RUNX1: A MicroRNA Hub in Normal and Malignant Hematopoiesis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Transcription Factor-MicroRNA Networks in Hematopoiesis
3. RUNX1 in Hematopoiesis and Leukemia
3.1. RUNX1: A Master Regulator of Hematopoiesis
3.2. RUNX1 Perturbations in Leukemia
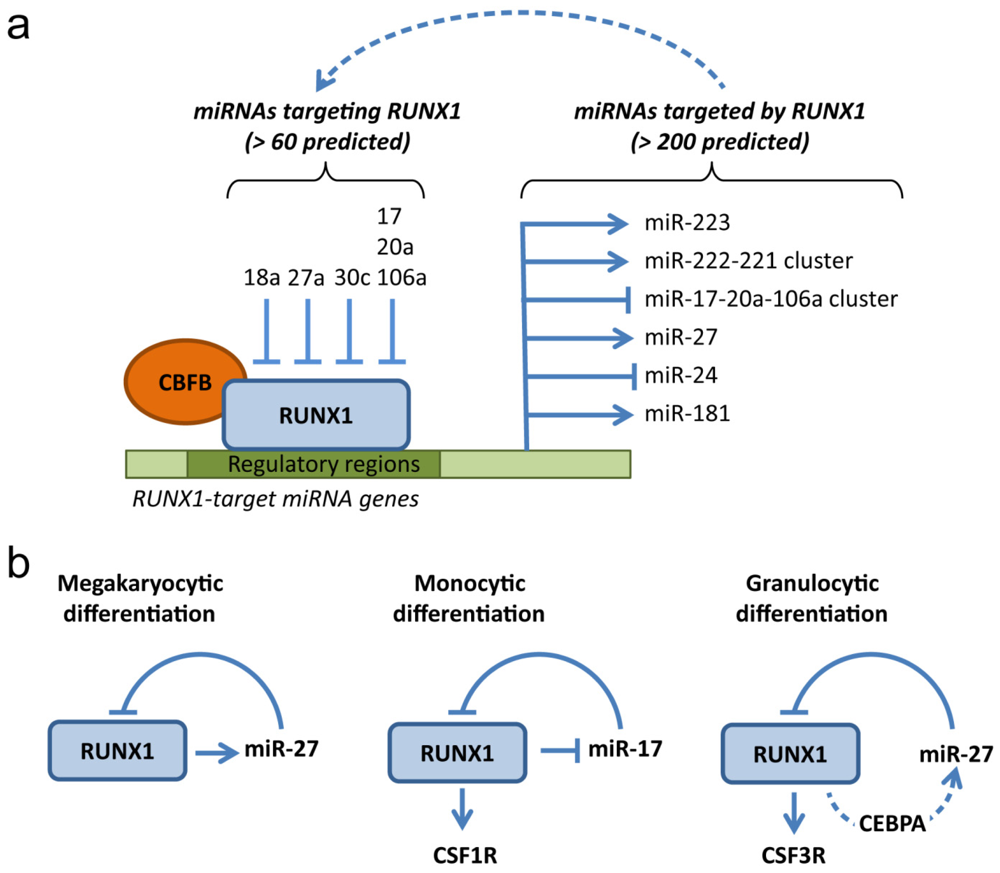
4. RUNX1: A MicroRNA Hub in Normal and Malignant Hematopoiesis
4.1. MicroRNAs Targeting RUNX1
4.2. MicroRNAs Targeted by RUNX1
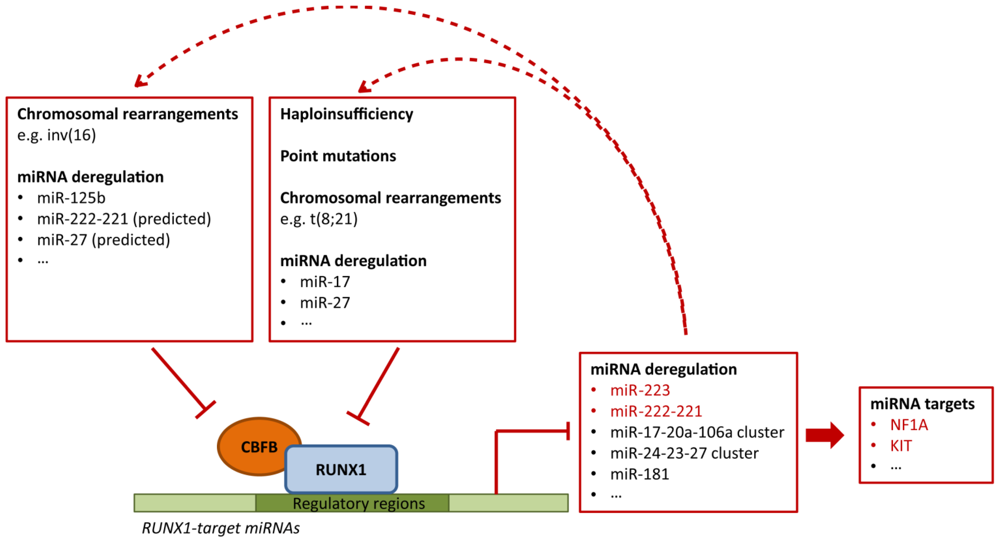
4.3. Deregulation of RUNX1-Related miRNAs in Leukemia
5. Concluding Remarks
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar]
- Wang, L.D.; Wagers, A.J. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nat. Rev. Mol. Cell. Biol 2011, 12, 643–655. [Google Scholar]
- Ottersbach, K.; Smith, A.; Wood, A.; Gottgens, B. Ontogeny of haematopoiesis: Recent advances and open questions. Br. J. Haematol 2009, 148, 343–355. [Google Scholar]
- Jaffredo, T.; Nottingham, W.; Liddiard, K.; Bollerot, K.; Pouget, C.; de Bruijn, M. From hemangioblast to hematopoietic stem cell: An endothelial connection? Exp. Hematol 2005, 33, 1029–1040. [Google Scholar]
- Mercer, E.M.; Lin, Y.C.; Murre, C. Factors and networks that underpin early hematopoiesis. Semin. Immunol 2011, 23, 317–325. [Google Scholar]
- Zhu, J.; Emerson, S.G. Hematopoietic cytokines, transcription factors and lineage commitment. Oncogene 2002, 21, 3295–3313. [Google Scholar]
- Spyropoulos, D.D.; Pharr, P.N.; Lavenburg, K.R.; Jackers, P.; Papas, T.S.; Ogawa, M.; Watson, D.K. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the fli1 transcription factor. Mol. Cell. Biol 2000, 20, 5643–5652. [Google Scholar]
- Lancrin, C.; Sroczynska, P.; Stephenson, C.; Allen, T.; Kouskoff, V.; Lacaud, G. The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature 2009, 457, 892–895. [Google Scholar]
- D’Souza, S.L.; Elefanty, A.G.; Keller, G. Scl/tal-1 is essential for hematopoietic commitment of the hemangioblast but not for its development. Blood 2005, 105, 3862–3870. [Google Scholar]
- Chen, M.J.; Yokomizo, T.; Zeigler, B.M.; Dzierzak, E.; Speck, N.A. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 2009, 457, 887–891. [Google Scholar]
- North, T.; Gu, T.L.; Stacy, T.; Wang, Q.; Howard, L.; Binder, M.; Marin-Padilla, M.; Speck, N.A. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development 1999, 126, 2563–2575. [Google Scholar]
- Yokomizo, T.; Ogawa, M.; Osato, M.; Kanno, T.; Yoshida, H.; Fujimoto, T.; Fraser, S.; Nishikawa, S.; Okada, H.; Satake, M.; et al. Requirement of runx1/aml1/pebp2alphab for the generation of haematopoietic cells from endothelial cells. Genes Cells 2001, 6, 13–23. [Google Scholar]
- DeKoter, R.P.; Singh, H. Regulation of b lymphocyte and macrophage development by graded expression of pu.1. Science 2000, 288, 1439–1441. [Google Scholar]
- Scott, E.W.; Simon, M.C.; Anastasi, J.; Singh, H. Requirement of transcription factor pu.1 in the development of multiple hematopoietic lineages. Science 1994, 265, 1573–1577. [Google Scholar]
- Scott, E.W.; Fisher, R.C.; Olson, M.C.; Kehrli, E.W.; Simon, M.C.; Singh, H. Pu.1 functions in a cell-autonomous manner to control the differentiation of multipotential lymphoid-myeloid progenitors. Immunity 1997, 6, 437–447. [Google Scholar]
- McKercher, S.R.; Torbett, B.E.; Anderson, K.L.; Henkel, G.W.; Vestal, D.J.; Baribault, H.; Klemsz, M.; Feeney, A.J.; Wu, G.E.; Paige, C.J.; et al. Targeted disruption of the pu.1 gene results in multiple hematopoietic abnormalities. EMBO J 1996, 15, 5647–5658. [Google Scholar]
- Rosenbauer, F.; Owens, B.M.; Yu, L.; Tumang, J.R.; Steidl, U.; Kutok, J.L.; Clayton, L.K.; Wagner, K.; Scheller, M.; Iwasaki, H.; et al. Lymphoid cell growth and transformation are suppressed by a key regulatory element of the gene encoding pu.1. Nat. Genet 2006, 38, 27–37. [Google Scholar]
- Radomska, H.S.; Huettner, C.S.; Zhang, P.; Cheng, T.; Scadden, D.T.; Tenen, D.G. Ccaat/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol. Cell. Biol 1998, 18, 4301–4314. [Google Scholar]
- Zhang, D.E.; Zhang, P.; Wang, N.D.; Hetherington, C.J.; Darlington, G.J.; Tenen, D.G. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in ccaat enhancer binding protein alpha-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 569–574. [Google Scholar]
- Starnes, L.M.; Sorrentino, A.; Ferracin, M.; Negrini, M.; Pelosi, E.; Nervi, C.; Peschle, C. A transcriptome-wide approach reveals the key contribution of nfi-a in promoting erythroid differentiation of human cd34(+) progenitors and cml cells. Leukemia 2010, 24, 1220–1223. [Google Scholar]
- Starnes, L.M.; Sorrentino, A.; Pelosi, E.; Ballarino, M.; Morsilli, O.; Biffoni, M.; Santoro, S.; Felli, N.; Castelli, G.; De Marchis, M.L.; et al. Nfi-a directs the fate of hematopoietic progenitors to the erythroid or granulocytic lineage and controls beta-globin and g-csf receptor expression. Blood 2009, 114, 1753–1763. [Google Scholar]
- Suzuki, H.; Forrest, A.R.; van Nimwegen, E.; Daub, C.O.; Balwierz, P.J.; Irvine, K.M.; Lassmann, T.; Ravasi, T.; Hasegawa, Y.; de Hoon, M.J.; et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat. Genet 2009, 41, 553–562. [Google Scholar]
- Pimanda, J.E.; Gottgens, B. Gene regulatory networks governing haematopoietic stem cell development and identity. Int. J. Dev. Biol 2010, 54, 1201–1211. [Google Scholar]
- Fazi, F.; Nervi, C. Microrna: Basic mechanisms and transcriptional regulatory networks for cell fate determination. Cardiovasc. Res 2008, 79, 553–561. [Google Scholar]
- El Gazzar, M.; McCall, C.E. Micrornas regulatory networks in myeloid lineage development and differentiation: Regulators of the regulators. Immunol. Cell Biol 2012, 90, 587–593. [Google Scholar]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for micrornas in the immune system. Nat. Rev. Immunol 2010, 10, 111–122. [Google Scholar]
- Alemdehy, M.F.; Erkeland, S.J. Micrornas: Key players of normal and malignant myelopoiesis. Curr. Opin. Hematol 2012, 19, 261–267. [Google Scholar]
- Bissels, U.; Bosio, A.; Wagner, W. Micrornas are shaping the hematopoietic landscape. Haematologica 2011, 97, 160–167. [Google Scholar]
- O’Connell, R.M.; Zhao, J.L.; Rao, D.S. Microrna function in myeloid biology. Blood 2011, 118, 2960–2969. [Google Scholar]
- Havelange, V.; Garzon, R. Micrornas: Emerging key regulators of hematopoiesis. Am. J. Hematol 2010, 85, 935–942. [Google Scholar]
- Shalgi, R.; Brosh, R.; Oren, M.; Pilpel, Y.; Rotter, V. Coupling transcriptional and post-transcriptional mirna regulation in the control of cell fate. Aging 2009, 1, 762–770. [Google Scholar]
- Tsang, J.; Zhu, J.; van Oudenaarden, A. Microrna-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell 2007, 26, 753–767. [Google Scholar]
- Shalgi, R.; Lieber, D.; Oren, M.; Pilpel, Y. Global and local architecture of the mammalian microrna-transcription factor regulatory network. PLoS Comput. Biol 2007, 3, e131. [Google Scholar]
- Starnes, L.M.; Sorrentino, A. Regulatory circuitries coordinated by transcription factors and micrornas at the cornerstone of hematopoietic stem cell self-renewal and differentiation. Curr. Stem. Cell. Res. Ther 2011, 6, 142–161. [Google Scholar]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; de Marchis, M.L.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of microrna-223 and transcription factors nfi-a and c/ebpalpha regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar]
- Rosa, A.; Ballarino, M.; Sorrentino, A.; Sthandier, O.; De Angelis, F.G.; Marchioni, M.; Masella, B.; Guarini, A.; Fatica, A.; Peschle, C.; et al. The interplay between the master transcription factor pu.1 and mir-424 regulates human monocyte/macrophage differentiation. Proc. Natl. Acad. Sci. USA 2007, 104, 19849–19854. [Google Scholar]
- Fontana, L.; Pelosi, E.; Greco, P.; Racanicchi, S.; Testa, U.; Liuzzi, F.; Croce, C.M.; Brunetti, E.; Grignani, F.; Peschle, C. Micrornas 17–5p-20a-106a control monocytopoiesis through aml1 targeting and m-csf receptor upregulation. Nat. Cell Biol 2007, 9, 775–787. [Google Scholar]
- Chen, J.; Odenike, O.; Rowley, J.D. Leukaemogenesis: More than mutant genes. Nat. Rev. Cancer 2010, 10, 23–36. [Google Scholar]
- Link, K.A.; Chou, F.S.; Mulloy, J.C. Core binding factor at the crossroads: Determining the fate of the hsc. J. Cell. Physiol 2009, 222, 50–56. [Google Scholar]
- Lam, K.; Zhang, D.E. Runx1 and runx1-eto: Roles in hematopoiesis and leukemogenesis. Front. Biosci 2012, 17, 1120–1139. [Google Scholar]
- Friedman, A.D. Cell cycle and developmental control of hematopoiesis by runx1. J. Cell. Physiol 2009, 219, 520–524. [Google Scholar]
- Swiers, G.; de Bruijn, M.; Speck, N.A. Hematopoietic stem cell emergence in the conceptus and the role of runx1. Int. J. Dev. Biol 2010, 54, 1151–1163. [Google Scholar]
- Miyoshi, H.; Shimizu, K.; Kozu, T.; Maseki, N.; Kaneko, Y.; Ohki, M. T(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, aml1. Proc. Natl. Acad. Sci. USA 1991, 88, 10431–10434. [Google Scholar]
- Erickson, P.; Gao, J.; Chang, K.S.; Look, T.; Whisenant, E.; Raimondi, S.; Lasher, R.; Trujillo, J.; Rowley, J.; Drabkin, H. Identification of breakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusion transcript, aml1/eto, with similarity to drosophila segmentation gene, runt. Blood 1992, 80, 1825–1831. [Google Scholar]
- Nisson, P.E.; Watkins, P.C.; Sacchi, N. Transcriptionally active chimeric gene derived from the fusion of the aml1 gene and a novel gene on chromosome 8 in t(8;21) leukemic cells [published erratum appears in cancer genet cytogenet 1993 mar;66(1):81]. Cancer Genet. Cytogenet 1992, 63, 81–88. [Google Scholar]
- Miyoshi, H.; Kozu, T.; Shimizu, K.; Enomoto, K.; Maseki, N.; Kaneko, Y.; Kamada, N.; Ohki, M. The t(8;21) translocation in acute myeloid leukemia results in production of an aml1-mtg8 fusion transcript. EMBO J 1993, 12, 2715–2721. [Google Scholar]
- Okuda, T.; van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R. Aml1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330. [Google Scholar]
- Wang, Q.; Stacy, T.; Binder, M.; Marin-Padilla, M.; Sharpe, A.H.; Speck, N.A. Disruption of the cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc. Natl. Acad. Sci. USA 1996, 93, 3444–3449. [Google Scholar]
- North, T.E.; Stacy, T.; Matheny, C.J.; Speck, N.A.; de Bruijn, M.F. Runx1 is expressed in adult mouse hematopoietic stem cells and differentiating myeloid and lymphoid cells, but not in maturing erythroid cells. Stem Cell 2004, 22, 158–168. [Google Scholar]
- Lorsbach, R.B.; Moore, J.; Ang, S.O.; Sun, W.; Lenny, N.; Downing, J.R. Role of runx1 in adult hematopoiesis: Analysis of runx1-ires-gfp knock-in mice reveals differential lineage expression. Blood 2004, 103, 2522–2529. [Google Scholar]
- Putz, G.; Rosner, A.; Nuesslein, I.; Schmitz, N.; Buchholz, F. Aml1 deletion in adult mice causes splenomegaly and lymphomas. Oncogene 2006, 25, 929–939. [Google Scholar]
- Growney, J.D.; Shigematsu, H.; Li, Z.; Lee, B.H.; Adelsperger, J.; Rowan, R.; Curley, D.P.; Kutok, J.L.; Akashi, K.; Williams, I.R.; et al. Loss of runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005, 106, 494–504. [Google Scholar]
- Ichikawa, M.; Asai, T.; Saito, T.; Seo, S.; Yamazaki, I.; Yamagata, T.; Mitani, K.; Chiba, S.; Ogawa, S.; Kurokawa, M.; et al. Aml-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat. Med 2004, 10, 299–304. [Google Scholar]
- Ichikawa, M.; Goyama, S.; Asai, T.; Kawazu, M.; Nakagawa, M.; Takeshita, M.; Chiba, S.; Ogawa, S.; Kurokawa, M. Aml1/runx1 negatively regulates quiescent hematopoietic stem cells in adult hematopoiesis. J. Immunol 2008, 180, 4402–4408. [Google Scholar]
- Meyers, S.; Downing, J.R.; Hiebert, S.W. Identification of aml-1 and the (8;21) translocation protein (aml- 1/eto) as sequence-specific DNA-binding proteins: The runt homology domain is required for DNA binding and protein-protein interactions. Mol. Cell. Biol 1993, 13, 6336–6345. [Google Scholar]
- Ogawa, E.; Inuzuka, M.; Maruyama, M.; Satake, M.; Naito-Fujimoto, M.; Ito, Y.; Shigesada, K. Molecular cloning and characterization of pebp2 beta, the heterodimeric partner of a novel drosophila runt-related DNA binding protein pebp2 alpha. Virology 1993, 194, 314–331. [Google Scholar]
- Ogawa, E.; Maruyama, M.; Kagoshima, H.; Inuzuka, M.; Lu, J.; Satake, M.; Shigesada, K.; Ito, Y. Pebp2/pea2 represents a family of transcription factors homologous to the products of the drosophila runt gene and the human aml1 gene. Proc. Natl. Acad. Sci. USA 1993, 90, 6859–6863. [Google Scholar]
- Wang, Q.; Stacy, T.; Miller, J.D.; Lewis, A.F.; Gu, T.L.; Huang, X.; Bushweller, J.H.; Bories, J.C.; Alt, F.W.; Ryan, G.; et al. The cbfbeta subunit is essential for cbfalpha2 (aml1) function in vivo. Cell 1996, 87, 697–708. [Google Scholar]
- Sasaki, K.; Yagi, H.; Bronson, R.T.; Tominaga, K.; Matsunashi, T.; Deguchi, K.; Tani, Y.; Kishimoto, T.; Komori, T. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc. Natl. Acad. Sci. USA 1996, 93, 12359–12363. [Google Scholar]
- Kitabayashi, I.; Yokoyama, A.; Shimizu, K.; Ohki, M. Interaction and functional cooperation of the leukemia-associated factors aml1 and p300 in myeloid cell differentiation. EMBO J 1998, 17, 2994–3004. [Google Scholar]
- Liu, H.; Carlsson, L.; Grundstrom, T. Identification of an n-terminal transactivation domain of runx1 that separates molecular function from global differentiation function. J. Biol. Chem 2006, 281, 25659–25669. [Google Scholar]
- Kanno, T.; Kanno, Y.; Chen, L.F.; Ogawa, E.; Kim, W.Y.; Ito, Y. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Mol. Cell. Biol 1998, 18, 2444–2454. [Google Scholar]
- Kitabayashi, I.; Aikawa, Y.; Nguyen, L.A.; Yokoyama, A.; Ohki, M. Activation of aml1-mediated transcription by moz and inhibition by the moz-cbp fusion protein. EMBO J 2001, 20, 7184–7196. [Google Scholar]
- Imai, Y.; Kurokawa, M.; Tanaka, K.; Friedman, A.D.; Ogawa, S.; Mitani, K.; Yazaki, Y.; Hirai, H. Tle, the human homolog of groucho, interacts with aml1 and acts as a repressor of aml1-induced transactivation. Biochem. Biophys. Res. Commun 1998, 252, 582–589. [Google Scholar]
- Levanon, D.; Goldstein, R.E.; Bernstein, Y.; Tang, H.; Goldenberg, D.; Stifani, S.; Paroush, Z.; Groner, Y. Transcriptional repression by aml1 and lef-1 is mediated by the tle/groucho corepressors. Proc. Natl. Acad. Sci. USA 1998, 95, 11590–11595. [Google Scholar]
- Fenrick, R.; Amann, J.M.; Lutterbach, B.; Wang, L.; Westendorf, J.J.; Downing, J.R.; Hiebert, S.W. Both tel and aml-1 contribute repression domains to the t(12;21) fusion protein. Mol. Cell. Biol 1999, 19, 6566–6574. [Google Scholar]
- Yu, M.; Mazor, T.; Huang, H.; Huang, H.T.; Kathrein, K.L.; Woo, A.J.; Chouinard, C.R.; Labadorf, A.; Akie, T.E.; Moran, T.B.; et al. Direct recruitment of polycomb repressive complex 1 to chromatin by core binding transcription factors. Mol. Cell 2012, 45, 330–343. [Google Scholar]
- Wang, L.; Huang, G.; Zhao, X.; Hatlen, M.A.; Vu, L.; Liu, F.; Nimer, S.D. Post-translational modifications of runx1 regulate its activity in the cell. Blood Cell. Mol. Dis 2009, 43, 30–34. [Google Scholar]
- Niebuhr, B.; Fischer, M.; Tager, M.; Cammenga, J.; Stocking, C. Gatekeeper function of the runx1 transcription factor in acute leukemia. Blood Cell. Mol. Dis 2008, 40, 211–218. [Google Scholar]
- Lichtinger, M.; Ingram, R.; Hannah, R.; Muller, D.; Clarke, D.; Assi, S.A.; Lie, A.L.M.; Noailles, L.; Vijayabaskar, M.S.; Wu, M.; et al. Runx1 reshapes the epigenetic landscape at the onset of haematopoiesis. EMBO J 2012, 31, 4318–4333. [Google Scholar]
- Huang, G.; Zhang, P.; Hirai, H.; Elf, S.; Yan, X.; Chen, Z.; Koschmieder, S.; Okuno, Y.; Dayaram, T.; Growney, J.D.; et al. Pu.1 is a major downstream target of aml1 (runx1) in adult mouse hematopoiesis. Nat. Genet 2008, 40, 51–60. [Google Scholar]
- Guo, H.; Ma, O.; Speck, N.A.; Friedman, A.D. Runx1 deletion or dominant inhibition reduces cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood 2012, 119, 4408–4418. [Google Scholar]
- Nuchprayoon, I.; Meyers, S.; Scott, L.M.; Suzow, J.; Hiebert, S.; Friedman, A.D. Pebp2/cbf, the murine homolog of the human myeloid aml1 and pebp2 beta/cbf beta proto-oncoproteins, regulates the murine myeloperoxidase and neutrophil elastase genes in immature myeloid cells. Mol. Cell. Biol 1994, 14, 5558–5568. [Google Scholar]
- Zhang, D.E.; Fujioka, K.; Hetherington, C.J.; Shapiro, L.H.; Chen, H.M.; Look, A.T.; Tenen, D.G. Identification of a region which directs the monocytic activity of the colony-stimulating factor 1 (macrophage colony-stimulating factor) receptor promoter and binds pebp2/cbf (aml1). Mol. Cell. Biol 1994, 14, 8085–8095. [Google Scholar]
- Takahashi, A.; Satake, M.; Yamaguchi-Iwai, Y.; Bae, S.C.; Lu, J.; Maruyama, M.; Zhang, Y.W.; Oka, H.; Arai, N.; Arai, K.; et al. Positive and negative regulation of granulocyte-macrophage colony-stimulating factor promoter activity by aml1-related transcription factor, pebp2. Blood 1995, 86, 607–616. [Google Scholar]
- Cameron, S.; Taylor, D.S.; TePas, E.C.; Speck, N.A.; Mathey-Prevot, B. Identification of a critical regulatory site in the human interleukin-3 promoter by in vivo footprinting. Blood 1994, 83, 2851–2859. [Google Scholar]
- De Braekeleer, E.; Douet-Guilbert, N.; Morel, F.; Le Bris, M.J.; Ferec, C.; De Braekeleer, M. Runx1 translocations and fusion genes in malignant hemopathies. Future Oncol 2011, 7, 77–91. [Google Scholar]
- Engel, M.E.; Hiebert, S.W. Proleukemic runx1 and cbfbeta mutations in the pathogenesis of acute leukemia. Cancer Treat. Res 2010, 145, 127–147. [Google Scholar]
- Rossetti, S.; Hoogeveen, A.T.; Sacchi, N. The mtg proteins: Chromatin repression players with a passion for networking. Genomics 2004, 84, 1–9. [Google Scholar]
- Gamou, T.; Kitamura, E.; Hosoda, F.; Shimizu, K.; Shinohara, K.; Hayashi, Y.; Nagase, T.; Yokoyama, Y.; Ohki, M. The partner gene of aml1 in t(16;21) myeloid malignancies is a novel member of the mtg8(eto) family. Blood 1998, 91, 4028–4037. [Google Scholar]
- Lutterbach, B.; Westendorf, J.J.; Linggi, B.; Patten, A.; Moniwa, M.; Davie, J.R.; Huynh, K.D.; Bardwell, V.J.; Lavinsky, R.M.; Rosenfeld, M.G.; et al. Eto, a target of t(8;21) in acute leukemia, interacts with the n-cor and msin3 corepressors. Mol. Cell. Biol 1998, 18, 7176–7184. [Google Scholar]
- Gelmetti, V.; Zhang, J.; Fanelli, M.; Minucci, S.; Pelicci, P.G.; Lazar, M.A. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner eto. Mol. Cell. Biol 1998, 18, 7185–7191. [Google Scholar]
- Wang, J.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. Eto, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human n-cor/msin3/hdac1 complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865. [Google Scholar]
- Kitabayashi, I.; Ida, K.; Morohoshi, F.; Yokoyama, A.; Mitsuhashi, N.; Shimizu, K.; Nomura, N.; Hayashi, Y.; Ohki, M. The aml1-mtg8 leukemic fusion protein forms a complex with a novel member of the mtg8(eto/cdr) family, mtgr1. Mol. Cell. Biol 1998, 18, 846–858. [Google Scholar]
- Follows, G.A.; Tagoh, H.; Lefevre, P.; Hodge, D.; Morgan, G.J.; Bonifer, C. Epigenetic consequences of aml1-eto action at the human c-fms locus. EMBO J 2003, 22, 2798–2809. [Google Scholar]
- Linggi, B.; Muller-Tidow, C.; van de Locht, L.; Hu, M.; Nip, J.; Serve, H.; Berdel, W.E.; van der Reijden, B.; Quelle, D.E.; Rowley, J.D.; et al. The t(8;21) fusion protein, aml1 eto, specifically represses the transcription of the p14(arf) tumor suppressor in acute myeloid leukemia. Nat. Med 2002, 8, 743–750. [Google Scholar]
- Peterson, L.F.; Yan, M.; Zhang, D.E. The p21waf1 pathway is involved in blocking leukemogenesis by the t(8;21) fusion protein aml1-eto. Blood 2007, 109, 4392–4398. [Google Scholar]
- Rossetti, S.; Van Unen, L.; Touw, I.P.; Hoogeveen, A.T.; Sacchi, N. Myeloid maturation block by aml1-mtg16 is associated with csf1r epigenetic downregulation. Oncogene 2005, 24, 5325–5332. [Google Scholar]
- Klampfer, L.; Zhang, J.; Zelenetz, A.O.; Uchida, H.; Nimer, S.D. The aml1/eto fusion protein activates transcription of bcl-2. Proc. Natl. Acad. Sci. USA 1996, 93, 14059–14064. [Google Scholar]
- Shimizu, K.; Kitabayashi, I.; Kamada, N.; Abe, T.; Maseki, N.; Suzukawa, K.; Ohki, M. Aml1-mtg8 leukemic protein induces the expression of granulocyte colony- stimulating factor (g-csf) receptor through the up-regulation of ccaat/enhancer binding protein epsilon. Blood 2000, 96, 288–296. [Google Scholar]
- Okumura, A.J.; Peterson, L.F.; Okumura, F.; Boyapati, A.; Zhang, D.E. T(8;21)(q22;q22) fusion proteins preferentially bind to duplicated aml1/runx1 DNA-binding sequences to differentially regulate gene expression. Blood 2008, 112, 1392–1401. [Google Scholar]
- Yergeau, D.A.; Hetherington, C.J.; Wang, Q.; Zhang, P.; Sharpe, A.H.; Binder, M.; Marin-Padilla, M.; Tenen, D.G.; Speck, N.A.; Zhang, D.E. Embryonic lethality and impairment of haematopoiesis in mice heterozygous for an aml1-eto fusion gene. Nat. Genet 1997, 15, 303–306. [Google Scholar]
- Okuda, T.; Cai, Z.; Yang, S.; Lenny, N.; Lyu, C.J.; van Deursen, J.M.; Harada, H.; Downing, J.R. Expression of a knocked-in aml1-eto leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood 1998, 91, 3134–3143. [Google Scholar]
- Higuchi, M.; O’Brien, D.; Kumaravelu, P.; Lenny, N.; Yeoh, E.J.; Downing, J.R. Expression of a conditional aml1-eto oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell 2002, 1, 63–74. [Google Scholar]
- De Guzman, C.G.; Warren, A.J.; Zhang, Z.; Gartland, L.; Erickson, P.; Drabkin, H.; Hiebert, S.W.; Klug, C.A. Hematopoietic stem cell expansion and distinct myeloid developmental abnormalities in a murine model of the aml1-eto translocation. Mol. Cell. Biol 2002, 22, 5506–5517. [Google Scholar]
- Rhoades, K.L.; Hetherington, C.J.; Harakawa, N.; Yergeau, D.A.; Zhou, L.; Liu, L.Q.; Little, M.T.; Tenen, D.G.; Zhang, D.E. Analysis of the role of aml1-eto in leukemogenesis, using an inducible transgenic mouse model. Blood 2000, 96, 2108–2115. [Google Scholar]
- Yuan, Y.; Zhou, L.; Miyamoto, T.; Iwasaki, H.; Harakawa, N.; Hetherington, C.J.; Burel, S.A.; Lagasse, E.; Weissman, I.L.; Akashi, K.; et al. Aml1-eto expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc. Natl. Acad. Sci. USA 2001, 98, 10398–10403. [Google Scholar]
- Wiemels, J.L.; Xiao, Z.; Buffler, P.A.; Maia, A.T.; Ma, X.; Dicks, B.M.; Smith, M.T.; Zhang, L.; Feusner, J.; Wiencke, J.; et al. In utero origin of t(8;21) aml1-eto translocations in childhood acute myeloid leukemia. Blood 2002, 99, 3801–3805. [Google Scholar]
- Liu, P.; Tarle, S.A.; Hajra, A.; Claxton, D.F.; Marlton, P.; Freedman, M.; Siciliano, M.J.; Collins, F.S. Fusion between transcription factor cbf beta/pebp2 beta and a myosin heavy chain in acute myeloid leukemia. Science 1993, 261, 1041–1044. [Google Scholar]
- Liu, P.P.; Wijmenga, C.; Hajra, A.; Blake, T.B.; Kelley, C.A.; Adelstein, R.S.; Bagg, A.; Rector, J.; Cotelingam, J.; Willman, C.L.; et al. Identification of the chimeric protein product of the cbfb-myh11 fusion gene in inv(16) leukemia cells. Genes Chromosom. Cancer 1996, 16, 77–87. [Google Scholar]
- Shigesada, K.; van de Sluis, B.; Liu, P.P. Mechanism of leukemogenesis by the inv(16) chimeric gene cbfb/pebp2b-mhy11. Oncogene 2004, 23, 4297–4307. [Google Scholar]
- Castilla, L.H.; Wijmenga, C.; Wang, Q.; Stacy, T.; Speck, N.A.; Eckhaus, M.; Marin-Padilla, M.; Collins, F.S.; Wynshaw-Boris, A.; Liu, P.P. Failure of embryonic hematopoiesis and lethal hemorrhages in mouse embryos heterozygous for a knocked-in leukemia gene cbfb-myh11. Cell 1996, 87, 687–696. [Google Scholar]
- Kuo, Y.H.; Landrette, S.F.; Heilman, S.A.; Perrat, P.N.; Garrett, L.; Liu, P.P.; Le Beau, M.M.; Kogan, S.C.; Castilla, L.H. Cbf beta-smmhc induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell 2006, 9, 57–68. [Google Scholar]
- Song, W.J.; Sullivan, M.G.; Legare, R.D.; Hutchings, S.; Tan, X.; Kufrin, D.; Ratajczak, J.; Resende, I.C.; Haworth, C.; Hock, R.; et al. Haploinsufficiency of cbfa2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat. Genet 1999, 23, 166–175. [Google Scholar]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med 2011, 364, 2496–2506. [Google Scholar]
- Gaidzik, V.I.; Bullinger, L.; Schlenk, R.F.; Zimmermann, A.S.; Rock, J.; Paschka, P.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; et al. Runx1 mutations in acute myeloid leukemia: Results from a comprehensive genetic and clinical analysis from the aml study group. J. Clin. Oncol 2011, 29, 1364–1372. [Google Scholar]
- Tang, J.L.; Hou, H.A.; Chen, C.Y.; Liu, C.Y.; Chou, W.C.; Tseng, M.H.; Huang, C.F.; Lee, F.Y.; Liu, M.C.; Yao, M.; et al. Aml1/runx1 mutations in 470 adult patients with de novo acute myeloid leukemia: Prognostic implication and interaction with other gene alterations. Blood 2009, 114, 5352–5361. [Google Scholar]
- Mangan, J.K.; Speck, N.A. Runx1 mutations in clonal myeloid disorders: From conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit. Rev. Oncog 2011, 16, 77–91. [Google Scholar]
- Matheny, C.J.; Speck, M.E.; Cushing, P.R.; Zhou, Y.; Corpora, T.; Regan, M.; Newman, M.; Roudaia, L.; Speck, C.L.; Gu, T.L.; et al. Disease mutations in runx1 and runx2 create nonfunctional, dominant-negative, or hypomorphic alleles. EMBO J 2007, 26, 1163–1175. [Google Scholar]
- Mendler, J.H.; Maharry, K.; Radmacher, M.D.; Mrozek, K.; Becker, H.; Metzeler, K.H.; Schwind, S.; Whitman, S.P.; Khalife, J.; Kohlschmidt, J.; et al. Runx1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and microrna expression signatures. J. Clin. Oncol 2012, 30, 3109–3118. [Google Scholar]
- Cai, Z.; de Bruijn, M.; Ma, X.; Dortland, B.; Luteijn, T.; Downing, R.J.; Dzierzak, E. Haploinsufficiency of aml1 affects the temporal and spatial generation of hematopoietic stem cells in the mouse embryo. Immunity 2000, 13, 423–431. [Google Scholar]
- Nottingham, W.T.; Jarratt, A.; Burgess, M.; Speck, C.L.; Cheng, J.F.; Prabhakar, S.; Rubin, E.M.; Li, P.S.; Sloane-Stanley, J.; Kong, A.S.J.; et al. Runx1-mediated hematopoietic stem-cell emergence is controlled by a gata/ets/scl-regulated enhancer. Blood 2007, 110, 4188–4197. [Google Scholar]
- Pimanda, J.E.; Donaldson, I.J.; de Bruijn, M.F.; Kinston, S.; Knezevic, K.; Huckle, L.; Piltz, S.; Landry, J.R.; Green, A.R.; Tannahill, D.; et al. The scl transcriptional network and bmp signaling pathway interact to regulate runx1 activity. Proc. Natl. Acad. Sci. USA 2007, 104, 840–845. [Google Scholar]
- Ben-Ami, O.; Pencovich, N.; Lotem, J.; Levanon, D.; Groner, Y. A regulatory interplay between mir-27a and runx1 during megakaryopoiesis. Proc. Natl. Acad. Sci. USA 2009, 106, 238–243. [Google Scholar]
- Feng, J.; Iwama, A.; Satake, M.; Kohu, K. Microrna-27 enhances differentiation of myeloblasts into granulocytes by post-transcriptionally downregulating runx1. Br. J. Haematol 2009, 145, 412–423. [Google Scholar]
- Olive, V.; Jiang, I.; He, L. Mir-17–92, a cluster of mirnas in the midst of the cancer network. Int. J. Biochem. Cell Biol 2010, 42, 1348–1354. [Google Scholar]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased mir-17–92 expression in lymphocytes. Nat. Immunol 2008, 9, 405–414. [Google Scholar]
- Ivanovska, I.; Ball, A.S.; Diaz, R.L.; Magnus, J.F.; Kibukawa, M.; Schelter, J.M.; Kobayashi, S.V.; Lim, L.; Burchard, J.; Jackson, A.L.; et al. Micrornas in the mir-106b family regulate p21/cdkn1a and promote cell cycle progression. Mol. Cell. Biol 2008, 28, 2167–2174. [Google Scholar]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microrna polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar]
- Dixon-McIver, A.; East, P.; Mein, C.A.; Cazier, J.B.; Molloy, G.; Chaplin, T.; Andrew Lister, T.; Young, B.D.; Debernardi, S. Distinctive patterns of microrna expression associated with karyotype in acute myeloid leukaemia. PLoS One 2008, 3, e2141. [Google Scholar]
- Ota, A.; Tagawa, H.; Karnan, S.; Tsuzuki, S.; Karpas, A.; Kira, S.; Yoshida, Y.; Seto, M. Identification and characterization of a novel gene, c13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res 2004, 64, 3087–3095. [Google Scholar]
- Venturini, L.; Battmer, K.; Castoldi, M.; Schultheis, B.; Hochhaus, A.; Muckenthaler, M.U.; Ganser, A.; Eder, M.; Scherr, M. Expression of the mir-17–92 polycistron in chronic myeloid leukemia (cml) cd34+ cells. Blood 2007, 109, 4399–4405. [Google Scholar]
- Mi, S.; Li, Z.; Chen, P.; He, C.; Cao, D.; Elkahloun, A.; Lu, J.; Pelloso, L.A.; Wunderlich, M.; Huang, H.; et al. Aberrant overexpression and function of the mir-17–92 cluster in mll-rearranged acute leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 3710–3715. [Google Scholar]
- Tsuzuki, S.; Hong, D.; Gupta, R.; Matsuo, K.; Seto, M.; Enver, T. Isoform-specific potentiation of stem and progenitor cell engraftment by aml1/runx1. PLoS Med 2007, 4, e172. [Google Scholar]
- Ptasinska, A.; Assi, S.A.; Mannari, D.; James, S.R.; Williamson, D.; Dunne, J.; Hoogenkamp, M.; Wu, M.; Care, M.; McNeill, H.; et al. Depletion of runx1/eto in t(8;21) aml cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia 2012, 26, 1829–1841. [Google Scholar]
- Wu, J.Q.; Seay, M.; Schulz, V.P.; Hariharan, M.; Tuck, D.; Lian, J.; Du, J.; Shi, M.; Ye, Z.; Gerstein, M.; et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS Genet 2012, 8, e1002565. [Google Scholar]
- Fazi, F.; Racanicchi, S.; Zardo, G.; Starnes, L.M.; Mancini, M.; Travaglini, L.; Diverio, D.; Ammatuna, E.; Cimino, G.; Lo-Coco, F.; et al. Epigenetic silencing of the myelopoiesis regulator microrna-223 by the aml1/eto oncoprotein. Cancer Cell 2007, 12, 457–466. [Google Scholar]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microrna-223. Nature 2008, 451, 1125–1129. [Google Scholar]
- Pulikkan, J.A.; Dengler, V.; Peramangalam, P.S.; Peer Zada, A.A.; Muller-Tidow, C.; Bohlander, S.K.; Tenen, D.G.; Behre, G. Cell-cycle regulator e2f1 and microrna-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood 2010, 115, 1768–1778. [Google Scholar]
- Felli, N.; Fontana, L.; Pelosi, E.; Botta, R.; Bonci, D.; Facchiano, F.; Liuzzi, F.; Lulli, V.; Morsilli, O.; Santoro, S.; et al. Micrornas 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. USA 2005, 102, 18081–18086. [Google Scholar]
- Lennartsson, J.; Jelacic, T.; Linnekin, D.; Shivakrupa, R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cell 2005, 23, 16–43. [Google Scholar]
- Kent, D.; Copley, M.; Benz, C.; Dykstra, B.; Bowie, M.; Eaves, C. Regulation of hematopoietic stem cells by the steel factor/kit signaling pathway. Clin. Cancer Res 2008, 14, 1926–1930. [Google Scholar]
- Brioschi, M.; Fischer, J.; Cairoli, R.; Rossetti, S.; Pezzetti, L.; Nichelatti, M.; Turrini, M.; Corlazzoli, F.; Scarpati, B.; Morra, E.; et al. Down-regulation of micrornas 222/221 in acute myelogenous leukemia with deranged core-binding factor subunits. Neoplasia 2010, 12, 866–876. [Google Scholar]
- Gao, X.N.; Lin, J.; Gao, L.; Li, Y.H.; Wang, L.L.; Yu, L. Microrna-193b regulates c-kit proto-oncogene and represses cell proliferation in acute myeloid leukemia. Leuk. Res 2011, 35, 1226–1232. [Google Scholar]
- Gao, X.N.; Lin, J.; Li, Y.H.; Gao, L.; Wang, X.R.; Wang, W.; Kang, H.Y.; Yan, G.T.; Wang, L.L.; Yu, L. Microrna-193a represses c-kit expression and functions as a methylation-silenced tumor suppressor in acute myeloid leukemia. Oncogene 2011, 30, 3416–3428. [Google Scholar]
- Kim, W.K.; Park, M.; Kim, Y.K.; Tae, Y.K.; Yang, H.K.; Lee, J.M.; Kim, H. Microrna-494 downregulates kit and inhibits gastrointestinal stromal tumor cell proliferation. Clin. Cancer Res 2011, 17, 7584–7594. [Google Scholar]
- Zaidi, S.K.; Dowdy, C.R.; van Wijnen, A.J.; Lian, J.B.; Raza, A.; Stein, J.L.; Croce, C.M.; Stein, G.S. Altered runx1 subnuclear targeting enhances myeloid cell proliferation and blocks differentiation by activating a mir-24/mkp-7/mapk network. Cancer Res 2009, 69, 8249–8255. [Google Scholar]
- Li, X.; Zhang, J.; Gao, L.; McClellan, S.; Finan, M.A.; Butler, T.W.; Owen, L.B.; Piazza, G.A.; Xi, Y. Mir-181 mediates cell differentiation by interrupting the lin28 and let-7 feedback circuit. Cell. Death Differ 2012, 19, 378–386. [Google Scholar]
- De Marchis, M.L.; Ballarino, M.; Salvatori, B.; Puzzolo, M.C.; Bozzoni, I.; Fatica, A. A new molecular network comprising pu.1, interferon regulatory factor proteins and mir-342 stimulates atra-mediated granulocytic differentiation of acute promyelocytic leukemia cells. Leukemia 2009, 23, 856–862. [Google Scholar]
- Ghani, S.; Riemke, P.; Schonheit, J.; Lenze, D.; Stumm, J.; Hoogenkamp, M.; Lagendijk, A.; Heinz, S.; Bonifer, C.; Bakkers, J.; et al. Macrophage development from hscs requires pu.1-coordinated microrna expression. Blood 2011, 118, 2275–2284. [Google Scholar]
- Pulikkan, J.A.; Peramangalam, P.S.; Dengler, V.; Ho, P.A.; Preudhomme, C.; Meshinchi, S.; Christopeit, M.; Nibourel, O.; Muller-Tidow, C.; Bohlander, S.K.; et al. C/ebpalpha regulated microrna-34a targets e2f3 during granulopoiesis and is down-regulated in aml with cebpa mutations. Blood 2010, 116, 5638–5649. [Google Scholar]
- Eyholzer, M.; Schmid, S.; Wilkens, L.; Mueller, B.U.; Pabst, T. The tumour-suppressive mir-29a/b1 cluster is regulated by cebpa and blocked in human aml. Br. J. Cancer 2010, 103, 275–284. [Google Scholar]
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.T.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microrna expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540. [Google Scholar]
- Bousquet, M.; Nguyen, D.; Chen, C.; Shields, L.; Lodish, H.F. Microrna-125b transforms myeloid cell lines by repressing multiple mrna. Haematologica 2012, 97, 1713–1721. [Google Scholar]
- Lin, K.Y.; Zhang, X.J.; Feng, D.D.; Zhang, H.; Zeng, C.W.; Han, B.W.; Zhou, A.D.; Qu, L.H.; Xu, L.; Chen, Y.Q. Mir-125b, a target of cdx2, regulates cell differentiation through repression of the core binding factor in hematopoietic malignancies. J. Biol. Chem 2011, 286, 38253–38263. [Google Scholar]
- Taniuchi, I.; Osato, M.; Ito, Y. Runx1: No longer just for leukemia. EMBO J 2012, 31, 4098–4099. [Google Scholar]
- Scheitz, C.J.; Tumbar, T. New insights into the role of runx1 in epithelial stem cell biology and pathology. J. Cell. Biochem. 2012. [Epub ahead of print]. [Google Scholar]
- Chimge, N.O.; Frenkel, B. The runx family in breast cancer: Relationships with estrogen signaling. Oncogene 2012. [Epub ahead of print]. [Google Scholar]
- Janes, K.A. Runx1 and its understudied role in breast cancer. Cell Cycle 2011, 10, 3461–3465. [Google Scholar]
- Wang, L.; Brugge, J.S.; Janes, K.A. Intersection of foxo- and runx1-mediated gene expression programs in single breast epithelial cells during morphogenesis and tumor progression. Proc. Natl. Acad. Sci. USA 2011, 108, E803–812. [Google Scholar]
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012, 486, 353–360. [Google Scholar]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 2005, 4, 1920–1932. [Google Scholar]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. TCGA-Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet 2003, 33, 49–54. [Google Scholar]
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol. Cell. Biol 2010, 30, 3943–3955. [Google Scholar]
- Di Leva, G.; Gasparini, P.; Piovan, C.; Ngankeu, A.; Garofalo, M.; Taccioli, C.; Iorio, M.V.; Li, M.; Volinia, S.; Alder, H.; et al. Microrna cluster 221–222 and estrogen receptor alpha interactions in breast cancer. J. Natl. Cancer Inst 2010, 102, 706–721. [Google Scholar]

© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rossetti, S.; Sacchi, N. RUNX1: A MicroRNA Hub in Normal and Malignant Hematopoiesis. Int. J. Mol. Sci. 2013, 14, 1566-1588. https://doi.org/10.3390/ijms14011566
Rossetti S, Sacchi N. RUNX1: A MicroRNA Hub in Normal and Malignant Hematopoiesis. International Journal of Molecular Sciences. 2013; 14(1):1566-1588. https://doi.org/10.3390/ijms14011566
Chicago/Turabian StyleRossetti, Stefano, and Nicoletta Sacchi. 2013. "RUNX1: A MicroRNA Hub in Normal and Malignant Hematopoiesis" International Journal of Molecular Sciences 14, no. 1: 1566-1588. https://doi.org/10.3390/ijms14011566
APA StyleRossetti, S., & Sacchi, N. (2013). RUNX1: A MicroRNA Hub in Normal and Malignant Hematopoiesis. International Journal of Molecular Sciences, 14(1), 1566-1588. https://doi.org/10.3390/ijms14011566