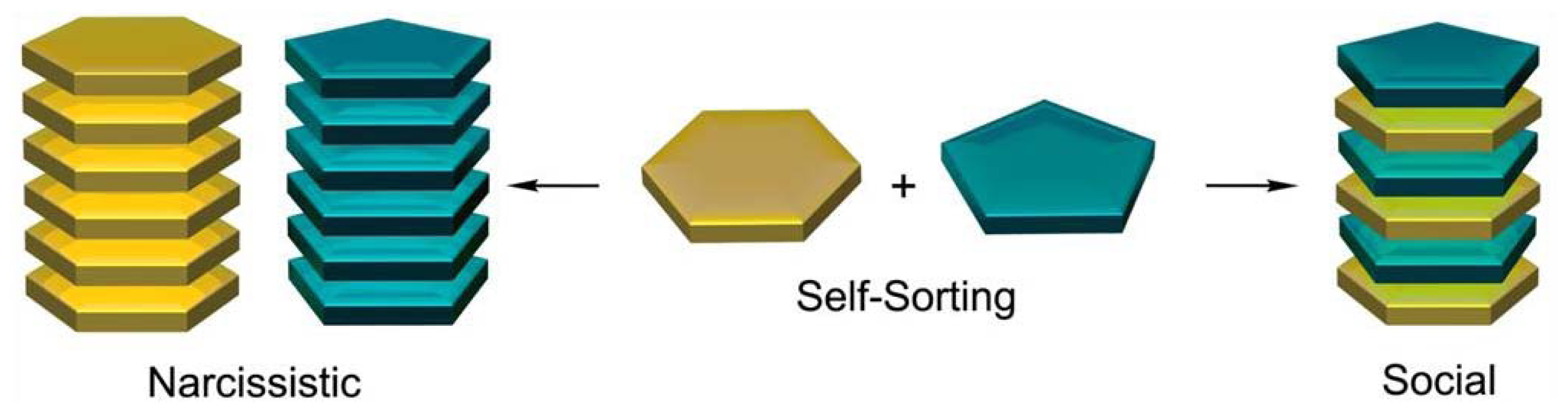
In the following sections, we have classified the examples described in the literature depending on the intermolecular force(s) that show a major contribution to the stability of the self-sorted architectures. Although hydrophobic forces play a significant role in most of the examples reported, other secondary interactions such as electrostatic forces and hydrogen bonding can also drive recognition processes if the influence of the hydrophobic effect is reduced.
2.1. Self-Sorting Systems Driven by Hydrophobic and π–π Interactions
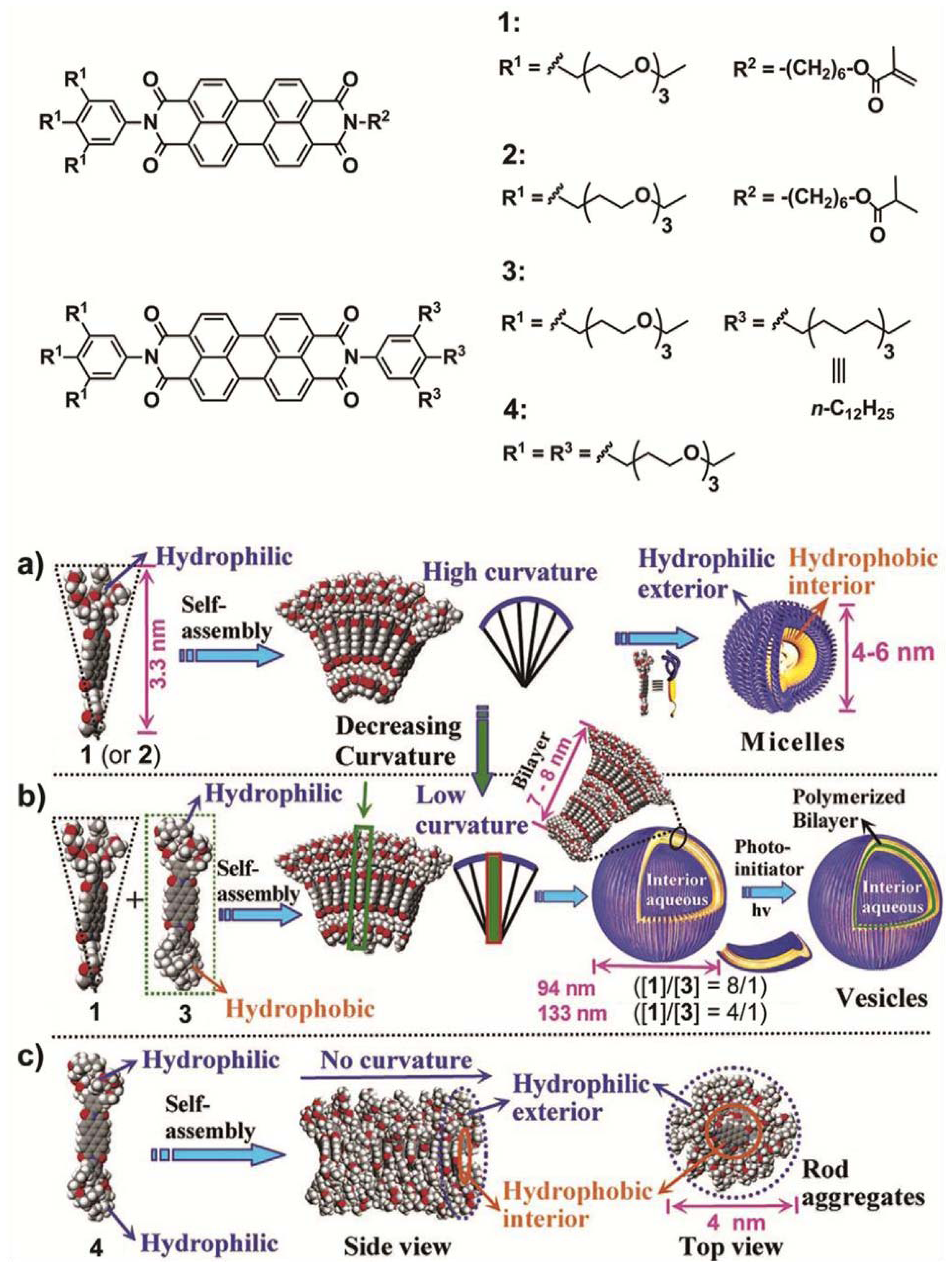
Würthner and co-workers designed four different wedge- and dumbbell-shaped amphiphilic perylene bisimides (PBIs) substituted with a variable number of alkyl and/or polar ethyleneglycol substituents and investigated the self-sorting behavior in a mixture of two of them in aqueous solution [
15]. Wedge-shaped PBI
1 and
2 self-assemble into spherical micelles with a diameter of 4–6 nm in THF-containing water (2%
v/
v), as visualized by transmission electron microscopy (TEM) studies (
Scheme 2a and
Figure 2a). This aggregation is driven by strong hydrophobic and aromatic interactions between the hydrophobic PBI cores supported by hydrophilic forces between the glycol chains, giving rise to fluorescent micellar objects with high curvature. In contrast to the behavior of derivatives
1 and
2, dumbbell-shaped PBI
4 self-assembles into rod-like assemblies driven by π–π and hydrophobic interactions between the PBI cores in a preferred direction with a minimum of curvature (
Scheme 2c and
Figure 2b). As a result, stiff aggregates with a regular diameter of 4 nm and several tens of nanometers long are formed (see model in
Scheme 2c). The authors then questioned what would happen if a wedge-shaped PBI
1 and a dumbbell-shaped PBI
3 were mixed in different ratios (
Scheme 2b). Surprisingly, for the mixtures of
1/
3 in THF-containing water (2%
v/
v) with ratios between 8:1 and 4:1 vesicles with average diameters of 94 nm and a bilayer membrane (7–8 nm) could be observed in TEM that exhibited an increasing diameter for higher contents of
3 (
Figure 2c,e). The varying size could be explained by taking into account that the higher content of dumbbell-shaped molecules caused a more demanding volume of hydrophobic moieties and consequently the arrangement between the building blocks has a lower curvature. These vesicular compartments have been more recently exploited to encapsulate donor bispyrene moieties in their interior [
16]. The donor-loaded vesicles display pH-dependent fluorescence resonance energy transfer from the encapsulated donors to the bilayer dye membrane, providing ultrasensitive pH information on their aqueous environment with fluorescence color changes covering the whole visible light range. These findings provide access to multifunctional nanocontainers with the ability to encapsulate a broad variety of guest molecules.
The research group of Myongsoo Lee has devoted intensive studies towards the creation of highly-organized supramolecular architectures in aqueous medium upon self- or co-assembly of a wide variety of amphiphilic systems featuring large hydrophobic segments [
17]. In an elegant example, these authors synthesized an amphiphilic dumbbell-shaped molecule (
5) consisting of a hexa-para-phenylene rod segment and aliphatic polyether dendrons based on a tetrahedral core and investigated whether the addition of aromatic guest molecules would influence the self-assembly behavior (
Scheme 3) [
18]. The aggregation of amphiphile
5 was first investigated through fluorescence experiments. In a “good” solvent like chloroform,
5 shows two emission maxima at 381 and 400 nm. However, the emission maximum in water is quenched and red-shifted compared to that in chloroform, which is indicative of the aggregation of the hydrophobic rod segments.
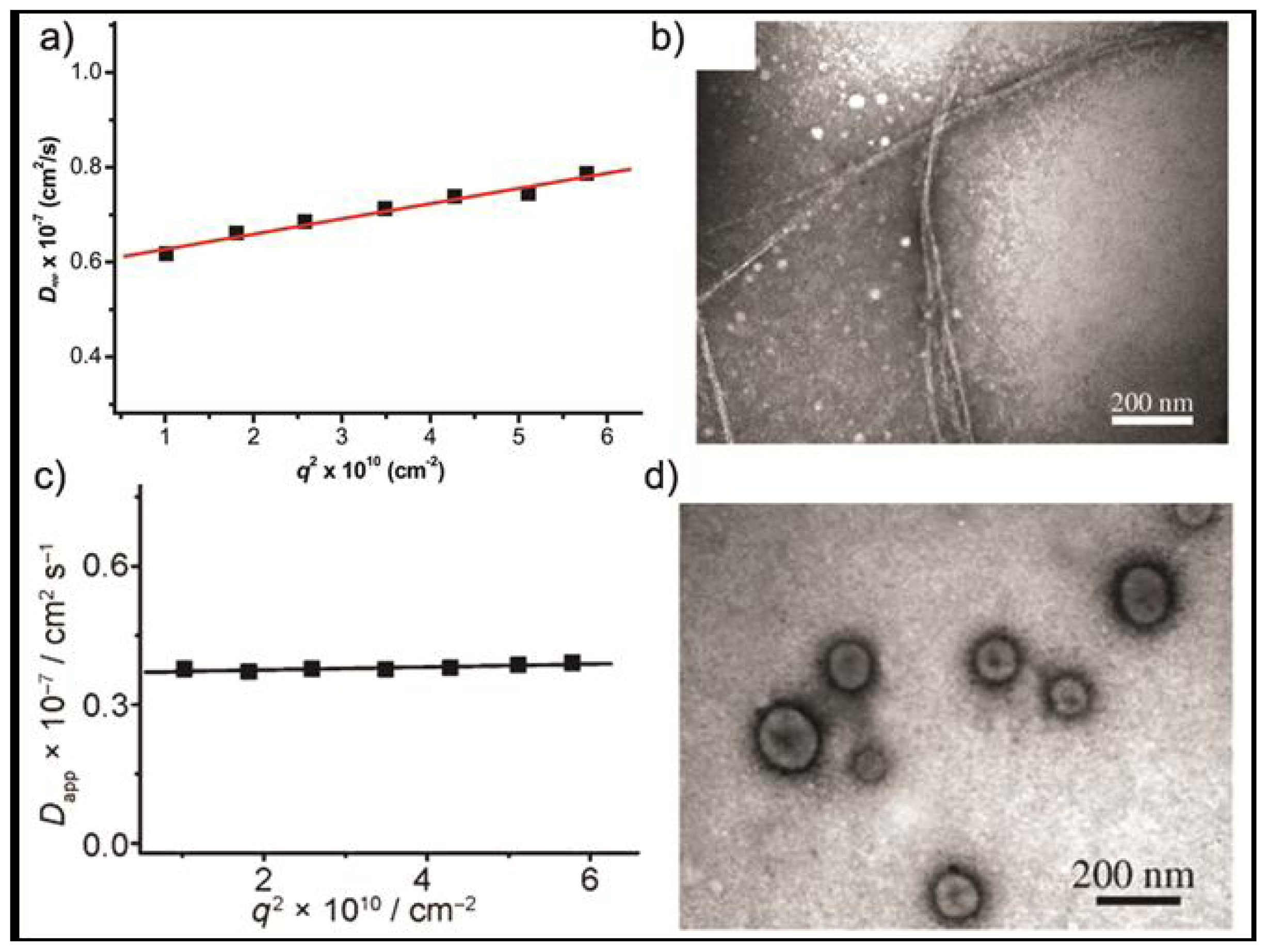
Dynamic light scattering (DLS) experiments on aqueous solutions show an aggregate size of around 60 nm with a slope of the angular dependence of the apparent diffusion coefficient (
Dapp) of 0.03, suggesting the formation of anisotropic objects (
Figure 3a). This was further confirmed by TEM imaging, in which helical cylinders with a uniform diameter of about 10 nm can be observed (
Figure 3b). In these aggregates, the elongated aromatic segments stack on top of each other with a slight rotation so that the sterical hindrance between the bulky dendrons is alleviated, while the dendritic substituents are oriented at the periphery exposed to the aqueous environment. By addition of aromatic guest molecules such as 4-bromo-nitrobenzene to an aqueous solution of
5, a structural rearrangement of the aggregates takes place. In contrast to the behavior of
5 in isolation, the gradient of the slope of the angular dependence of
Dapp is approximately zero, which suggests the formation of spherical aggregates (
Figure 3c). TEM experiments reveal that the addition of 4-bromo-nitrobenzene induces a parallel orientation of the rod segments in which the guest molecules are intercalated between the hydrophobic units (
Scheme 3). In this way a spherical capsule consisting of a unilamellar membrane was formed (
Figure 3d). Evidence supporting the formation of hollow capsules was provided by the encapsulation of the hydrophilic fluorescent guest Calcein. When Calcein was encapsulated into these structures its fluorescence was suppressed whereas by heating the solution to 60 °C the fluorescence intensity increased indicating the release of the dye molecules.
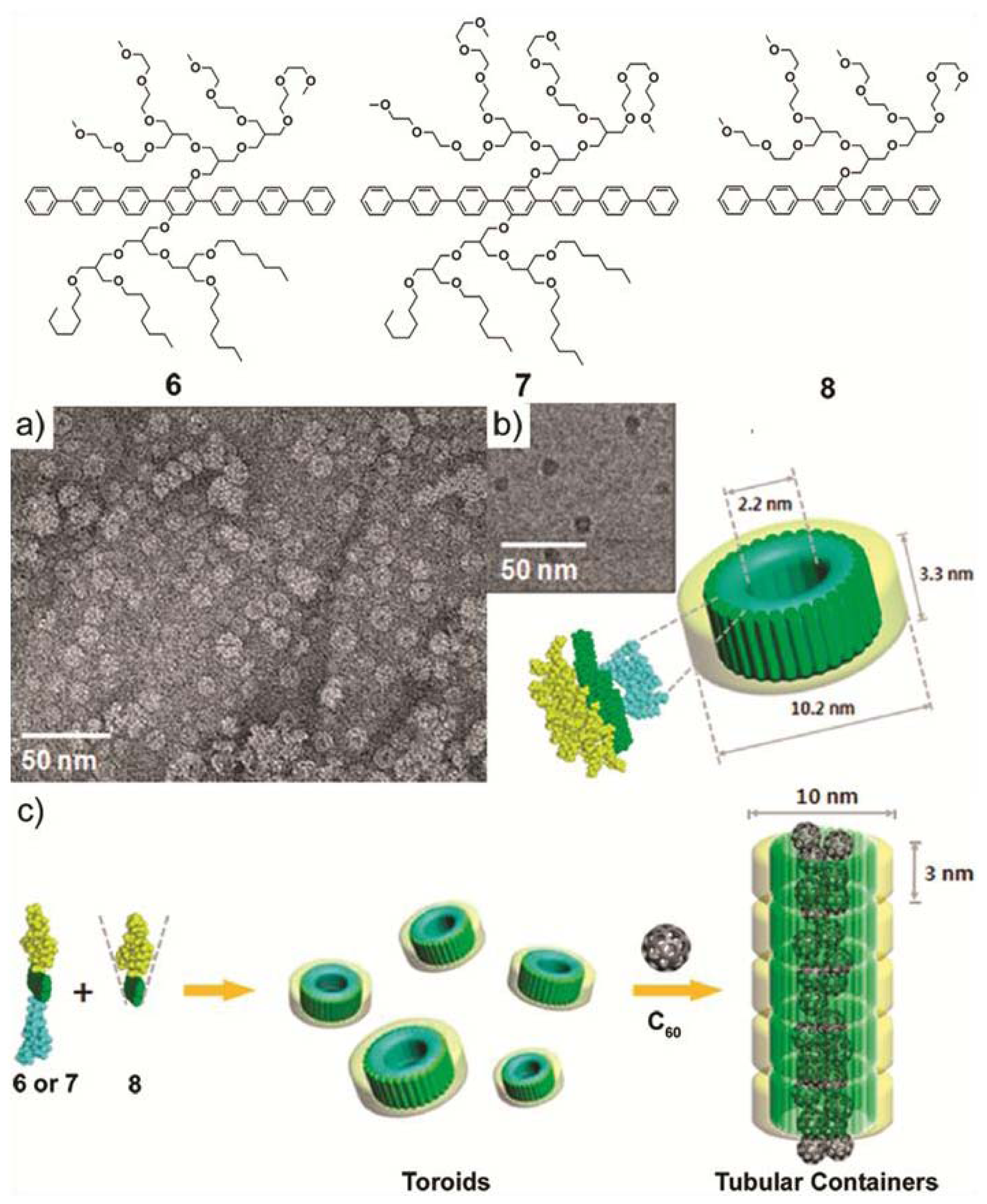
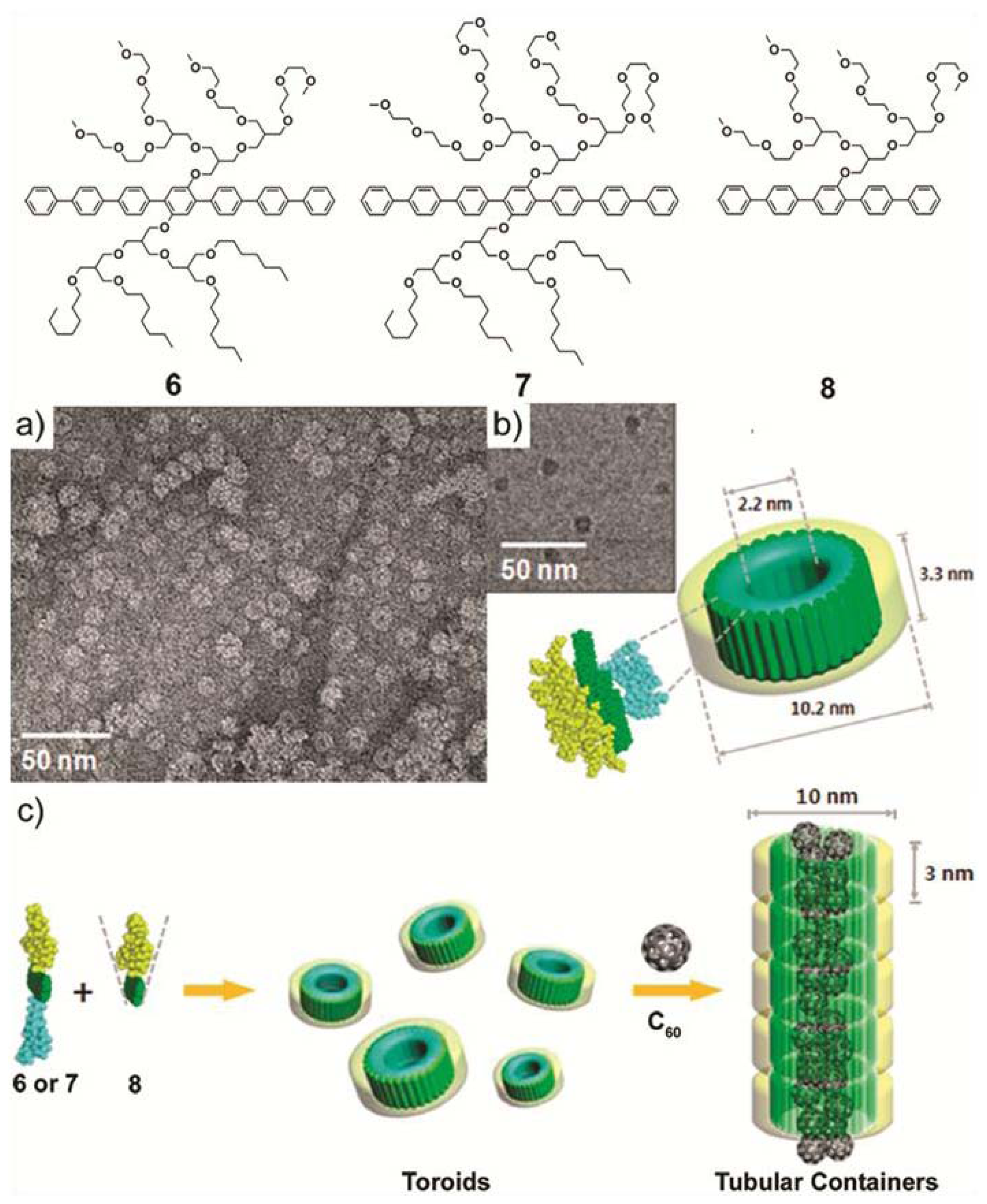
In a more recent example, M. Lee and co-workers synthesized two novel laterally substituted amphiphilic molecules (
6 and
7) that consist of a hepta(
para-phenylene) core bearing hydrophilic oligoether dendrons on one side and hydrophobic branched heptyl side chains on the opposite side of the aromatic rod [
19]. In aqueous solution,
6 and
7 self-assemble forming planar and ribbon-like arrangements, respectively. This structural diversity motivated the authors to investigate whether an increase of the volume fraction of the hydrophilic side chains would induce the formation of more curved structures. To that end, a new amphiphilic derivative
8 featuring a hydrophilic branched chain on only one side was synthesized and its co-assembly with
6 and
7 investigated. DLS experiments indicate that the size of the co-assemblies decreased with increasing content of
8, whereas TEM pictures demonstrate the reorganization of the molecules from planar sheets for pure
6 to discrete ribbon-like aggregates for the binary mixture (
6:8) up to a content of
8 of 40 mol%. Further addition of
8 (70 mol%) forced the ribbon structure to eventually convert at 80 mol% into discrete separate toroids (
Figure 4a,b). These co-assembled nanostructures consist of a single layer wall where the hydrophobic chains cover the inner part whereas the hydrophilic dendrons are oriented at the periphery. The interior of the co-aggregates of
1 and
8 is hydrophobic and possesses a cavity of 1–2 nm that can be exploited to incorporate [60] fullerene units and solubilize them in aqueous media. First evidence for this encapsulation was provided by fluorescence experiments performed on a mixture of C
60 and a co-assembled solution of
6 and
8. The quenching of the fluorescence intensity of the co-assembled mixture upon addition of fullerene demonstrates an effective encapsulation of fullerene units in the cavity of the toroidal objects up to a mole percent of 30%. The encapsulation of fullerenes also influenced the structure of the aggregates. DLS and TEM studies revealed that the aggregates became longer upon addition of C
60, giving rise to tubular cylindrical structures of 10 nm in diameter and several hundreds of nanometers in length (
Figure 4c). The suggested model implies the stacking of the toroids on top of each other stabilized by the fullerene skeleton in the hydrophobic interior. The ability to organize fullerenes at the nanometer scale is an important factor to optimize in the field of optoelectronics [
20]. The achievements of Lee and co-workers represent an important contribution with potential application in these fields.
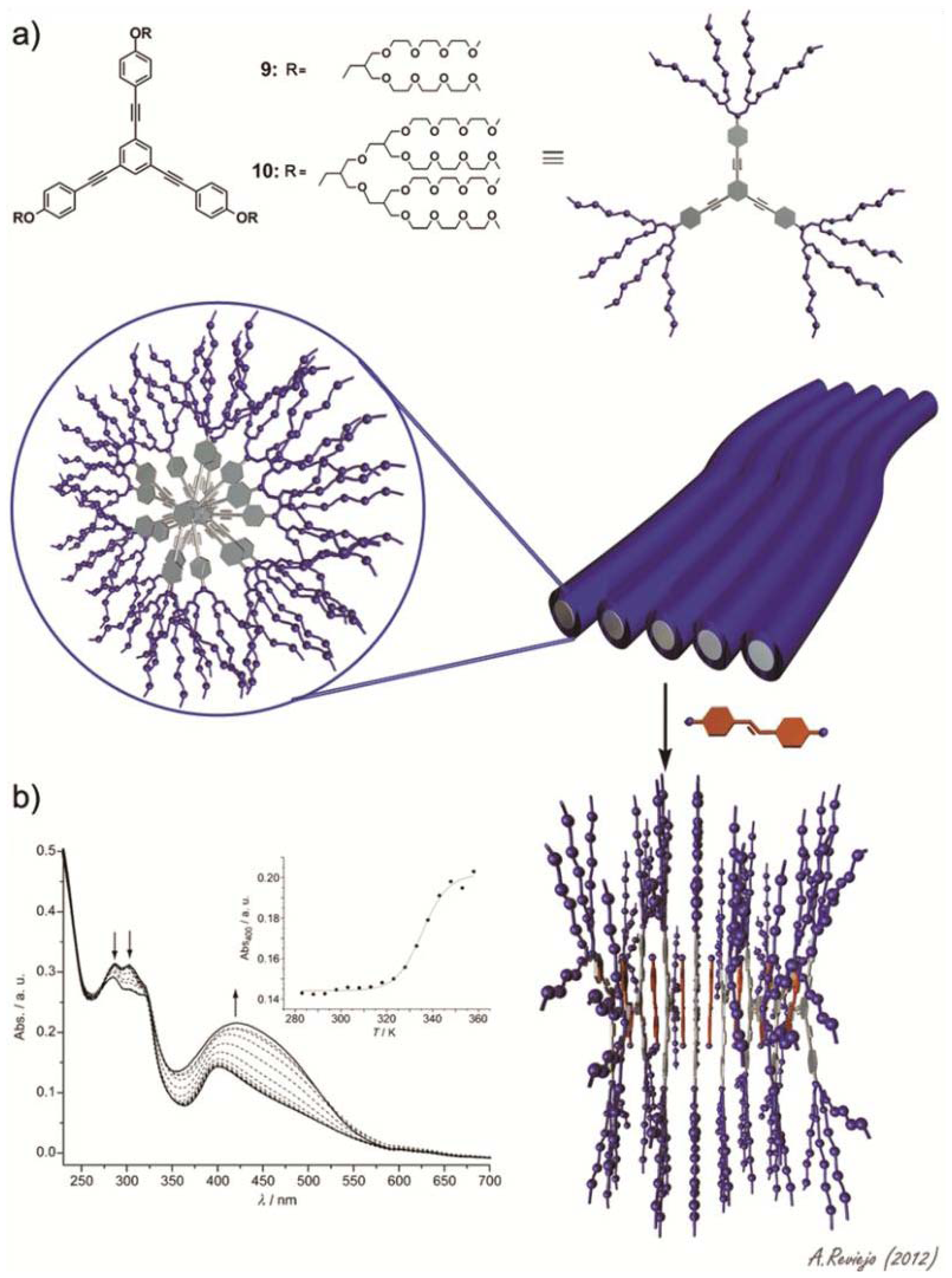
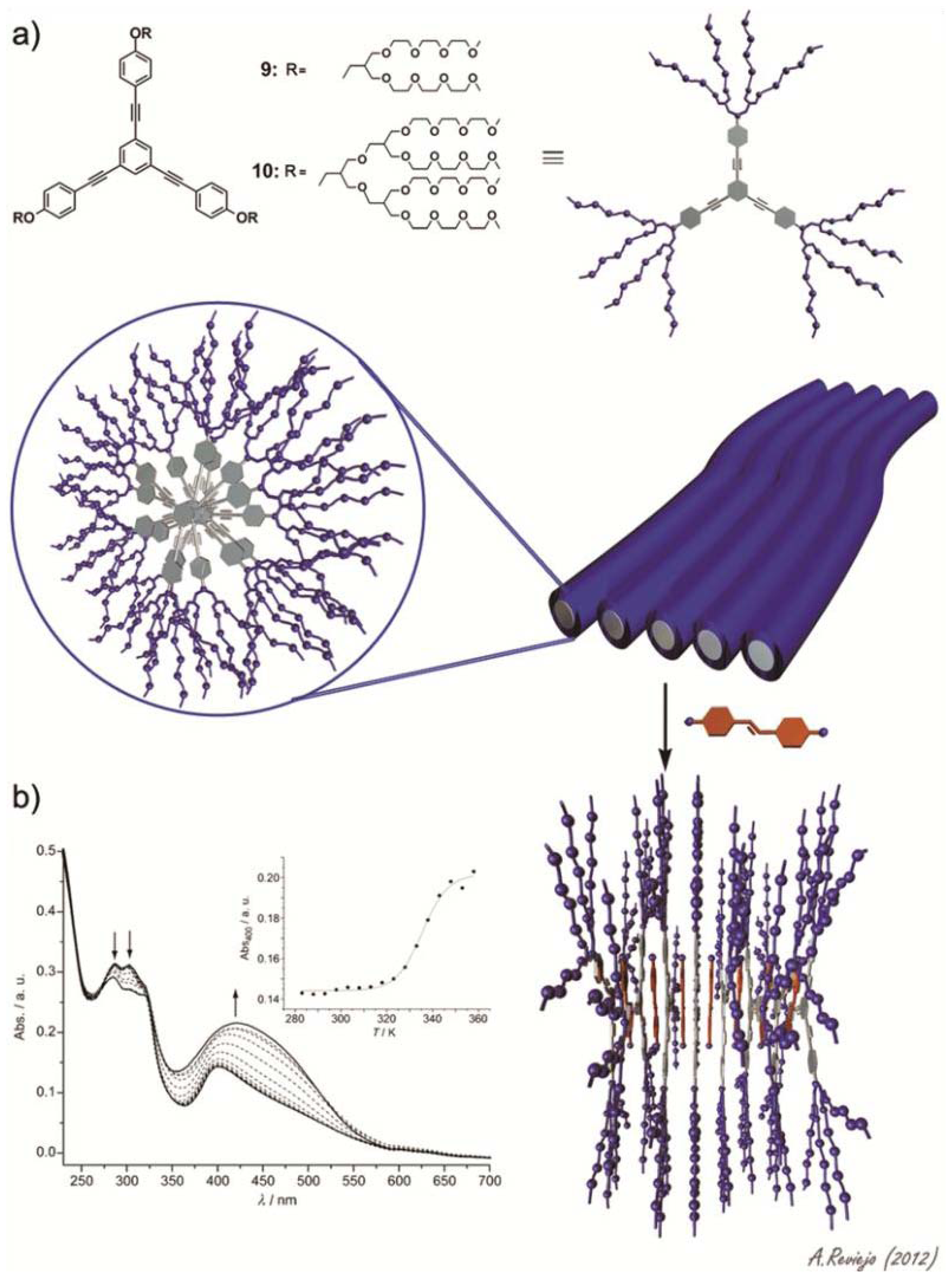
Sánchez and co-workers have investigated the self-assembly behavior of two amphiphilic triangular-shaped oligophenyleneethynylene (OPE) derivatives and their co-assembly with the hydrophobic dye Disperse Orange 3 [
21]. OPE building blocks are well-known, besides their remarkable optical and electronic properties, for their ability to self-assemble into a wide variety of supramolecular structures [
22–
31]. The amphiphiles feature a triangular OPE-based aromatic core bearing dendritic glycol chains on each terminal phenyl ring (compounds
9 and
10 in
Figure 5a). Concentration- and temperature-dependent experiments in acetonitrile demonstrate the isodesmic aggregation of both amphiphiles with binding constants of ~10
5 M
−1. In aqueous solutions, in which the propensity of the amphiphiles to aggregate is higher as a consequence of an increased hydrophobic effect, values of binding constant larger than 10
7 M
−1 were predicted. Dynamic (DLS) and static (SLS) light scattering experiments along with TEM imaging demonstrate the formation of nanometer-long fiber-like structures for both dyes. Interestingly, the authors investigated the ability of these fibrillar associates to encapsulate and release a hydrophobic dye, 4-(4-nitrophenyl-azo)aniline (Disperse Orange 3). To that end, an excess of the dye (10 equivalents) was added to an aqueous solution of either
9 or
10 followed by sonication for three hours and removal of the dye excess through filtration. The solutions were then investigated by means of temperature-dependent UV-Vis studies. At room temperature, the absorption corresponding to both the OPE fragments at ~300 nm and the hydrophobic dye (400–550 nm) can be observed, indicating an effective encapsulation of the dye by the fibrillar structures driven by the hydrophobic effect (
Figure 5b). Upon increasing temperature, the diagnostic band of the dye gradually increases in intensity, while the maxima at 284 and 303 nm decrease (
Figure 5b). The absence of isosbestic points indicates the lack of a defined stoichiometry in the co-assemblies. The spectral changes at 400 nm against temperature are clearly nonlinear (inset in
Figure 5b), which is diagnostic of a reversible dye intercalation-release process. This reversible encapsulation-release process anticipates the potential applicability of these systems in drug-delivery processes.
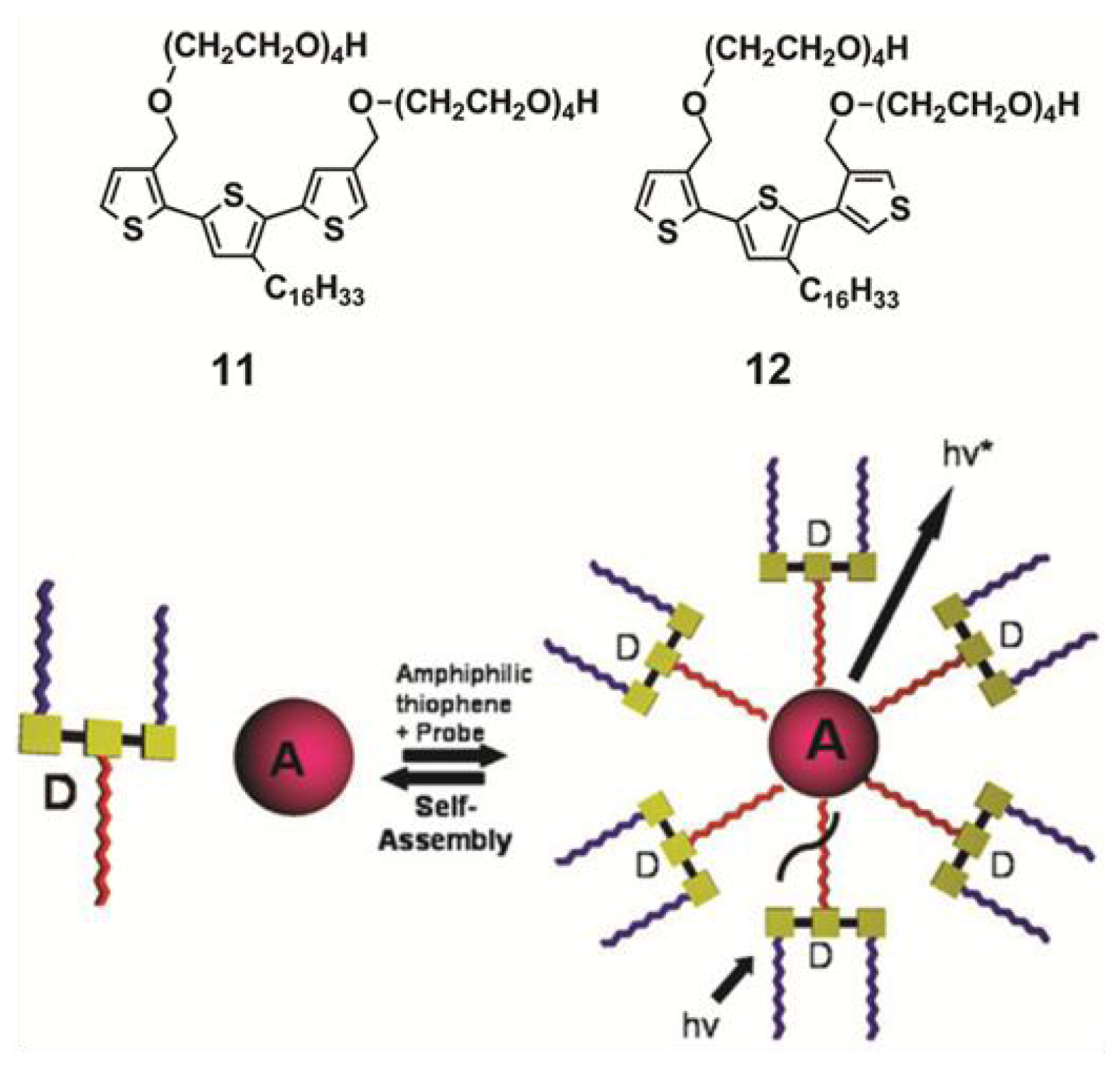
Van Esch
et al. have exploited a similar strategy for the encapsulation of suitable hydrophobic acceptor molecules into the interior of micellar associates based on donor amphiphiles, thus creating a stable self-assembled donor-acceptor ET-system [
32]. Two amphiphiles (compounds
11 and
12 in
Scheme 4) consisting of a hydrophobic terthiophene core substituted with a hydrophobic hexadecane chain in the central ring and hydrophilic tetraethylene glycol chains connected to the peripheral thiophene rings were synthesized. Above 1 mM, amphiphiles
11 and
12 self-assemble in water into spherical and cylindrical micelles, respectively, with diameters of 6 ± 2 nm for
11 and 21 ± 5 nm for
12, as demonstrated by cryogenic TEM and DLS experiments. More interestingly, the potential of the formed micelles to act as a host for hydrophobic chromophores creating an ET-system was investigated. To that end, two hydrophobic chromophores (Nile Red and Tetraphenylporphyrin) were chosen because of the overlap of their absorption spectra with the emission spectra of the amphiphilic terthiophenes. First evidence suggesting an encapsulation process is provided by the fact that both hydrophobic dyes become soluble in water upon addition of
11 or
12 above their critical micellar concentration. The encapsulation process was followed by fluorescence experiments. A simultaneous quenching of the emission of
11 or
12 upon addition of the corresponding acceptor molecules along with a gradual increase of the emission intensity of the acceptors support the creation of a self-assembling energy transfer system in water between the donor aggregates and the acceptor guest molecules, as depicted in
Scheme 4. More recently, Nandi and co-workers have exploited the gelation ability of melamine-quinazoniledione pairs to construct a similar light harvesting hydrogel with FRET emission upon encapsulation of riboflavin units [
33].
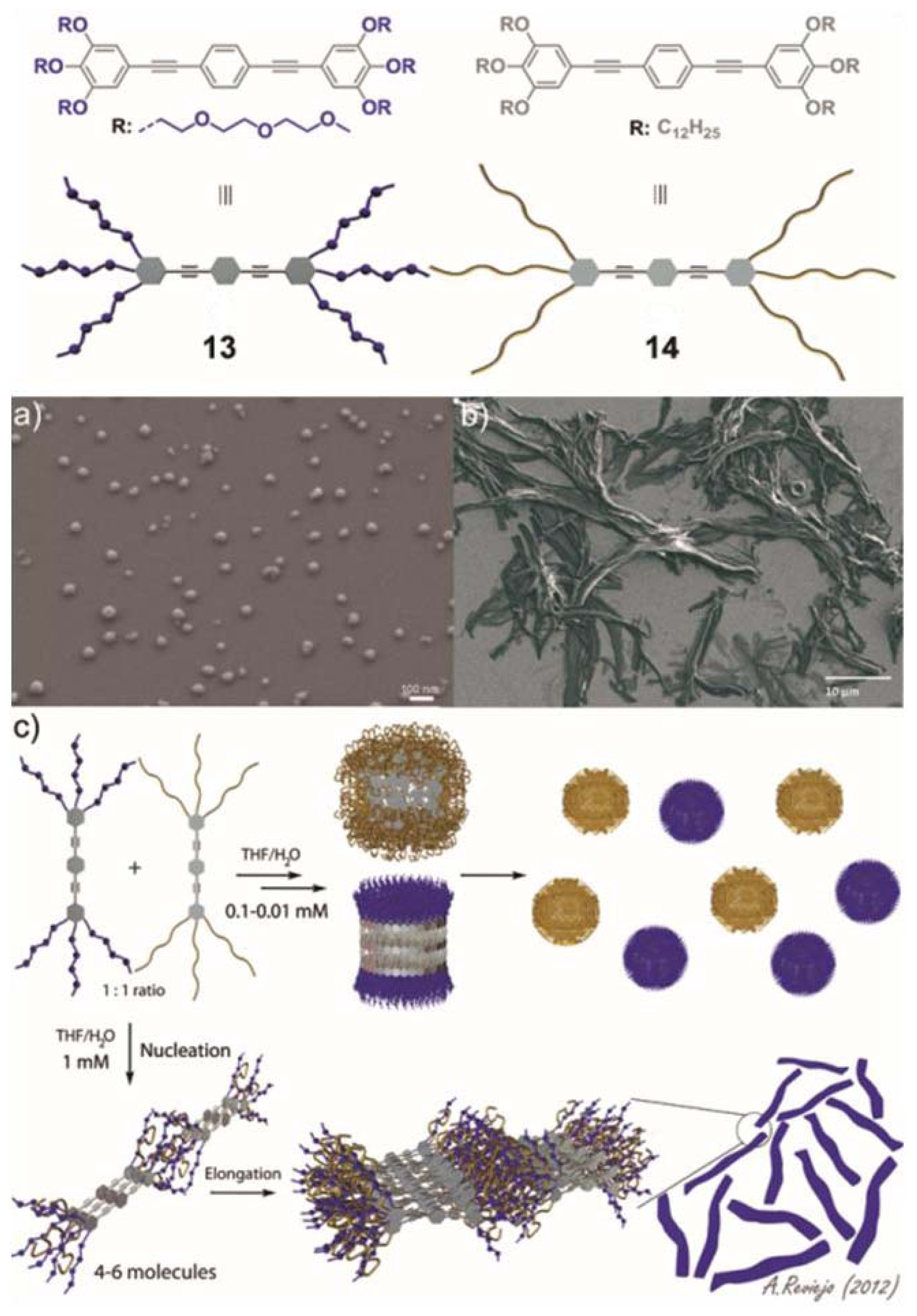
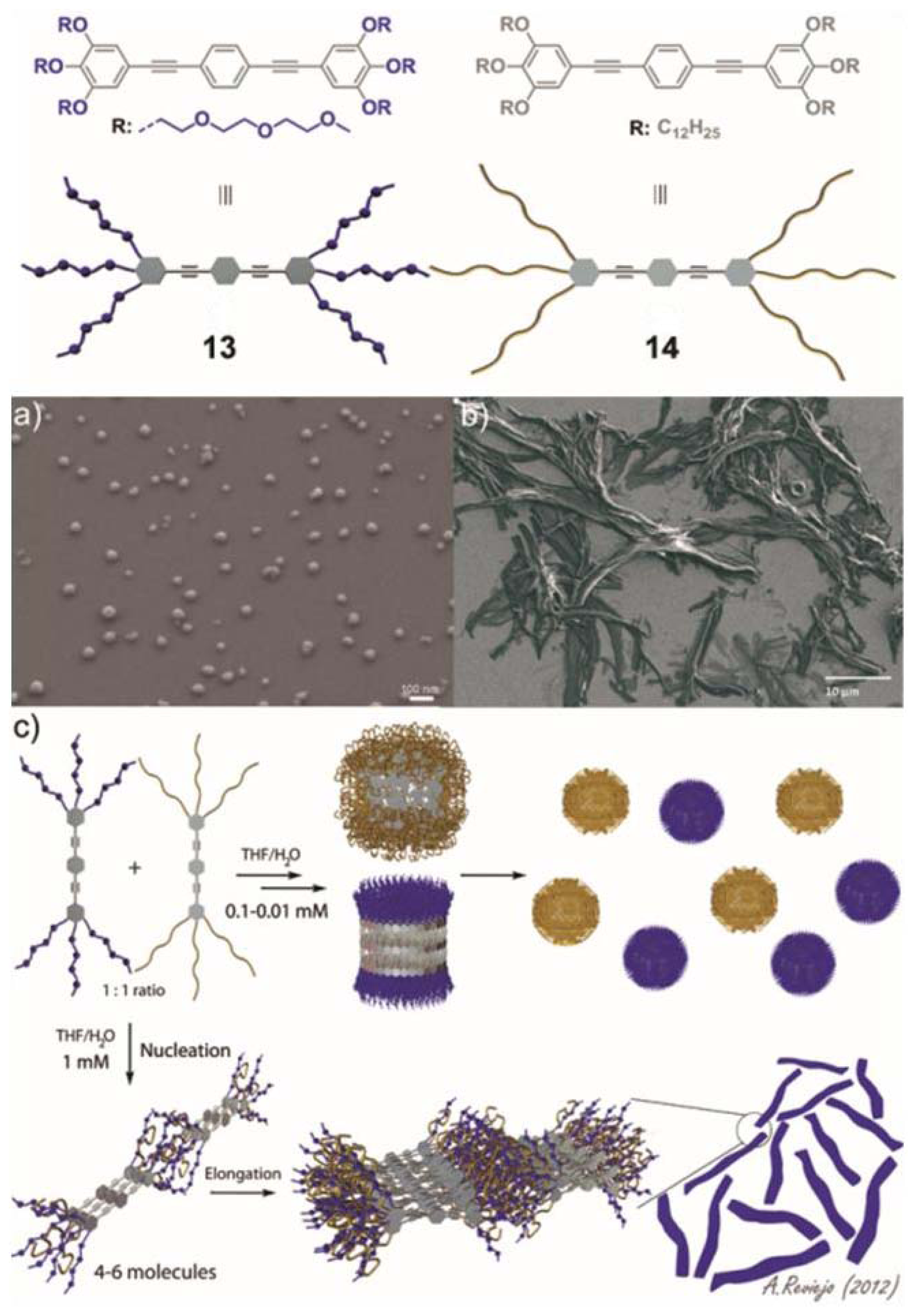
In our group, we are particularly interested in the behavior of small synthetic oligophenyleneethynylene (OPE)-based amphiphiles in multicomponent mixtures. Despite that the supramolecular properties of OPE derivatives are relatively well-understood, the ability of directing narcissistic or social self-sorting processes in these systems remains unexplored. Recently, we described the self-assembly of two structurally related OPE derivatives substituted with polar or nonpolar chains (
Figure 6) and their narcissistic
vs. social self-sorting behavior in aqueous media that can be tuned by concentration and solvent changes [
34].
The self-assembly mechanisms of OPEs
13 and
14 were studied by temperature-dependent UV-Vis experiments in THF/water mixtures suggesting a non-cooperative (isodesmic) self-assembly process between well-defined species. AFM and SEM imaging confirmed the appearance of spherical micelles with sizes between 3 nm and 10 nm for
13 and diameters ranging from 4 to 50 nm for
14, which were in agreement with the values observed by DLS. This remarkable propensity of both OPEs to aggregate was exploited to investigate their self-sorting behavior in aqueous mixtures driven by geometrical complementarity and the hydrophobic effect. Self-sorting experiments were carried out in 1:1 THF/water mixtures for an adequate comparison of the behavior of
13 and
14 in isolation and in their mixtures. At relatively low concentrations (0.01–0.1 mM) the 1:1 mixture of both compounds self-assembles into independent spherical micelles (
Figure 6a and model shown in
Figure 6c top). By simply raising the concentration to 1 mM the equilibrium is shifted towards the formation of social self-sorted systems, giving rise to micrometric-sized ribbon-like aggregates with widths of about 2 μM and lengths of several microns (
Figure 6b and model shown in
Figure 6c bottom). This unprecedented process is accompanied by a change in the aggregation mechanism from isodesmic to cooperative, as demonstrated by temperature-dependent UV-Vis experiments. The co-aggregates also showed lyotropic liquid crystal behavior—an emergent property of the mixture—that was not observed in any of the compounds in isolation. Our findings demonstrate that subtle changes in certain parameters such as solvent and concentration are efficiently capable of driving the system towards narcissistic or social self-sorting systems.
2.3. Self-Sorting Systems Driven by Interaction between Charged Groups and/or Hydrogen Bonding Complementarity
In contrast to most of the examples reported in the literature that exploit cooperative π–π and hydrophobic interactions to create self-sorted systems, less efforts have been devoted to the design and construction of extended co-aggregated systems based on other secondary interactions. A promising alternative approach in this regard makes use of strong interactions between positively and negatively charged groups. This strategy has the advantage that the introduction of charged groups enhances considerably the solubility in aqueous medium and, if polar substituents are also present, the influence of hydrophobic forces can be neglected. In this regard, Lemmers and co-workers have studied the co-assembly behavior in water of a triblock copolymer bearing a neutral hydrophilic core with two negatively charged terminal groups (
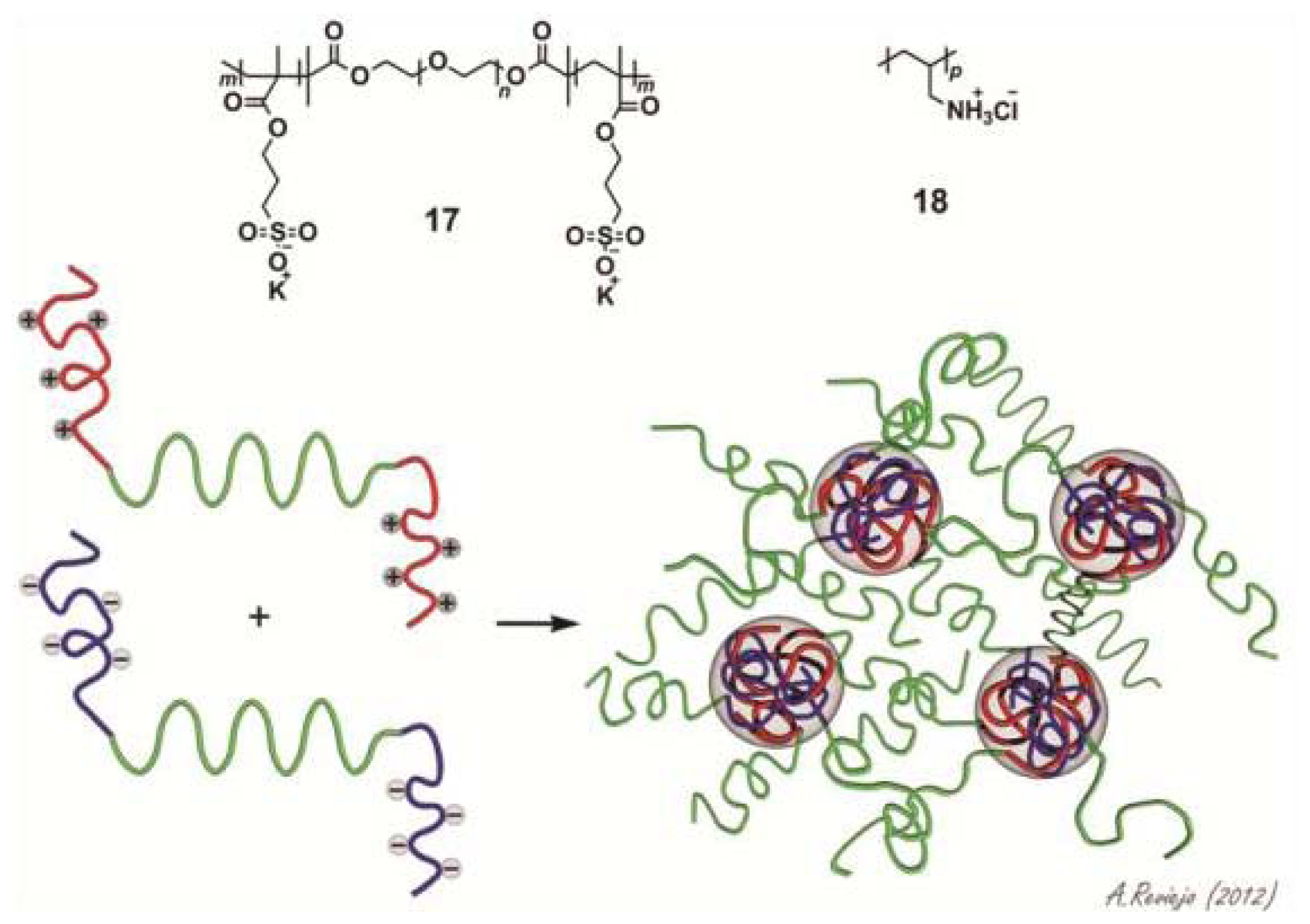
17) and a homopolymer PAH
160 bearing positively charged amino functions (
18) and investigated the influence of different variables (stoichiometry, temperature, ionic strength and pH variations) on the self-sorting processes [
38]. Diluted binary mixtures of
17 and
18 at different ratios were studied by DLS. An increase in the scattered light intensity was observed to reach a maximum when the positive and negative charges have been compensated, after which it decreases again. These results suggest the co-assembly of the two compounds forming flowerlike micelles with a hydrodynamic radius of ~20 nm (
Scheme 5). At higher concentration, a reversible gelation process takes place in which the micellar structures interconnect into a three-dimensional network. X-ray scattering (SAXS) measurements demonstrate that the micellar cores possess a radius of around 8 nm and an intermicellar distance of around 30 nm (
Scheme 5). Different studies revealed that these networks are responsive to changes in concentration, temperature, ionic strength and pH. For instance, rheometry investigations showed that the viscosity of the gel significantly grows with increasing concentration whereas an increase in the temperature showed the opposite effect. In addition, the influence of ionic strength was tested by addition of KCl. With a gradual increase of the salt concentration the scattered light intensity as well as the viscosity of the gel decreased indicating the dissolution of the gel. A similar effect is observed if the pH is raised. Overall, the ability of the hydrogels to respond to different external stimuli makes these materials interesting candidates for different applications in materials science or biomedicine.
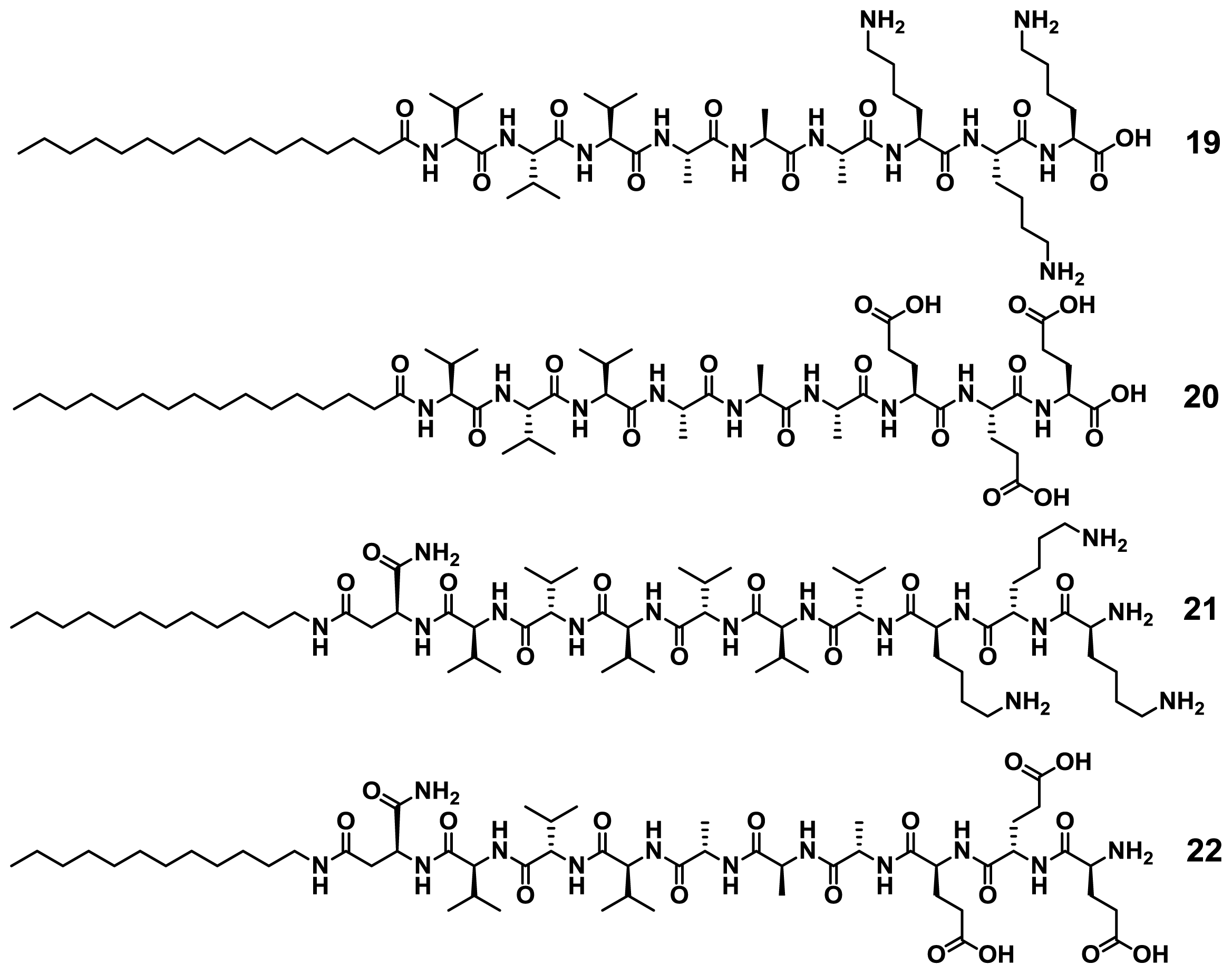
Stupp and co-workers have investigated the self-assembly of a wide variety of peptide amphiphiles (PA) into β-sheet-forming nanofibers driven by hydrogen bonding complementarity and secondary hydrophobic and/or electrostatic interactions [
39,
40]. Because of their high geometrical similarity and ability to aggregate in aqueous media, such molecules are expected to undergo narcissistic or social self-sorting processes when allowed to interact in multicomponent mixtures. In an elegant example, Stupp and co-workers synthesized four related PAs (
19–
22), two positively charged with a triple lysine sequence (
19 and
21) and two negatively charged with a triple glutamic acid sequence (
20 and
22) that can lead to co-assembled structures in which the peptide sequence has either normal or reverse polarity (
Chart 1) [
41].
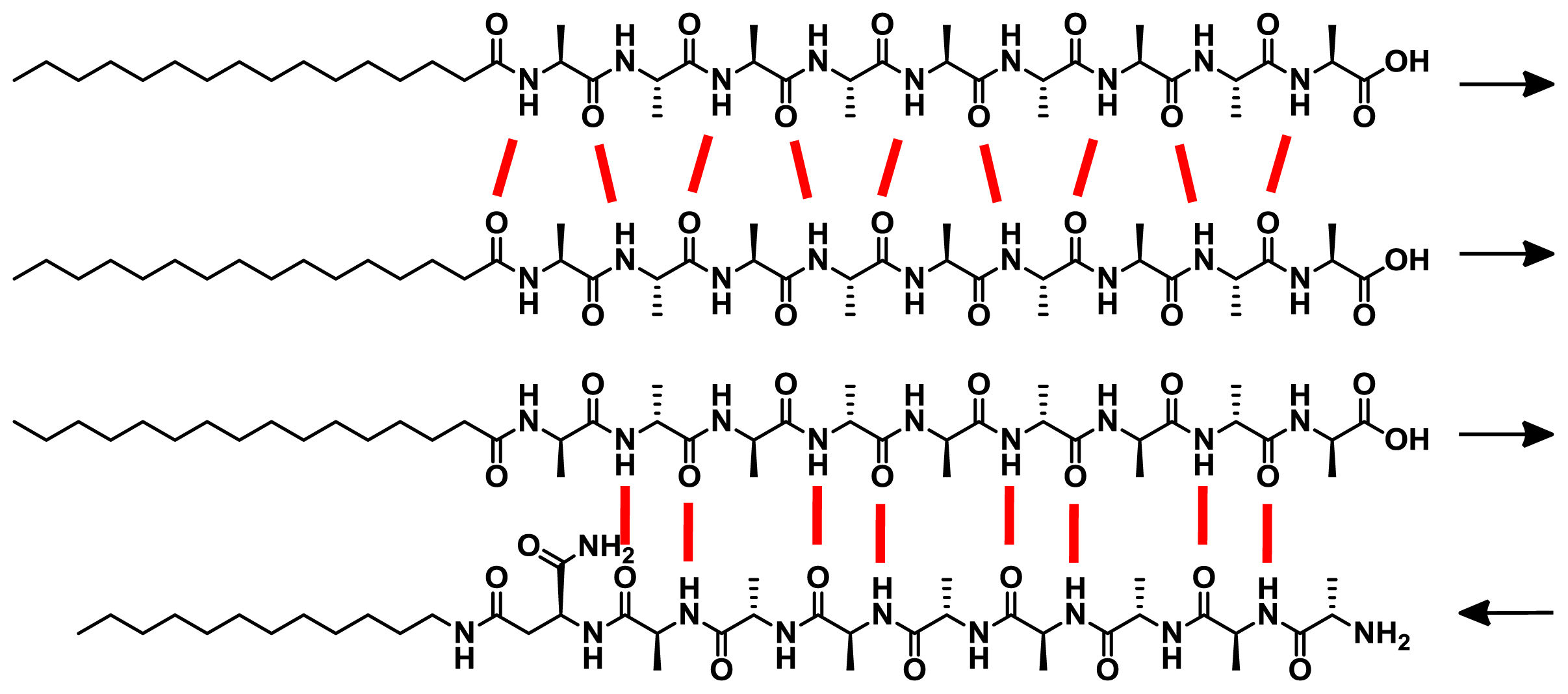
All peptides form β-sheet structures in isolation upon neutralizing the charges by changing the pH or adding calcium ions. Interestingly, when negatively charged peptides (
20 or
22) are mixed with complementary charged
19 or
21 residues, mixed nanofibers in which two molecules form a single aggregate structure are created (
Chart 2), as shown by circular dichroism (CD) studies. However, when two PAs of similar charge are mixed, less stable β-sheet structures or more disordered conformations are formed due to charge repulsion. Among the supramolecular structures formed upon co-assembly of PAs with identical (
19/20) or opposite polarities (
20/
21), it was shown that the arrangement of hydrogen bonding in the
20/
21 system is energetically more favorable than that in the
19/
20 arrangement, which results in an enhanced CD signal. This example clearly demonstrates that the appropriate sequence of hydrogen bonding groups can also efficiently drive co-aggregation processes in water when the molecules possess a good solubility in this medium. More recently, Nilsson and co-workers have investigated the self-sorting behavior in equimolar mixtures of enantiomeric amphipathic peptides [
42]. Fluorescence resonance energy transfer (FRET), CD, IR and TEM experiments revealed the formation of β-sheet fibrils formed by an alternating arrangement of
l- and
d-peptides also driven by hydrogen bonding complementarity in aqueous medium.
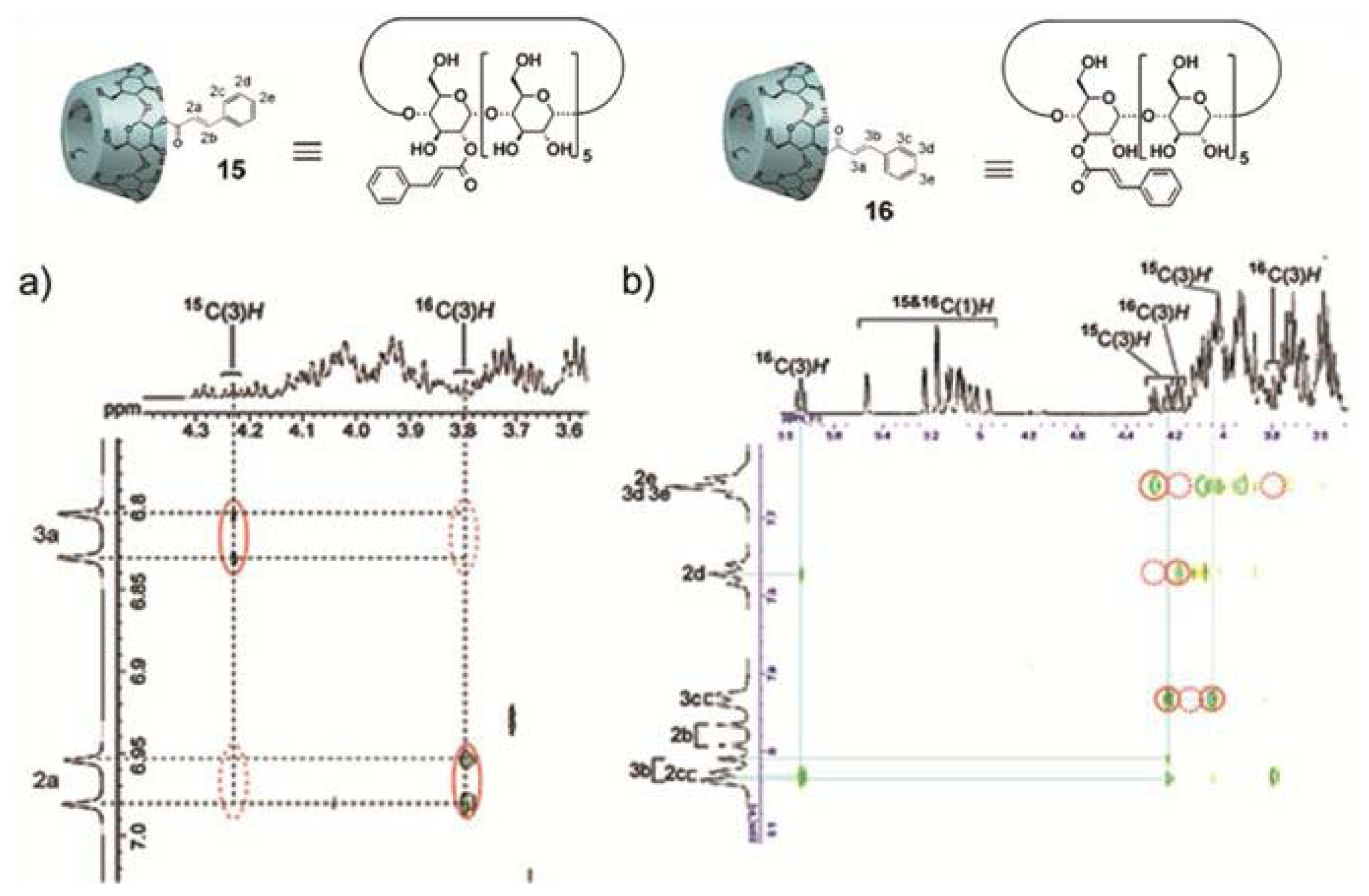
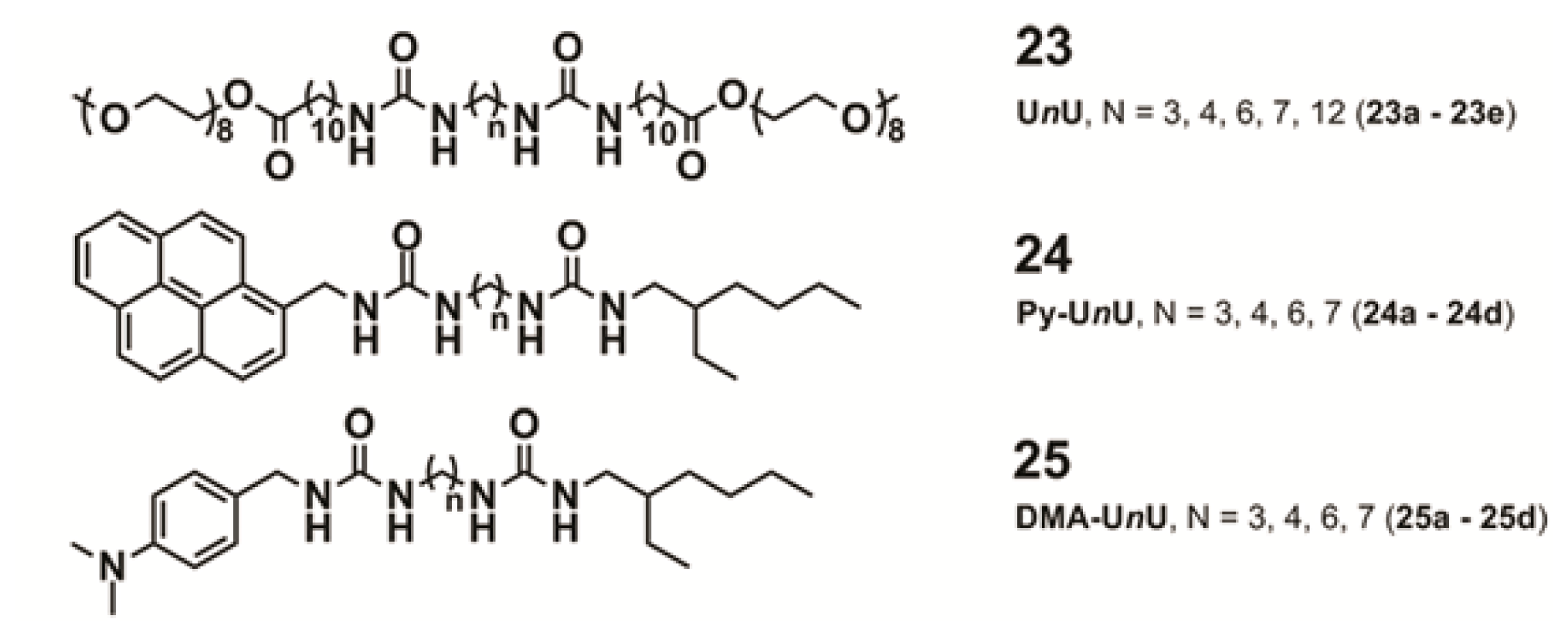
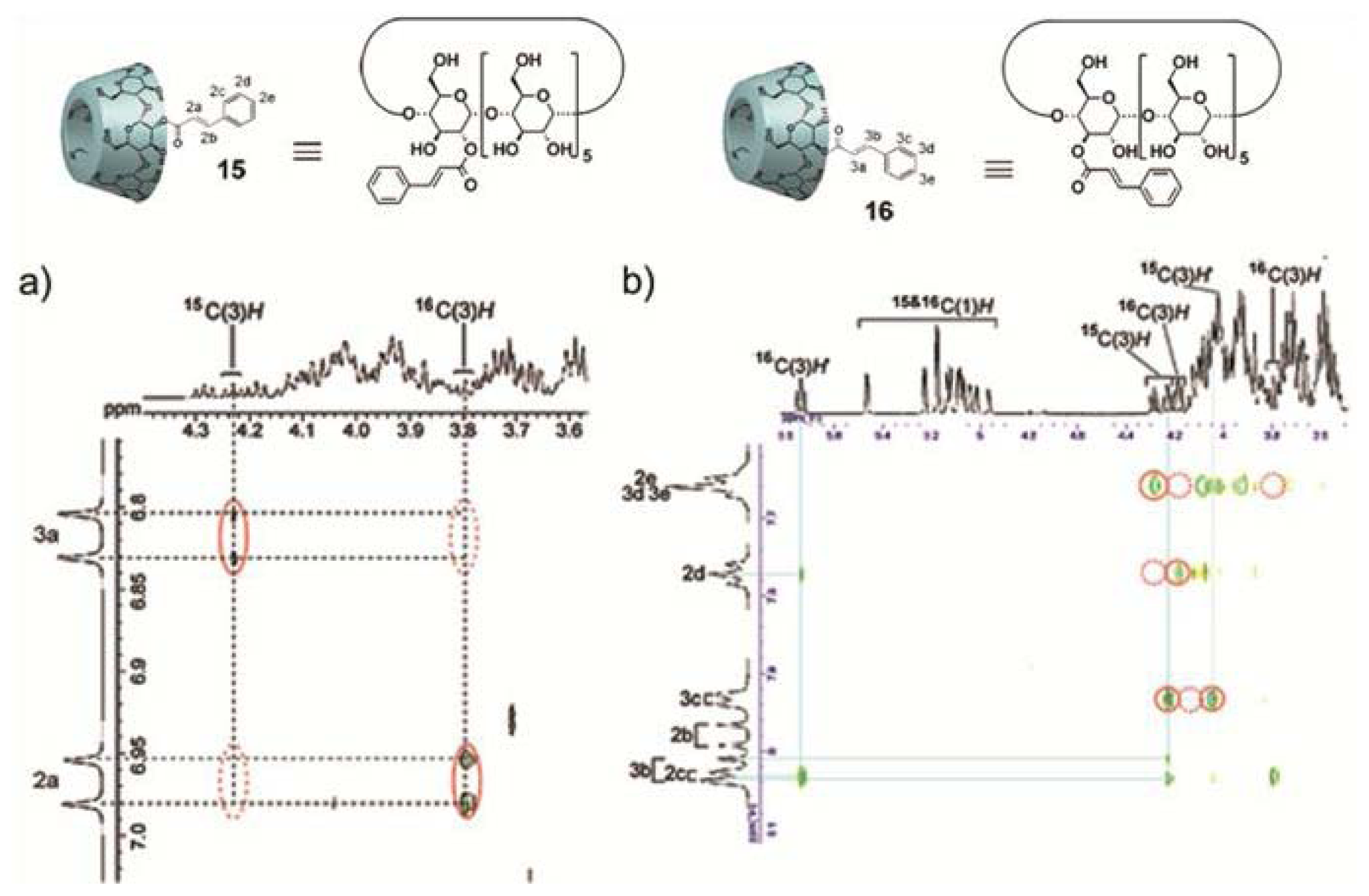
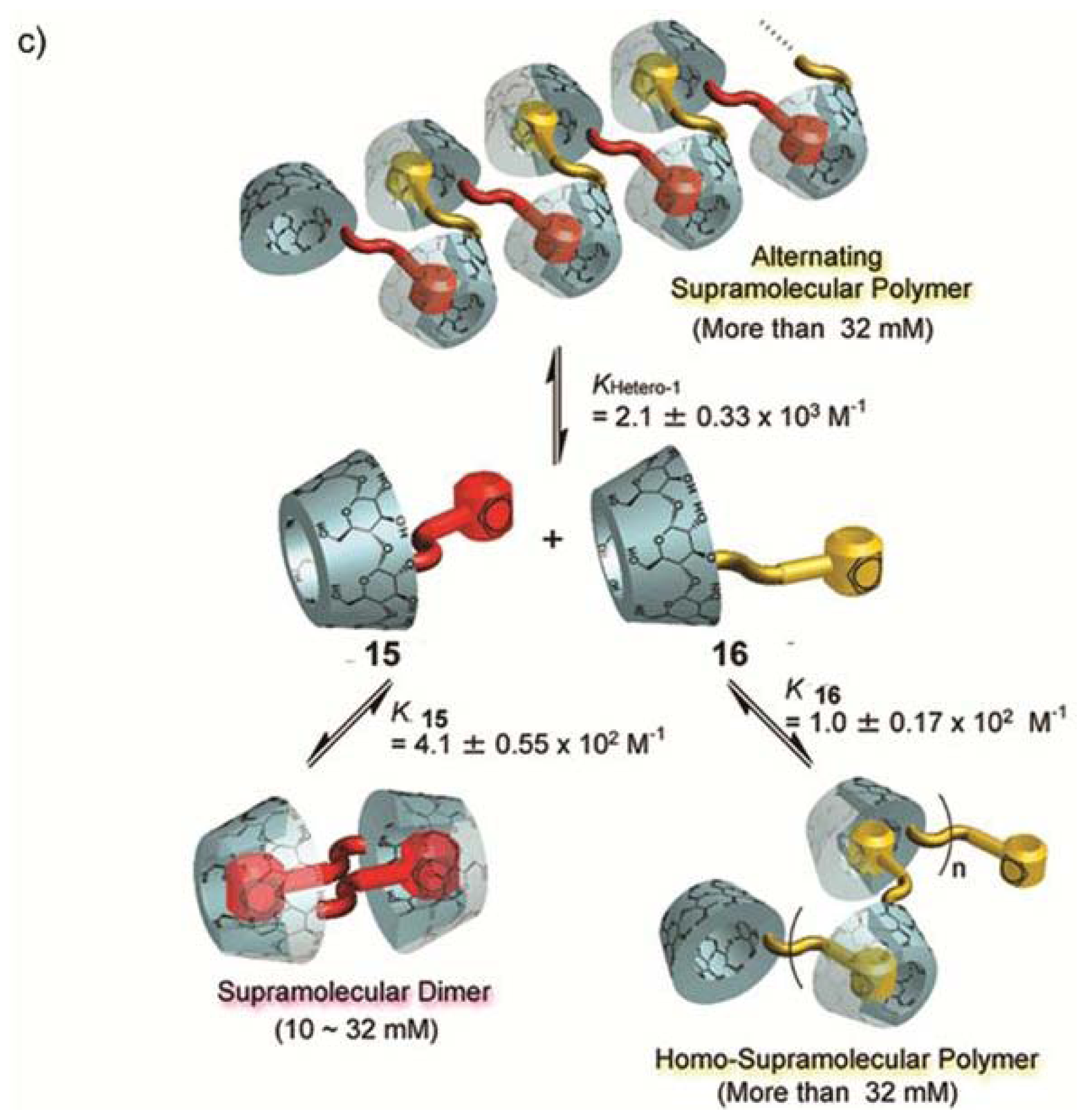
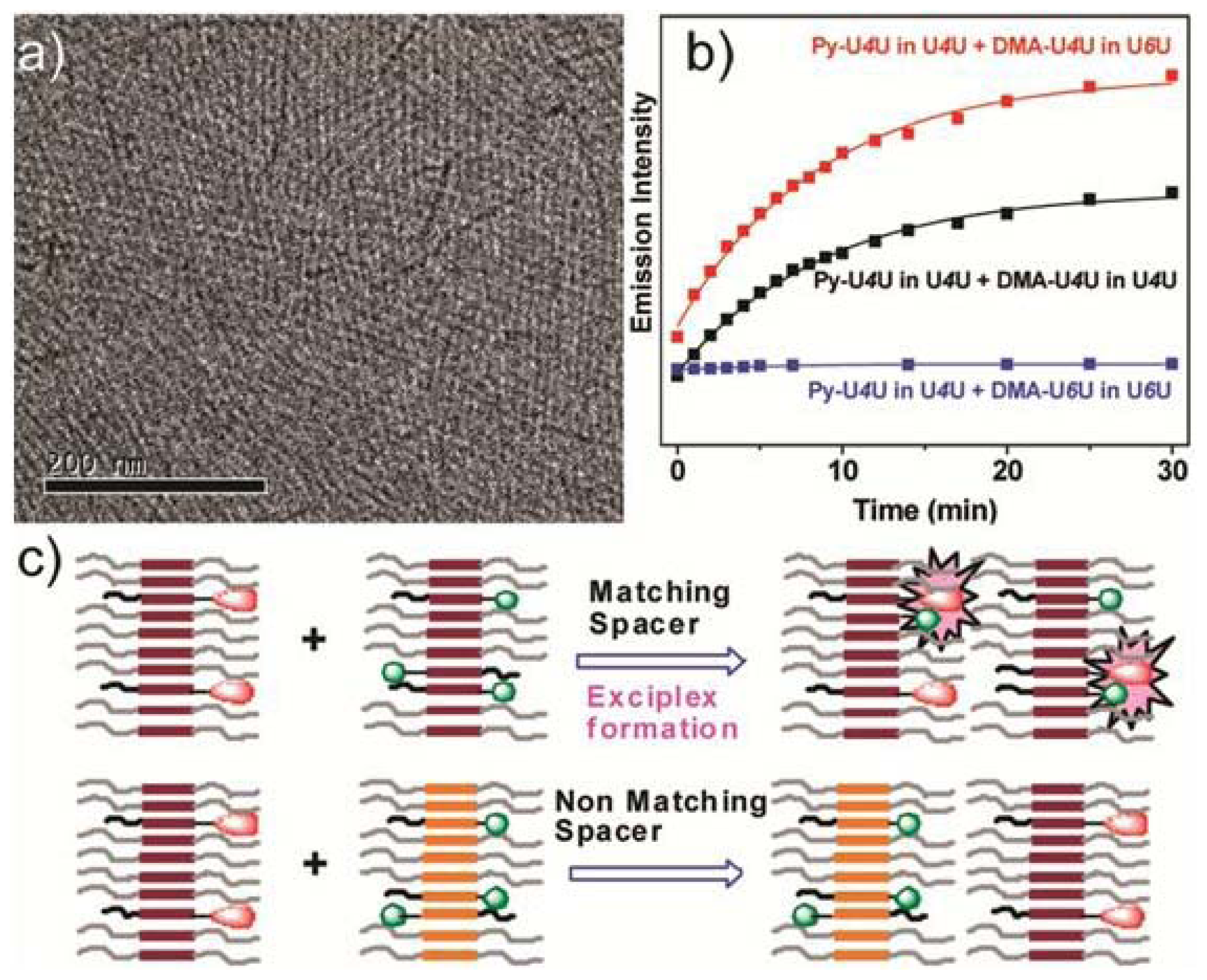
Perhaps one of the most illustrative examples highlighting the role of hydrogen bonding complementarity has recently been reported by Sijbesma and co-workers [
43,
44]. For their investigations, the authors synthesized a series of bisurea-based bolaamphiphiles (U
nU in
Figure 8) composed of two urea groups spaced by alkyl chains of variable length and similar derivatives substituted with fluorescent pyrene and dimethylamino groups (Py-U
nU and DMA-U
nU, respectively). These dyes were used as fluorescent probes to investigate whether a co-assembly process takes place, which would result in an exciplex band in the fluorescence spectra if both dyes are in spatial proximity. Bisurea bolaamphiphiles U3U (
23a), U4U (
23b), U6U (
23c) and U7U (
23d) form micrometer-long cylindrical micelles in water driven by hydrogen bonding between the urea groups, as revealed by Cryo-TEM images (
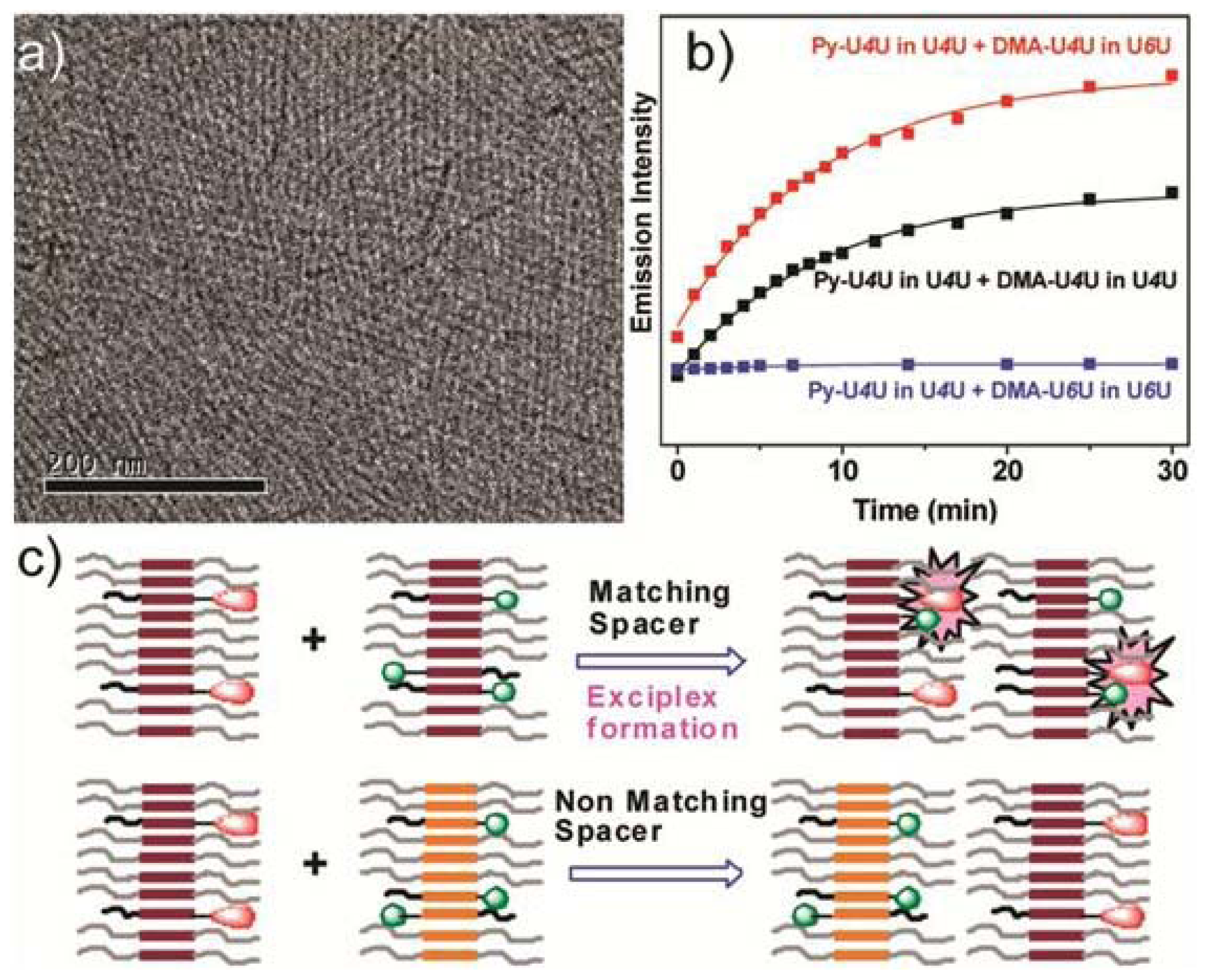
Figure 8a). The self-sorting behavior of U4U (
23b) was studied by fluorescence spectroscopy using the pyrene probe Py-U4U (
24b) and the dimethylaniline probe DMA-U4U (
25b). Upon mixing U4U (
23b) with either Py-U4U (
24b) (up to 0.01 eq) or DMA-U4U (
25b) (up to 0.15 eq), a co-assembly process driven by hydrogen-bonding complementarity takes place in which the pyrene or DMA units are randomly dispersed in the micelles of U4U (
23b) and no exciplex band is expected. However, upon mixing both solutions (U4U (
23b)-Py-U4U (
24b) and U4U (
23b)-DMA-U4U (
25b)), a band at 520 nm appeared in the fluorescence spectrum, indicating the molecular contact between Py- and DMA-USU upon co-assembly of three compounds into micellar structures (
Figure 8b). Interestingly, when nonmatching micelles U4U (
23b) (containing 0.01 eq Py-U4U (
24b)) and U6U (
23c) (containing 0.15 eq DMA-U6U (
25c)) were mixed, no exciplex band was observed, indicating that the probes form separate “narcissistic” micelles due to the fact that the distance between the urea groups in U4U (
23b)-Py-U4U (
24b) is smaller than that in U6U (
23c)-DMA-U6U (
25c) and the hydrogen bonding pattern in the co-assembly cannot take place (
Figure 8c). In an even more complex experiment, the authors created up to three separate micellar aggregates by mixing bisurea derivatives with different alkyl spacers. These interesting findings hold potential for the construction of a large number of assemblies that coexist independent from one another in the same medium, providing access to exciting nanomaterials that can mimic the operation of natural systems.
2.4. Enzyme-Responsive Self-Sorted Systems
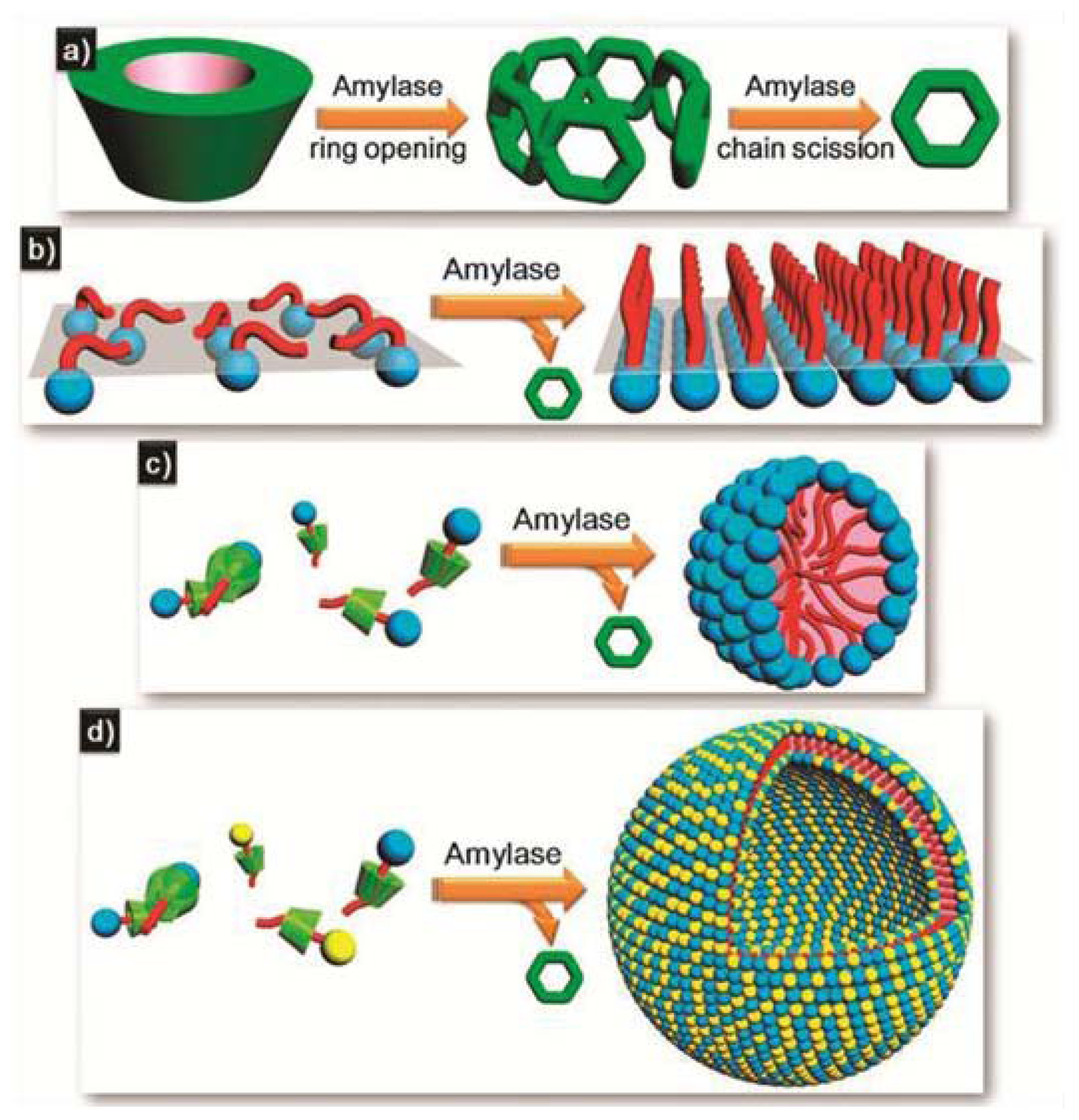
A new approach that has gained considerable attention in recent years is the proposal to interface supramolecular objects with biological systems such as enzymes. Enzyme-responsive supramolecular assemblies are a class of smart nanomaterials that undergo macroscopic transitions when triggered by selective catalytic actions of enzymes [
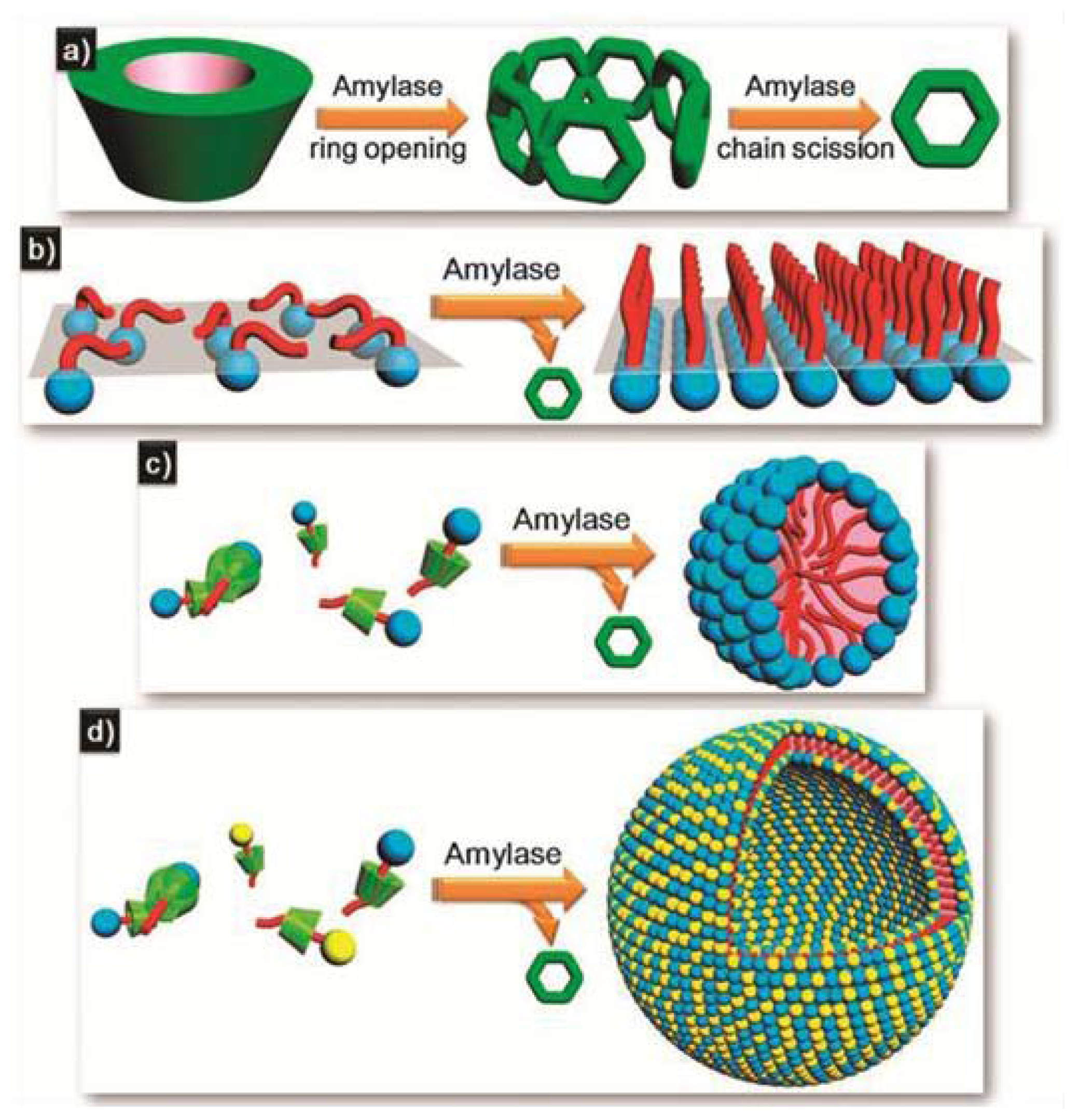
45]. Examples include bulk phase transitions (sol-to-gel and gel-to-sol) or the transformation, generation or disassembly of supramolecular structures induced by enzyme addition. Recently, Huang and co-workers have developed a host-guest strategy to construct enzyme-triggered self-sorted systems on the basis of surfactant-cyclodextrin (CD) complexes and α-amylase [
46]. CDs are well-known for their ability to encapsulate the hydrophobic moieties of surfactants into their cavities driven by hydrophobic interactions. On the other hand, the enzyme α-amylase has the ability to cleave the α-1,4 linkages between the glucose units of starch molecules including CDs, which ultimately leads to the degradation of the CD units, and the concomitant release and subsequent self-assembly of the surfactant molecules (
Figure 9a). The influence of β-CD on the organization of different non-ionic, ionic or zwitterionic surfactants was investigated. Prior to the addition of β-CD, the surfactants are forming adsorption monolayers. However, an excess of β-CD induces the breakdown of the monolayers, which can be again recovered upon subsequent treatment with α-amylase. This enzyme degrades the CD units and triggers the release and subsequent organization of the surfactant molecules into monolayers (
Figure 9b). A similar effect is observed for the surfactant tetradecyl dimethylammonium propane sulfonate (TDPS). This molecule self-assembles into micelles in aqueous medium, as revealed by DLS studies. After the addition of an excess of β-CD, the solution scattering is notably reduced, which implies the disassembly of the micelles upon complexation with the CD units. Different dosages of amylase were applied to the β-CD/TDPS co-assembly, the scattering of which is observed to become constant after 24 h. The size distributions calculated by DLS for different dosages are nearly the same and demonstrate the formation of spherical micelles with an average hydrodynamic radius of 2 nm (
Figure 9c). Finally, the authors investigated the influence of CD and amylase on vesicular structures formed upon co-assembly of two surfactants, sodium dodecyl sulfate (SDS) and DEAB (dodecyl triethyl ammonium bromide). The SDS/DEAB solution (0.4:0.6 mM) is dominated by vesicular assemblies, which can be degraded upon addition of 8 mM of β-CD. The significant enhancement observed in the scattering of the mixture SDS/DEAB/β-CD upon addition of amylase indicates the formation of large aggregates of several hundreds of nanometers. Cryo-TEM studies reveal the formation of hollow spherical aggregates ranging from 100 to 300 nm, as depicted in
Figure 9d. The enzyme α-amylase can therefore trigger the self-assembly of CD-surfactant self-sorted systems into different organized arrangements by degradation of the CD molecules and subsequent release and rearrangement of the surfactant molecules.
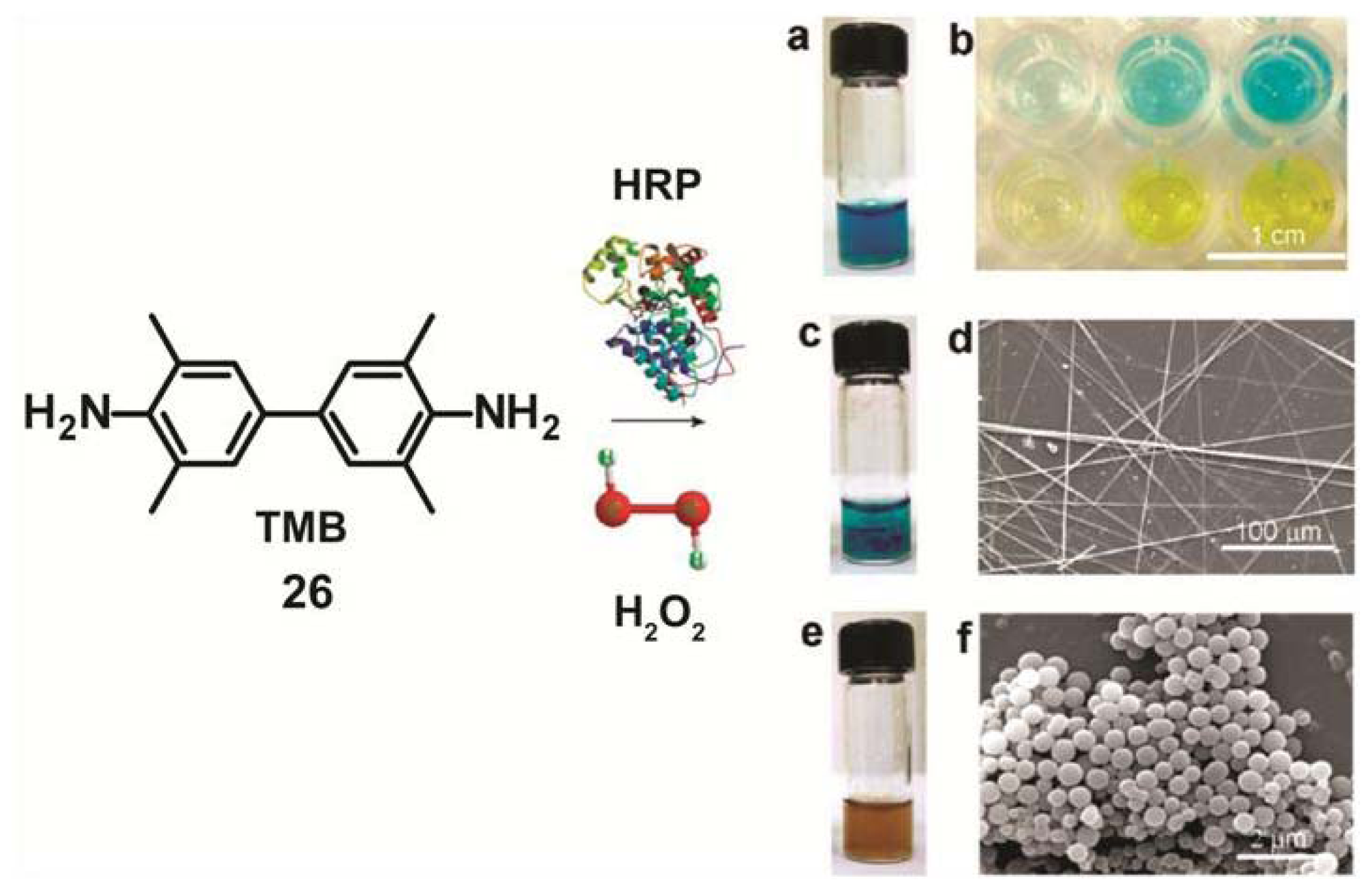
A similar strategy has been recently developed by Gao and co-workers for the self-assembly and transformation of nanostructures induced by enzymes in a tetramethylbenzidine (TMB)/horseradish peroxidase (HRP)/H
2O
2 system [
47]. TMB is the most common chromogenic substrate for HRP used as color reagent in enzyme-linked immunosorbent assay (ELISA).The oxidation of TMB (
26) by HRP/H
2O
2 at pH value of 4 produces a blue color that can change to yellow when the reaction is stopped by addition of sulfuric acid (
Figure 10a,b). However, when the blue solution reacts for 24 h at room temperature without stopping it with sulfuric acid, nanobelt-like structures with lengths up to 1 mm are formed (
Figure 10c,d). More interestingly, if an additional amount of the enzyme HRP is added to this mixture, the color of the system turns brown and ultimately the precipitation of uniform spherical nanoparticles with diameters of ~120–800 nm takes place (
Figure 10e,f). These findings indicate that this reaction enables the generation and transformation of different co-assembled structures under control of enzymatic kinetics. The concentration of the enzyme HRP was observed to play a more significant role than the concentration of TMB and H
2O
2 in the self-sorting phenomena in this system. These enzyme-responsive materials may find application in smart circuitry and bioresponsive devices.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}