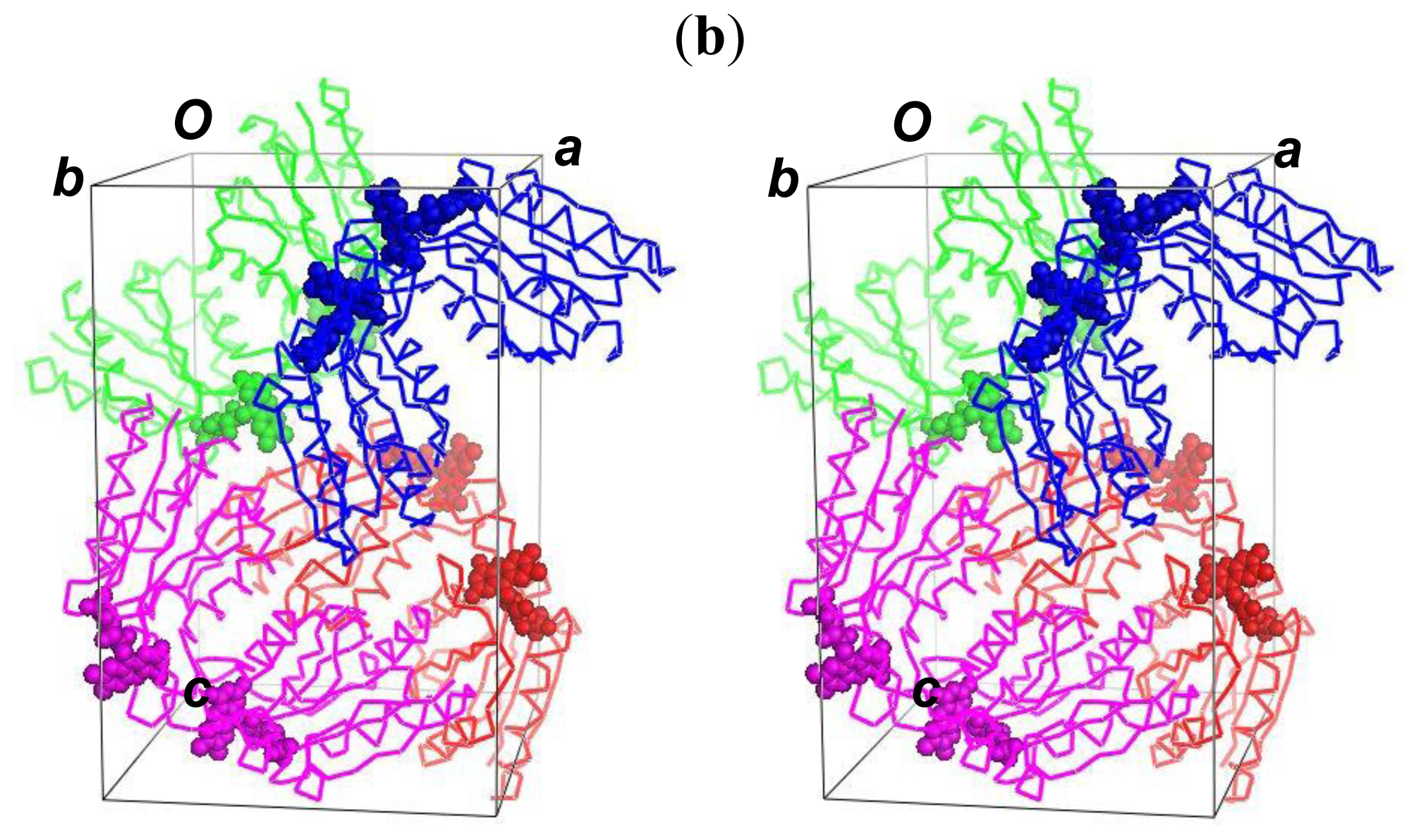
2.5. The Dimerization and Crystal Packing of Fld
2.6. Comparison with Fld Structures from Desulfovibrio sp
3. Experimental Section
3.1. Protein Purification
3.2. Crystallization
3.3. X-ray Data Collection and Processing
3.4. Structural Determination and Refinement
4. Conclusions
Supplementary Information
ijms-14-01667-s001.pdfAcknowledgments
References
- Meyer, T.E.; Cusanovich, M.A. Structure, function and distribution of soluble bacterial redox proteins. Biochim. Biophys. Acta 1989, 975, 1–28. [Google Scholar]
- Nogues, I.; Martinez-Julvez, M.; Navarro, J.A.; Hervas, M.; Armenteros, L.; delaRosa, M.A.; Brodie, T.B.; Hurley, J.K.; Tollin, G.; Gomez-Moreno, C.; et al. Role of hydrophobic interactions in the flavodoxin mediated electron transfer from photosystem I to ferredoxin-NADP+ reductase in anabaena PCC 7119. Biochemistry 2003, 42, 2036–2045. [Google Scholar]
- Nogues, I.; Hervas, M.; Peregrina, J.R.; Navarro, J.A.; delaRosa, M.A.; Gomez-Moreno, C.; Medina, M. Anabaena flavodoxin as an electron carrier from photosystem I to ferredoxin-NADP+ reductase. Role of flavodoxin residues in protein-protein interaction and electron transfer. Biochemistry 2005, 44, 97–104. [Google Scholar]
- Muhlenhoff, U.; Setif, P. Laser flash absorption spectroscopy study of flavodoxin reduction byphotosystem I in synechococcus sp. 2 PCC 700. Biochemistry 1996, 35, 1367–1374. [Google Scholar]
- Hall, D.A.; Jordan-Starck, T.C.; Loo, R.O.; Ludwig, M.L.; Matthews, R.G. Interaction of flavodoxin with cobalamin-dependent methionine synthase. Biochemistry 2000, 39, 10711–10719. [Google Scholar]
- Jarrett, J.T.; Huang, S.; Matthews, R.G. Methionine synthase exists in two distinct conformations that differ in reactivity toward methyltetrahydrofolate, adenosylmethionine and flavodoxin. Biochemistry 1998, 37, 5372–5382. [Google Scholar]
- Puan, K.J.; Wang, H.; Dairi, T.; Kuzuyama, T.; Morita, C.T. FldA is an essential gene required in the 2-C-methyl-d-erythritol 4-phosphate pathway for isoprenoid biosynthesis. FEBS Lett 2005, 579, 3802–3806. [Google Scholar]
- Lawson, R.J.; von Wachenfeldt, C.; Haq, I.; Perkins, J.; Munro, A.W. Expression and characterization of the two flavodoxin proteins of Bacillus subtilis, YkuN and YkuP: Biophysical properties and interactions with cytochrome P450 BioI. Biochemistry 2004, 43, 12390–123409. [Google Scholar]
- Gangeswaran, R.; Eady, R.R. Flavodoxin 1 of Azotobacter vinelandii: Characterization and role in electron donation to purified assimilatory nitrate reductase. Biochem. J 1996, 317, 103–108. [Google Scholar]
- Peelen, S.; Wijmenga, S.; Erbel, P.J.; Robson, R.L.; Eady, R.R.; Vervoort, J. Possible role of a short extra loop of the long-chain flavodoxin from Azotobacter chroococcum in electron transfer to nitrogenase: Complete 1H, 15N and 13C backbone assignments and secondary solution structure of the flavodoxin. J. Biomol. NMR 1996, 7, 315–330. [Google Scholar]
- Sawers, G.; Watson, G. A glycyl radical solution: Oxygen-dependent interconversion of pyruvate formate lyase. Mol. Microbiol 1998, 29, 945–954. [Google Scholar]
- Mulliez, E.; Padovani, D.; Atta, M.; Alcouffe, C.; Fontecave, M. Activation of class III ribonucleotide reductase by flavodoxin: A protein radical-Driven electron transfer to the iron-sulfur center. Biochemistry 2001, 40, 3730–3736. [Google Scholar]
- Deistung, J.R.; Thorneley, N.F. Electron transfer to nitrogenase. Characterization of flavodoxin from Azotobacter chroococcum and comparison of its redox potentials with those of flavodoxins from Azotobacter vinelandii and Klebsiella pneumoniae (nitF-gene Product). Biochem. J 1986, 239, 69–75. [Google Scholar]
- Gurley, G.P.; Carr, M.C.; O’Farell, P.A.; Mayhew, S.G.; Voordouw, G. Flavins and Flavoproteins; de Gruyter: Berlin, Germany, 1991; pp. 429–454. [Google Scholar]
- Mayhew, S.G.; Foust, G.P.; Massey, V. Oxidation-reduction properties of flavodoxin from Peptostreptococcus elsdenii. J. Biol. Chem 1969, 244, 803–810. [Google Scholar]
- Ludwig, M.L.; Luschinsky, C.L. Chemistry and Biochemistry of Flavoenzymes; Muller, F., Ed.; CRC Press: Boca Raton, FL, USA, 1992; pp. 427–466. [Google Scholar]
- Pochart, P.; Dore, J.; Lemann, F.; Goderel, I.; Rambaud, J.C. Interrelations between populations of methanogenic archaea and sulfate-reducing bacteria in the human colon. FEMS Microbiol. Lett 1992, 77, 225–228. [Google Scholar]
- Hamilton, W.A. Bioenergetics of sulphate-reducing bacteria in relation to their environmental impact. Biodegradation 1998, 9, 201–202. [Google Scholar]
- Dinh, H.T.; Kuever, J.; Mussmann, M.; Hassel, A.W.; Stratmann, M.; Widdel, F. Iron corrosion by novel anaerobic microorganisms. Nature 2004, 427, 829–832. [Google Scholar]
- Hammack, R.W.; Edenborn, H.M. The removal of nickel from mine waters using bacterial sulfate reduction. Appl. Microbiol. Biotech 1992, 37, 674–678. [Google Scholar]
- Haveman, S.A.; Greene, E.A.; Stilwell, C.P.; Voordouw, J.K.; Voordouw, G. Physiological and gene expression analysis of inhibition of Desulfovibrio vulgaris hildenborough by nitrite. J. Bacteriol 2004, 186, 7944–7950. [Google Scholar]
- Matias, P.M.; Pereira, I.A.; Soares, C.M.; Carrondo, M.A. Sulphate respiration from hydrogen in Desulfovibrio bacteria: A structural biology overview. Prog. Biophys. Mol. Biol 2005, 89, 292–329. [Google Scholar]
- Kim, J.H.; Akagi, J.M. Characterization of a trithionate reductase system from Desulfovibrio vulgaris. J. Bacterial 1985, 163, 472–475. [Google Scholar]
- Barata, B.A.; LeGall, J.; Moura, J.J. Aldehyde oxidoreductase activity in Desulfovibrio gigas: In vitro reconstitution of an electron-transfer chain from aldehydes to the production of molecular hydrogen. Biochemistry 1993, 32, 11559–11568. [Google Scholar]
- Chen, L.; Chen, M.Y.; LeGall, J. Isolation and characterization of flavoredoxin, a new flavoprotein that permits in vitro reconstitution of an electron transfer chain from molecular hydrogen to sulfite reduction in the cacterium Desulfovibrio gigas. Arch. Biochem. Biophys 1993, 303, 45–50. [Google Scholar]
- Palma, P.N.; Moura, I.; LeGall, J.; VanBeeumen, J.; Wampler, J.E.; Moura, J.J. Evidence for a ternary complexes formed between flavodoxin and cytochrome C3: 1H-NMR and molecular modeling studies. Biochemistry 1994, 33, 6394–6407. [Google Scholar]
- Watt, W.; Tulinsky, A.; Swenson, R.P.; Watenpaugh, K.D. Comparison of the crystal structure of a flavodoxin in its three oxidation states at cryogenic temperatures. J. Mol. Biol 1991, 218, 195–208. [Google Scholar]
- Guelker, M.; Stagg, L.; Wittung-Stafshede, P.; Shamoo, Y. Pseudosymmetry, high copy munber and twining complicate the structure determination of Desulfovibrio desulfuricans (ATCC 29677) flavodoxin. Acta Cryst. D Biol. Crystallogr 2009, 65, 523–534. [Google Scholar]
- O’Farrell, P.A.; Walsh, M.A.; McCarthy, A.A.; Higgins, T.M.; Voordouw, G.; Mayhew, S.G. Modulation of the redox potentials of FMN in Desulfovibrio vulgaris flavodoxin: Thermodynamic properties and crystal structure of glycine-61 mutants. Biochemistry 1998, 37, 8405–84016. [Google Scholar]
- McCarthy, A.A.; Walsh, M.A.; Verma, C.S.; O’Connell, D.P.; Reinhold, M.; Yalloway, G.N.; D’Arcy, D.; Higgins, T.M.; Voordouw, G.; Mayhew, S.G. Crystallographic investigation of the role of aspartate 95 in the modulation of the redox potentials of Desulfovibrio vulgaris flavodoxin. Biochemistry 2002, 41, 10950–10962. [Google Scholar]
- Mattews, B.W. Solvent content of protein crystals. J. Mol. Biol 1968, 33, 491–497. [Google Scholar]
- Sharkey, C.T.; Mayhew, S.G.; Higgins, T.M.; Walsh, M.A. Flavins and Flavoproteins; Stevenson, K.J., Massey, V., Williams, C.H., Eds.; University of Calgary Press: Calgary, AB, Canada, 1997; pp. 445–448. [Google Scholar]
- Smith, W.W.; Burnett, R.M.; Darling, G.D.; Ludwig, M.L. Structure of the semiquinone form of flavodoxin from Clostridum MP. Extension of 1.8 Å resolution and some comparisons with the oxidized state. J. Mol. Biol 1977, 117, 195–225. [Google Scholar]
- Orville, A.M.; Buono, R.; Cowan, M.; Heroux, A.; Shea-McCarthy, G.; Schneider, D.K.; Skinner, J.M.; Skinner, M.J.; Stoner-Ma, D.; Sweet, R.M. Correlated single-crystal electronic absorption spectroscopy and X-ray crystallography at NSLS beamline X26-C. J. Synchrotron Rad 2011, 18, 358–366. [Google Scholar]
- Røhr, Å.K.; Hersleth, H.P.; Andersson, K.K. Tracking flavin conformations in protein crystal structures with raman spectroscopy and QM/MM calculations. Angew. Chem. Int. Ed. 2010, 49, 2324–2327. [Google Scholar]
- Fantuzzi, A.; Artali, R.; Bombieri, G.; Marchini, N.; Meneghetti, F.; Gilardi, G.; Sadeghi, S.; Gavazzini, D.; Rossi, G.L. Redox properties and crystal structures of a Desulfovibrio vulgaris flavodoxin mutant in the monomeric and homodimeric forms. Biochim. Biophys. Acta 2009, 1794, 496–505. [Google Scholar]
- Hall, D.A.; Vander Kooi, C.W.; Stasik, C.N.; Stevens, S.Y.; Zuiderweg, E.R.; Matthews, R.G. Mapping the interactions between flavodoxin and its physiological partners flavodoxin reductase and cobalamin-dependent methionine synthase. Proc. Natl. Acad. Sci. USA 2001, 98, 951–956. [Google Scholar]
- Skyring, G.W.; Jones, H.E. Variations in the spectrum of desulfoviridin from Desulfovibrio gigas. Aust. J. Biol. Sci 1976, 29, 291–299. [Google Scholar]
- Moura, I.; Moura, J.J.; Bruschi, M.; LeGall, J. Flavodoxin and rebredoxin from Desulphovibrio salexigens. Biochi. Biophys. Acta 1980, 591, 1–8. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [Google Scholar]
- Brünger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr. D 1998, 54, 905–921. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [Google Scholar]
- Collaborative computational project number 4: The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D 1994, 50, 760–763.
- Gead, R.J. Improved fourier coefficients for maps using phases from partial structures with errors. Acta Cryst 1986, A42, 140–149. [Google Scholar]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cβ geometry: ϕ/ψ and Cβ deviation. Proteins 2002, 50, 437–450. [Google Scholar]
- PyMOL, version 1.3. Available online: http://www.rcsb.org accessed on 7 January 2013.



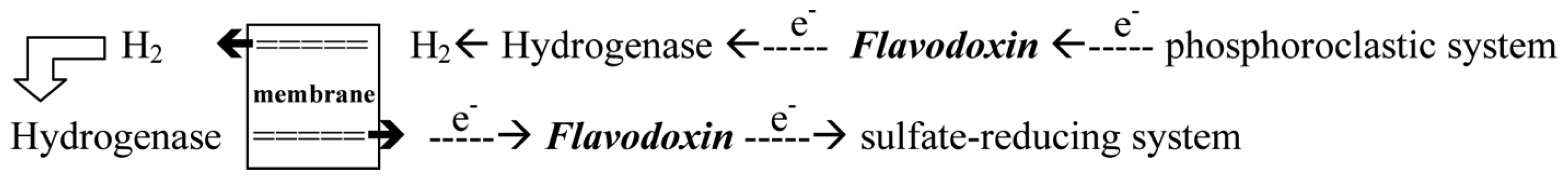

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
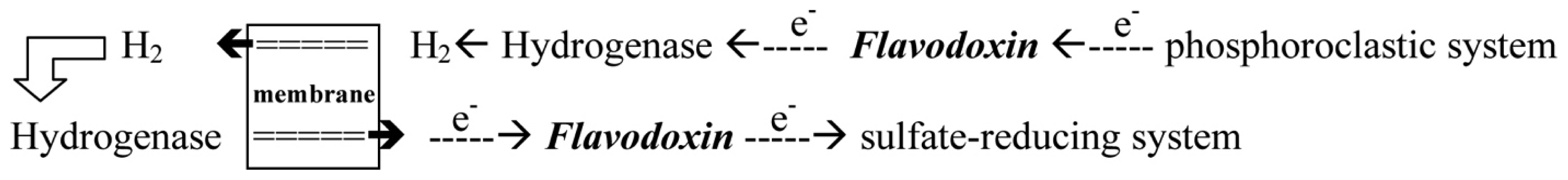{kind=link}
| Data collection | |
|---|---|
| Wavelength (Å) | 1.00 |
| Temperature (K) | 110 |
| Space group | P212121 |
| Resolution Range (Å) | 30.0–1.21 (1.25–1.21) a |
| Cell dimensions (Å) | |
| a | 50.20 |
| b | 60.37 |
| c | 76.25 |
| Unique reflections | 71,508 (7049) a |
| Completeness (%) | 99.9 (99.7) a |
| <I/σ(I)> | 33.9 (3.4) a |
| Average redundancy | 7.1 (6.8) a |
| Rsymb (%) | 8.7 (71.8%) a |
| Mosaicity | 0.28 |
| No. of molecules per asymmetric unit | 2 |
| Matthews coefficient (Å3 Da−1) | 2.06 |
| Solvent content (%) | 40.4 |
| Refinement | |
| Resolution range (Å) | 30.0–1.3 |
| Rworkc/Rfreed (%) | 18.0/21.1 |
| No. of atoms | |
| Protein | 2152 |
| Ligand (FMN) | 61 |
| Water molecules | 346 |
| B-factors (Å2) | |
| Protein | 11.5 |
| Ligand (FMN) | 8.0 |
| Water molecules | 21.1 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.027 |
| Bond angles (°) | 2.460 |
| FMN | Contact | Atoms | Distance (Å) (monomer A) | Distance (Å) (monomer B) |
|---|---|---|---|---|
| O3P [O] | 12(THR) | N [N] | 2.93 | 2.88 |
| 14(ASN) | N [N] | 2.93 | 2.92 | |
| 12(THR) | OG1 [O] | 2.56 | 2.57 | |
| O1P [O] | 60(TRP) | NE1[N] | 2.82 | 3.2 |
| 58(SER) | OG [O] | 2.74 | 2.71 | |
| 11(THR) | N [N] | 2.82 | 2.78 | |
| O2P [O] | 15(THR) | N [N] | 2.73 | 2.71 |
| 15(THR) | OG1 [O] | 2.75 | 2.76 | |
| 10(SER) | OG [O] | 2.71 | 2.69 | |
| O4′ [O] | 14(ASN) | ND2 [N] | 2.84 | 2.86 |
| O2′ [O] | 59(THR) | O [O] | 2.72 | 2.7 |
| O2 [O] | 95(ASP) | N [N] | 2.94 | 2.91 |
| 102(CYS) | N [N] | 2.78 | 2.78 | |
| Source (chain/residue) | Atoms | Target (chain/residue) | Atoms | Distance (Å) |
|---|---|---|---|---|
| A/64(GLU) | CB [C] | B/65(ILE) | O [O] | 3.30 |
| A/65(ILE) | N [N] | B/65(ILE) | O [O] | 3.20 |
| A 65(ILE) | O [O] | B/64(GLU) | CA [C] | 3.35 |
| B/65(ILE) | N [N] | 2.82 | ||
| A/66(GLU) | CG [C] | B/63(ASP) | O [O] | 3.45 |
| A/66(GLU) | CD [C] | B/63(ASP) | O [O] | 3.35 |
| A/67(LEU) | N [N] | B/100(TYR) | OH [O] | 3.03 |
| A/72(VAL) | CG2 [C] | B/100(TYR) | OH [O] | 3.49 |
| A/76(GLU) | OE2 [O] | B/99(THR) | CG2 [C] | 3.44 |
| B/99(THR) | OG1 [O] | 2.62 | ||
| A/100(TYR) | CD1 [C] | B/75(TYR) | CE2 [C] | 3.49 |
| A/106(ASP) | OD1 [O] | B/110(LYS) | CG [C] | 3.16 |
| A/109(GLU) | OE1 [O] | B/110(LYS) | CE [C] | 3.33 |
| B/110(LYS) | NZ [N] | 2.87 | ||
| A/110(LYS) | CB [C] | B/106(ASP) | OD1 [O] | 3.40 |
| A/110(LYS) | CD [C] | B/109(GLU) | OE1 [O] | 3.18 |
| A/110(LYS) | CE [C] | B/109(GLU) | OE1 [O] | 3.12 |
| A/110(LYS) | NZ [N] | B/109(GLU) | CD [C] | 3.45 |
| B/109(GLU) | OE1 [O] | 2.74 | ||
| B/113(GLU) | CD [C] | 3.25 | ||
| B/113(GLU) | OE1 [O] | 2.60 | ||
| B/113(GLU) | OE2 [O] | 3.20 | ||
| A/114(GLU) | OE2 [O] | B/125(LYS) | NZ [N] | 2.85 |
| A/125(LYS) | NZ [N] | B/114(GLU) | OE2 [O] | 2.83 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hsieh, Y.-C.; Chia, T.S.; Fun, H.-K.; Chen, C.-J. Crystal Structure of Dimeric Flavodoxin from Desulfovibrio gigas Suggests a Potential Binding Region for the Electron-Transferring Partner. Int. J. Mol. Sci. 2013, 14, 1667-1683. https://doi.org/10.3390/ijms14011667
Hsieh Y-C, Chia TS, Fun H-K, Chen C-J. Crystal Structure of Dimeric Flavodoxin from Desulfovibrio gigas Suggests a Potential Binding Region for the Electron-Transferring Partner. International Journal of Molecular Sciences. 2013; 14(1):1667-1683. https://doi.org/10.3390/ijms14011667
Chicago/Turabian StyleHsieh, Yin-Cheng, Tze Shyang Chia, Hoong-Kun Fun, and Chun-Jung Chen. 2013. "Crystal Structure of Dimeric Flavodoxin from Desulfovibrio gigas Suggests a Potential Binding Region for the Electron-Transferring Partner" International Journal of Molecular Sciences 14, no. 1: 1667-1683. https://doi.org/10.3390/ijms14011667